Article Text

Abstract
Background This phase Ib study evaluated the safety, clinical activity, pharmacokinetics, and pharmacodynamics (PD) of emactuzumab (anti-colony stimulating factor 1 receptor monoclonal antibody (mAb)) in combination with selicrelumab (agonistic cluster of differentiation 40 mAb) in patients with advanced solid tumors.
Methods Both emactuzumab and selicrelumab were administered intravenously every 3 weeks and doses were concomitantly escalated (emactuzumab: 500 to 1000 mg flat; selicrelumab: 2 to 16 mg flat). Dose escalation was conducted using the product of independent beta probabilities dose-escalation design. PD analyzes were performed on peripheral blood samples and tumor/skin biopsies at baseline and on treatment. Clinical activity was evaluated using investigator-based and Response Evaluation Criteria In Solid Tumors V.1.1-based tumor assessments.
Results Three dose-limiting toxicities (all infusion-related reactions (IRRs)) were observed at 8, 12 and 16 mg of selicrelumab together with 1000 mg of emactuzumab. The maximum tolerated dose was not reached at the predefined top doses of emactuzumab (1000 mg) and selicrelumab (16 mg). The most common adverse events were IRRs (75.7%), fatigue (54.1%), facial edema (37.8%), and increase in aspartate aminotransferase and creatinine phosphokinase (35.1% both). PD analyzes demonstrated an increase of Ki67+-activated CD8+ T cells accompanied by a decrease of B cells and the reduction of CD14Dim CD16bright monocytes in peripheral blood. The best objective clinical response was stable disease in 40.5% of patients.
Conclusion Emactuzumab in combination with selicrelumab demonstrated a manageable safety profile and evidence of PD activity but did not translate into objective clinical responses.
Trialregistration number NCT02760797.
- clinical trials as topic
- tumor biomarkers
- tumor microenvironment
- translational medical research
- myeloid-derived suppressor cells
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- clinical trials as topic
- tumor biomarkers
- tumor microenvironment
- translational medical research
- myeloid-derived suppressor cells
Background
Intratumoral immune infiltrates consist of a variety of lymphoid and myeloid cells. Among others, tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) are thought to contribute to the escape from immune surveillance and checkpoint blockade therapy.1 Therefore, for adaptive immune responses to persist, tumor-infiltrating lymphocytes (TILs) must overcome a suppressive cytokine milieu and mechanisms of tolerance propagated by TAMs to successfully attack cancer cells. TAMs have been described as either proinflammatory and anti-tumor (M1) or tumor-promoting (M2), depending on their functional phenotype and cytokine profile.2 3 Anti-colony stimulating factor 1 receptor (CSF-1R) signaling supports recruitment, differentiation and maintenance of immune suppressive macrophages within the tumor.4 CSF-1R-positive TAMs in the tumor microenvironment (TME) correlate with immune dysfunction and increased immune suppression,5 as well as poor prognosis in a variety of solid malignancies such as breast, ovarian and pancreatic cancers, as well as in leiomyosarcoma, mantle cell and Hodgkin lymphoma.6–10
Cluster of differentiation 40 (CD40) is a co-stimulatory molecule of the tumor necrosis factor receptor superfamily. CD40 is expressed on antigen-presenting cells (APCs), for example dendritic cells and myeloid cells, with B cells showing highest expression, and is critical for their activation and proliferation.11 12 Activation of CD40 is triggered by binding of CD40 ligand (CD40L or CD154), which is primarily expressed by activated T cells, but can also be found on B cells and platelets.12 The CD40/CD40L axis is essential to initiate effective T-cell-specific immune responses. CD40 expression on the surface of APCs greatly increases their antigen presentation and co-stimulatory capacity, resulting in a more effective activation of cytotoxic T cells even in the absence of T helper cell signals.11–13 Hence, the main mode of action of agonistic cluster of differentiation 40 (aCD40) monoclonal antibodies (mAbs) may be the induction of increased tumor-specific antigen presentation via activation of APCs, resulting in the production of cytotoxic T cells directed against the tumor.14–16 B cells are also highly dependent on CD40-CD40L interaction, which stimulates formation of germinal centers, immunoglobulin isotype switching, somatic hypermutation and the production of plasma cells and memory B cells.12 It is thought that the activation of B cells by aCD40 mAbs might contribute to their antitumor effect as shown in vitro.17
Selicrelumab is a fully human and selective aCD40 mAb, which has been tested clinically as monotherapy,18 19 and together with tremelimumab20 or chemotherapy.21 22 Emactuzumab is a recombinant, humanized mAb directed against CSF-1R expressed by macrophages.4 23 Emactuzumab has been studied clinically as monotherapy in patients with diffuse-type giant cell tumor and demonstrated a profound anti-tumor effect through interference of the CSF-1/CSF-1R axis.4 24 It has also been investigated as a single agent and in combination with chemotherapy in solid tumors.25 In preclinical experiments, it was shown that the inhibition of CSF-1R signaling by various CSF-1R inhibitors acts as an amplifier of aCD40-regulated general immune activation via TAM reprogramming and T-cell activation.26–28 In the presence of CSF-1R signaling inhibition, the reprogramming of TAMs resulted in their hyperactivation and concomitant release of proinflammatory cytokines and chemokines. Despite their short-lived nature, these macrophages were sufficient to reinvigorate effective anti-tumor T-cell responses in transplanted tumors. Notably, transplanted tumor models that did no longer respond to immune checkpoint blockade remained sensitive to the myeloid-directed combination therapy.26 27
Here, we report for the first time the clinical and immunological impact of simultaneously activating CD40 and blocking CSF-1R in patients with advanced or metastatic solid tumors.
Methods
Study design
This was a phase Ib, open-label, non-randomized, dose escalation, multicenter study investigating the safety, pharmacokinetics (PK), pharmacodynamics (PD) and clinical activity of emactuzumab in combination with selicrelumab. The study was designed as a dose-escalation phase (Part 1) using the product of independent beta probabilities dose-escalation (PIPE) design and an extension part to further evaluate the highest dose level tested for the combination. The PIPE design recommended the dose combination at the latest estimate of the maximum tolerated dose combinations contour (product of), with the greatest uncertainty as to whether it lied above or below the MTDCC.29 As per sponsor decision, the extension phase (Part 2) has not been executed.
The study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki in six centers in Belgium, France and the USA.
Patients
For Part I, patients had a histologically confirmed diagnosis of locally advanced and/or metastatic triple-negative breast cancer (TNBC), ovarian cancer, pancreatic cancer, gastric cancer, colorectal cancer (CRC), melanoma (MEL) and mesothelioma, all of which were not amenable to standard treatment. Eligible patients were ≥18 years of age, had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1, and had adequate hematology, blood chemistry, as well as renal and liver function. Patients continued treatment until disease progression, unacceptable toxicity or consent withdrawal.
Study drug administration
Emactuzumab was administered as an intravenous infusion every 3 weeks (q3w) over 90 min if well-tolerated. The initial starting-dose of emactuzumab was 500 mg and the top dose was predefined as 1000 mg (ie, the optimal biological dose of emactuzumab monotherapy).25 The starting dose of emactuzumab was selected to assure systemic exposure above 90% target saturation, to warrant the estimated target exposure of 100 μg/mL for macrophage depletion and for being safe and tolerable as shown for treatment with emactuzumab as monotherapy.25 30 The selicrelumab infusion was started at least 1 hour after the emactuzumab infusion had ended. Selicrelumab was administered as an intravenous infusion q3w with a starting dose of 2 mg and a top dose of 16 mg (ie, the MTD of selicrelumab monotherapy.18 The starting dose of selicrelumab was selected to assure appropriate risk mitigation for systemic side effects (including infusion-relatedreaction (IRR), transaminase elevations and thrombocytopenia) that were seen for monotherapy with doses of ≥4 mg.18 19
Tumor response and safety
Investigator-based tumor response assessment using Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST V.1.131 was conducted at screening and every 6 weeks thereafter). Safety assessments included physical (ECOG performance status and vital signs) and laboratory examinations and ECG. Adverse events (AEs) were defined according to the Common Terminology Criteria for Adverse Events, V.4.0 (CTCAEv4.0). A dose-limiting toxicity (DLT) was defined as a related ≥grade 3 AE occurring during the assessment window of 6 weeks from the first administration of emactuzumab and selicrelumab (online supplemental material).
Supplemental material
Pharmacokinetic and immunogenicity assessments
Blood samples for PK assessments were taken for cycle 1 at predose, end of infusion, 5 hours, 24 hours, 96 hours, 168 hours, 264 hours, 336 hours and 432 hours post infusion for emactuzumab, and at predose, 15 min after infusion start, end of infusion, 4 hours, 8 hours, 10 hours, 24 hours, 30 hours, 48 hours and 168 hours post infusion for selicrelumab.
Analysis of emactuzumab in human serum was done with an ELISA assay as described previously.30 Similarly, analysis of selicrelumab was done using an ELISA assay (online supplemental material).
Non-compartmental analysis was conducted on emactuzumab and selicrelumab serum concentration data.
Blood samples for anti-dug antibody (ADA) assessments were taken predose for cycle 1 to 7. ADAs against emactuzumab and selicrelumab were measured in human serum using an ELISA assay (online supplemental material).
Pharmacodynamic assessments
Paired tumor and skin biopsies were collected during screening and on day 1 of cycle 2. Analyzes included immunohistochemistry (IHC) staining and quantitative assessment of TAMs and TILs in paired-tumor biopsies and dermal macrophages in paired skin biopsies.
Consecutive 2.5 μm thickness sections of formalin-fixed paraffin-embedded tumor tissues were stained with the following in-house developed IHC assays using Ventana Benchmark XT or Discovery Ultra automated platforms (Ventana Medical Systems, Tucson, Arizona, USA): Ki67/CD8, CD163/CD68, CSF1R and FOXP3.
For Ki67/CD8 assay, the RUO Discovery Universal procedure on Discovery Ultra was used. The tissue sections were treated with Cell Conditioner 1 for 64 min and then incubated in primary antibody CD8 (SP239, 1:12.5, Spring Biosciences, for 32 min at 38°C). Bound CD8 antibody was detected with UltraMap anti-rabbit alkaline phosphatase (AP) secondary antibody and Discovery Yellow detection kit (Ventana Medical Systems). Subsequently, after heat denaturation, slides were incubated in primary antibody Ki67 (30–9, RTU, Ventana Medical Systems) for 8 min at 38°C. Bound primary antibody was detected with hapten-linked multimer anti-rabbit hydroquinone (HQ) and anti-HQ horseradish peroxidase secondary antibody, followed by Discovery Purple detection kit (Ventana Medical Systems).
For CD163/CD68 assay, the XT IHC DS oDAB-uRed v4 procedure on Benchmark XT was used. The tissue sections were treated with Cell Conditioner 1 for 32 min and then incubated in primary antibody, CD163 (MRQ-26, RTU, Ventana Medical Systems), for 16 min at 37°C. Bound primary antibody was detected by the OptiView DAB IHC detection kit (Ventana Medical Systems). Subsequently, slides were incubated in primary antibody CD68 (KP-1, RTU, Ventana Medical Systems) for 16 min at 37°C. Bound primary antibody was detected by the UltraView Universal AP Red detection kit (Ventana Medical Systems).
For CSF1R assay, the XT Optiview DAB IHC v4 procedure on Benchmark XT was used. The tissue sections were treated with Cell Conditioner 1 for 32 min and then incubated in primary antibody CSF1R (clone 1A10, RTU, Roche) for 32 min at 37°C. Positive staining was detected with OptiView DAB detection kit (Ventana Medical Systems).
For FOXP3 assay, the XT Optiview DAB IHC v4 procedure on Benchmark XT was used. The tissue sections were treated with Cell Conditioner 1 for 32 min and then incubated in primary antibody FOXP3 (236A-E7, 1:100, Abcam) for 60 min at 37°C and positive staining was detected with OptiView DAB detection kit (Ventana Medical Systems).
All sections were counterstained with hematoxylin II (Ventana Medical Systems) for 8 min, bluing solution for 8 min and then dehydrated and cover-slipped.
For all assays, appropriate negative and positive controls were performed.
Algorithms for the detection and classification of IHC-stained objects on a whole slide basis were written in MATLAB. The results of automated digital slide analysis on the IRIS platform were reported for tumor areas as follows: Ki67−/CD8+, Ki67+/CD8+, total CD8+ and FOXP3 cell densities (number of cell counts per 1 mm2), CD68+/CD163+, CD68+/CD163−, total CD163+ and CSF1R percent of area coverage (area coverage in relation to the whole tumor area).
Peripheral whole blood was taken at baseline (prior to cycle 1) and then for each cycle at various time points prior to and several hours after study drug infusion. Analyzes included monocytes and circulating lymphocyte populations in peripheral blood (eg, CD4 and CD8 T cells, and B cells). Multicolor flow cytometry assays were performed for immunophenotyping analysis of circulating lymphocyte populations in whole blood. The main lymphocyte populations (CD3, CD4 and CD8 T cells, NK and B cells) were evaluated by assay 1 (Cyto-Chex) using the following antibodies: CD3_FITC (Clone SK7); CD4_BV510 (SK3); CD8_APC (SK1); CD19_BV421 (HIB19); CD16_PE (B73.1); CD56_PE (NCAM16.2); CD14_APC-H7 (MφP9); CD45_PerCP-Cy5.5 (2D1) (BD Biosciences, San Jose, California, USA). The proliferation and activation status of T cells were evaluated by assay 2 (sodium heparin) using the following antibodies: CD3_BV421 (SK7); CD4_FITC (SK3 +SK4); HLA-DR_PE (G46-6); CD8_PerCP-Cy5.5 (RPA-T4); Ki67_AF647 (B56) (BD Biosciences). The profile and activation status of dendritic cells and monocyte subsets were evaluated by assay 3 (Cyto-Chex) using the following antibodies: CD45_APC-H7 (2D1); CD16_BV421 (3G8); CD123 BV605 (7G3); HLA-DR_FITC (L243); CD80_PE (L307.4); CD83_PE-Cy5 (HB15e); CD86_APC (2331/FUN-1) (BD Biosciences); CD11c_APC-AF700 (BU15) (Beckman Coulter); CD14_PE-Cy7 (HCD14) (BioLegend); lineage exclusion: CD3 BV510 (UCHT1); CD19_BV510 (SJ25C1); CD56_BV510 (NCAM16.2) (BD Biosciences).
Briefly, 100 µL of whole blood were aliquoted and added to appropriately labeled tubes. Surface and intracellular markers were subsequently stained by adding fluorochrome-conjugates mAbs according to the specific panel; to facilitate population identification, respective isotype/‘fluorescence minus’ conditions were included as control tubes in panels 2 and 3. Tubes were incubated 20 min in the dark at room temperature. After washing with phosphate-buffered saline bovine serum albumin buffer, the pellets were resuspended in 100 µL of cold cell-staining buffer and kept at 4°C to 8°C till acquisition on a BD FACSCanto II (8 colors/3 lasers) or BD FACSCanto (10 colors/3 lasers) instruments (BD Biosciences) using BD FACSDiva software V.8.0 for analysis (BD Biosciences). Application settings were checked daily using cytometer performance checks with CS&T beads (BD Biosciences). Compensation values for each fluorochrome were determined using panel-specific antibodies. A minimal of 50,000 CD3+ events were acquired per sample. For flow cytometry measurements from dendritic cells and monocytes populations, it was considered as lower limit of quantification any measurement where the number of cell events acquired for the specific population was less than 50.
Absolute cell counts for each reportable were calculated indirectly by dual platform using white blood cell count (WBC) during the clinical trial, and applying the following calculation: Absolute cell count=% of captured events (%lymphs)×lymphocyte: leukocyte ratio×WBC. In the case, CD14+ absolute cell counts were calculated by applying the following calculation: Absolute cell count=% of captured events (%leuks)×WBC.
Statistical considerations
All patients who received at least one dose of study medication were included in the safety and efficacy analyzes. Descriptive statistics were used for demographics, safety, PK, PD and clinical activity.
Progression-free survival (PFS) was defined as the time from study treatment initiation (cycle 1, day 1) to the first occurrence of documented disease progression based on RECIST V.1.1 according to the investigator’s assessment or death from any cause, whichever occurred first. For patients who did not have documented progressive disease or death during the study, PFS was censored at the day of the last tumor assessment.
Results
Patients
Patient demographics and baseline characteristics are presented in table 1. Altogether, 37 patients were enrolled into six dose cohorts for dose escalation (figure 1). Women constituted 56.8% of patients, with a median age of 58 years. CRC (29.7%), ovarian cancer and pancreatic carcinoma (18.9% for both) were the most prevalent tumor types.

Flow diagram of patient enrolment and emactuzumab/selicrelumab dose cohorts a One patient was planned to be dosed in the 1000 mg emactuzumab/12 mg selicrelumab cohort but died in the screening phase due to a biopsy-related hemorrhage.
Baseline patient demographics and characteristics
Overall, 33 patients (89.2%) discontinued the study due to progressive disease and one patient (2.7%) each for pregnancy, an AE (grade 4 IRR related to selicrelumab), physician’s decision and withdrawal of consent.
Safety
Patients received a median of three treatment cycles (range: 1 to 16) of emactuzumab and selicrelumab. Three patients experienced DLTs. One patient of the selicrelumab 8 mg+emactuzumab 1000 mg cohort (grade 4 IRR considered resolved and grade 3 proteinuria considered unresolved); one patient of the selicrelumab 12 mg+emactuzumab 1000 mg cohort (grade 4 IRR considered resolved); and one patients of the selicrelumab 16 mg+emactuzumab 1000 mg cohort (grade 3 IRR considered resolved). The MTD was not reached based on the incidence of DLTs and the previously defined top doses of 1000 mg of emactuzumab (ie, the optimal biological dose (OBD) of emactuzumab monotherapy25 and 16 mg of selicrelumab (ie, the MTD of selicrelumab monotherapy).18
Thirty-six out of 37 patients (97.3%) experienced at least one AE during the study (table 2). The most frequently AEs reported (in ≥50% of patients) were IRR (75.7%; related: 75.7%) and fatigue (54.1%; related: 37.8%). Most IRRs were considered related to selicrelumab only and only 9 of 55 events (16%) were considered related to both study drugs. The most common IRR signs and symptoms were chills (67.6%) and fever (43.2%). Although the majority of AEs was of grade 1 or 2 of severity, 23 of 37 patients (62.2%) experienced at least one AE of grade 3 or 4, irrespective of relationship, and 16 patients (43.2%) had grade 3 or 4 events related to study treatments. No grade 5 AEs were reported. The most common grade ≥3 AEs (occurring in ≥10% of patients) were: fatigue (13.5%; related: 8.1%), hypertension (13.5%; related: 5.4%) and anemia (10.8%; related: 2.7%). Grade ≥3 lab abnormalities were frequently seen, including increase in creatinine phosphokinase (CPK; 16.2%; related: 16.2%), hypophosphatemia (13.5%; related: none), increase in aspartate aminotransferase (AST; 10.8%; related: 5.4%) and increase in gamma glutamyl transferase (10.8%; related: 2.7%). There was no dose-dependency in the incidence of grade≥3 AEs except the occurrence of IRRs with increasing doses of selicrelumab with an overall incidence of grade ≥3 IRRs of 8.1%. One patient deceased during the screening period before receiving the first study drug dose due to a biopsy-related hemorrhage.
Summary of adverse events of any grade and of grade ≥3 adverse events irrespective of relationship and events related to study treatments
Pharmacokinetic and immunogenicity analysis
Emactuzumab systemic exposure (area under the curvelast) showed a greater than dose-proportional increase from 500 mg to 1000 mg of emactuzumab, accompanied by an overall trend of total clearance decline (range: 346 to 752 mL/day), indicating that the elimination of emactuzumab was predominantly target-mediated following 500 mg and 750 mg q3w dosing in combination with selicrelumab (online supplemental figure 1 and table 1). Systemic exposure was above the 90% target saturation and resembles the one for monotherapy with emactuzumab as shown previously.30
Serum levels for selicrelumab were below the limit of detection (ie, 50.0 ng/mL) following administration of a 2 mg dose but were detectable following doses of 4 mg and 8 mg for some patients. PK profiles showed serum concentrations increasing more than dose proportionally from 4 mg to 8 mg doses (online supplemental figure 2a). High variability was observed in selicrelumab systemic exposure across the different doses used in the dose escalation (online supplemental figure 2b and table 2). Overlapping systemic exposure across the different selicrelumab doses was also observed, nevertheless the exposure resembles the one seen for monotherapy with selicrelumab.19
No patients had detectable positive ADA titers to emactuzumab. Five patients had detectable ADA titers to selicrelumab. Of these, two patients (both in the selicrelumab 2 mg/emactuzumab 500 mg cohort) reached maximum titers of 16 on cycle 4 day 1 and cycle 3 day 1, respectively. The remaining three patients with anti-selicrelumab titers (one in the selicrelumab 4 mg/emactuzumab 750 mg cohort, one in the selicrelumab 8 mg/emactuzumab 1000 mg cohort and one in the selicrelumab 16 mg/emactuzumab 1000 mg cohort) did not show rises of anti-selicrelumab titers above 1.
Antitumor activity
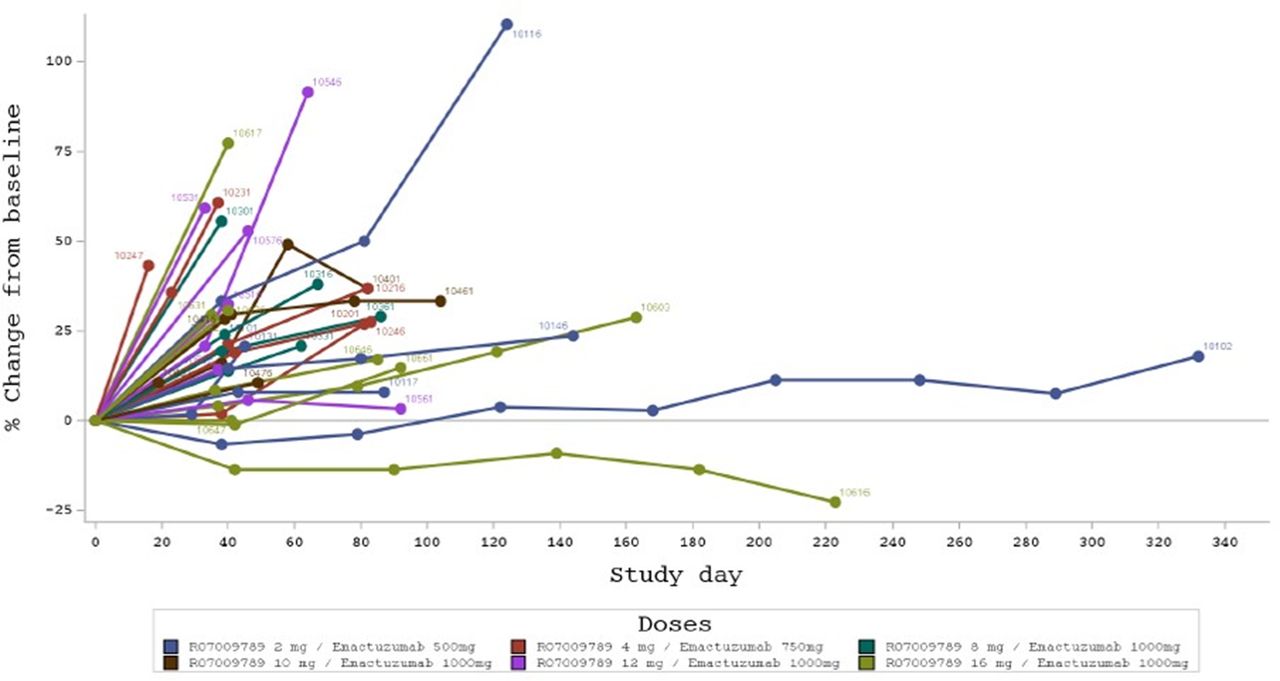
No objective clinical responses according to investigator-based RECIST assessment were observed (figure 2). The best overall confirmed response was stable disease (SD) in 15 patients (40.5%) across all dose levels tested. Two patients showed a tumor decrease during treatment, one patient in the 500 mg emactuzumab/2 mg selicrelumab cohort (maximum target lesion decrease by 9%) and one in the 1000 mg emactuzumab/16 mg selicrelumab cohort (maximum target lesion decrease by 23%); however, both patients discontinued the study for progressive disease. Overall, the median PFS across all dose levels tested was 42 days (90% CI: 40 to 58) with no discernible differences between dose cohorts.

Spider plot indicating the percentage change from baseline in sum of target lesion diameters per patient.
Pharmacodynamic analyses
Administration of emactuzumab plus selicrelumab resulted in a transient, dose-dependent trend for CD3+CD19+ B-cell reduction in peripheral blood (figure 3A,B). Concurrently, there was a transient increase of CD3+CD8+Ki67+ T cells with a peak on day 8 after administration of study treatments confirming published data19 (figure 3C,D). A trend for a dose-dependent increase in CD3+CD8+Ki67+ T cells can only be assumed at the highest selicrelumab dose tested (16 mg). As shown for emactuzumab monotherapy,25 we observed a peripheral CD14dimCD16bright monocyte reduction in the periphery after emactuzumab and selicrelumab treatment (figure 4). However, the kinetic of the monocyte reduction appeared to be different compared with what has been observed for emactuzumab single-agent treatment.30 The peripheral blood analysis showed a less pronounced reduction kinetic of CD14dimCD16bright monocytes with increasing doses of selicrelumab.
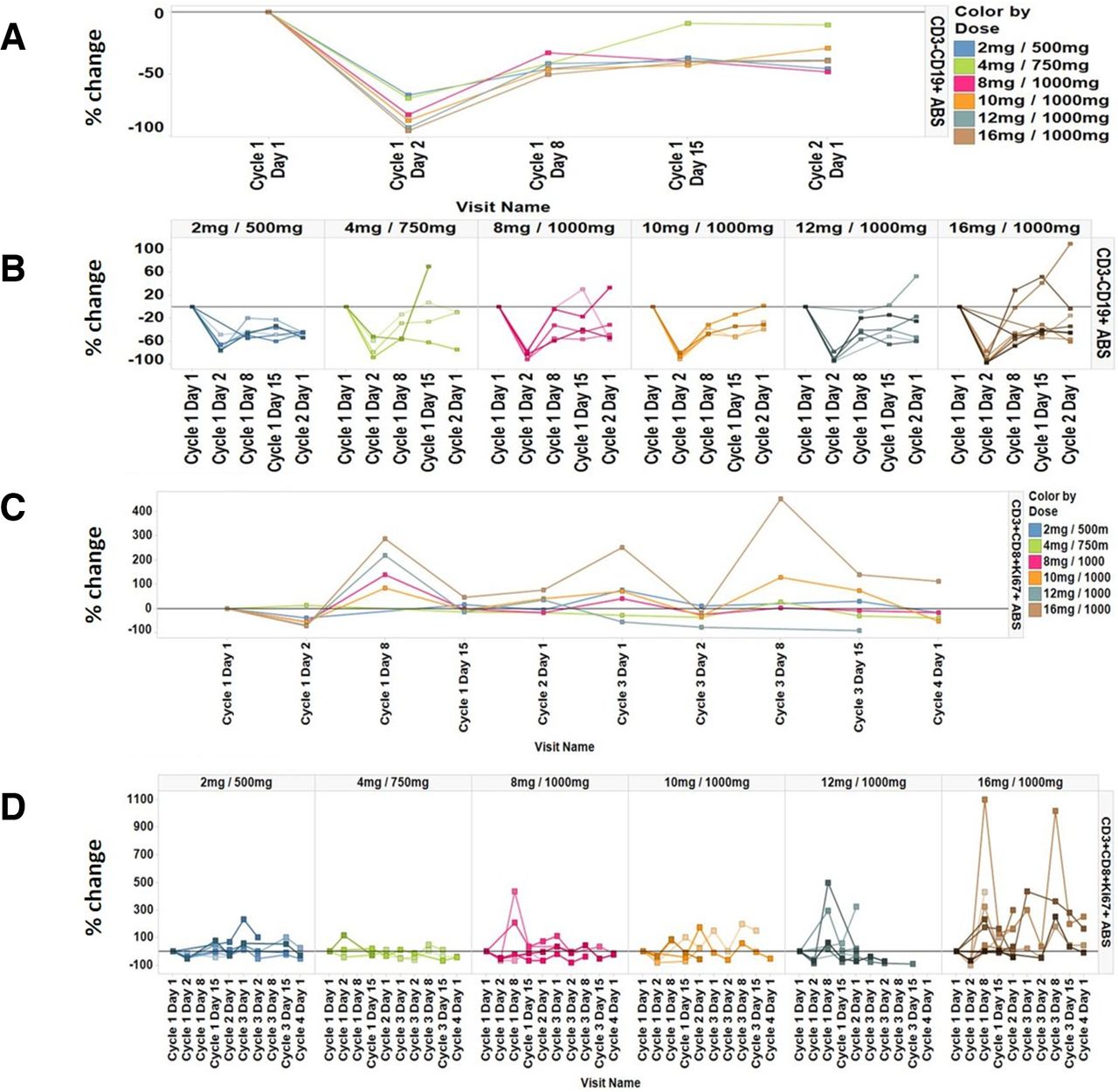
Percent change of peripheral B cells from baseline (a) per dose cohort and (b) for individual patients per dose cohort and percent change of peripheral proliferating CD3+CD8+Ki67+ T cells from baseline; (c) per dose cohort and (d) for individual patients per dose cohort.
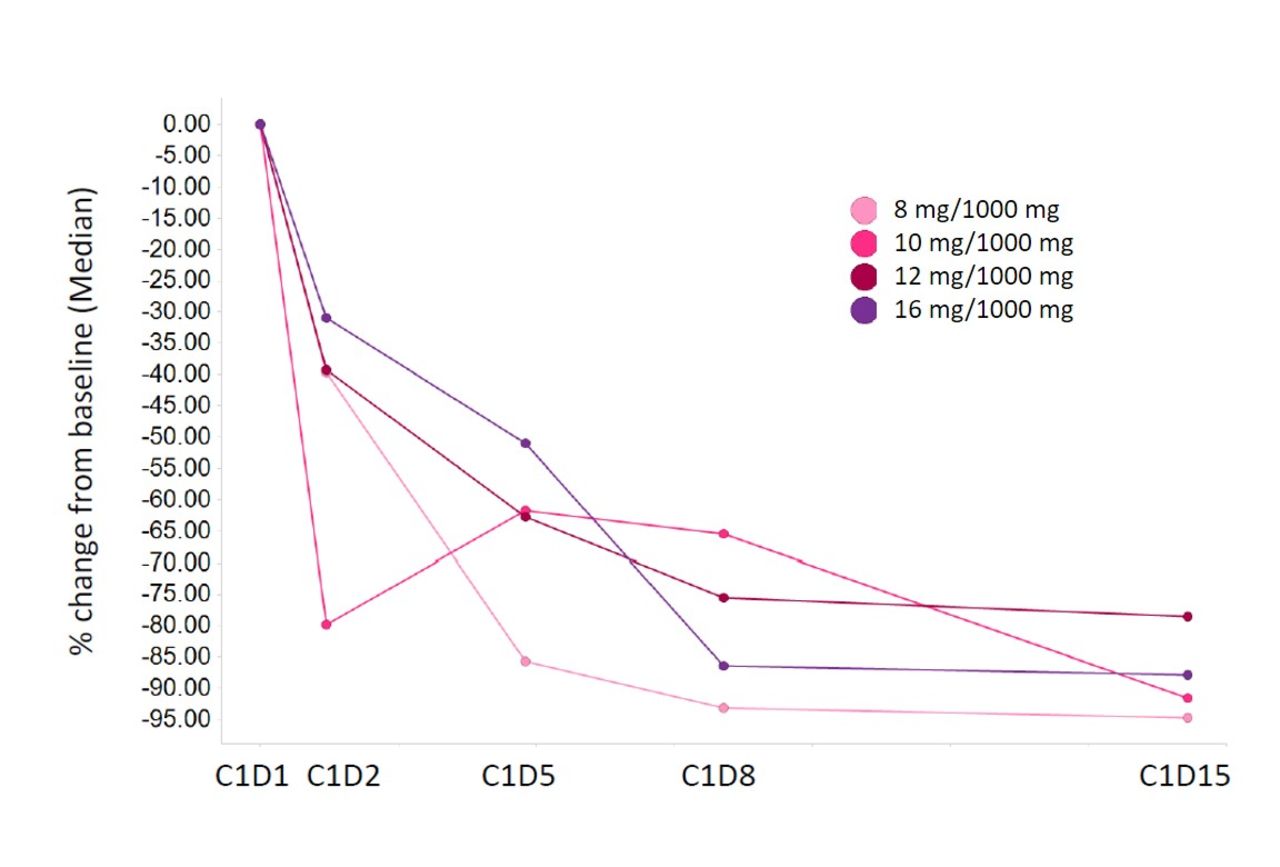
Percent change of peripheral CD14dim CD16high monocytes from baseline per dose cohort.
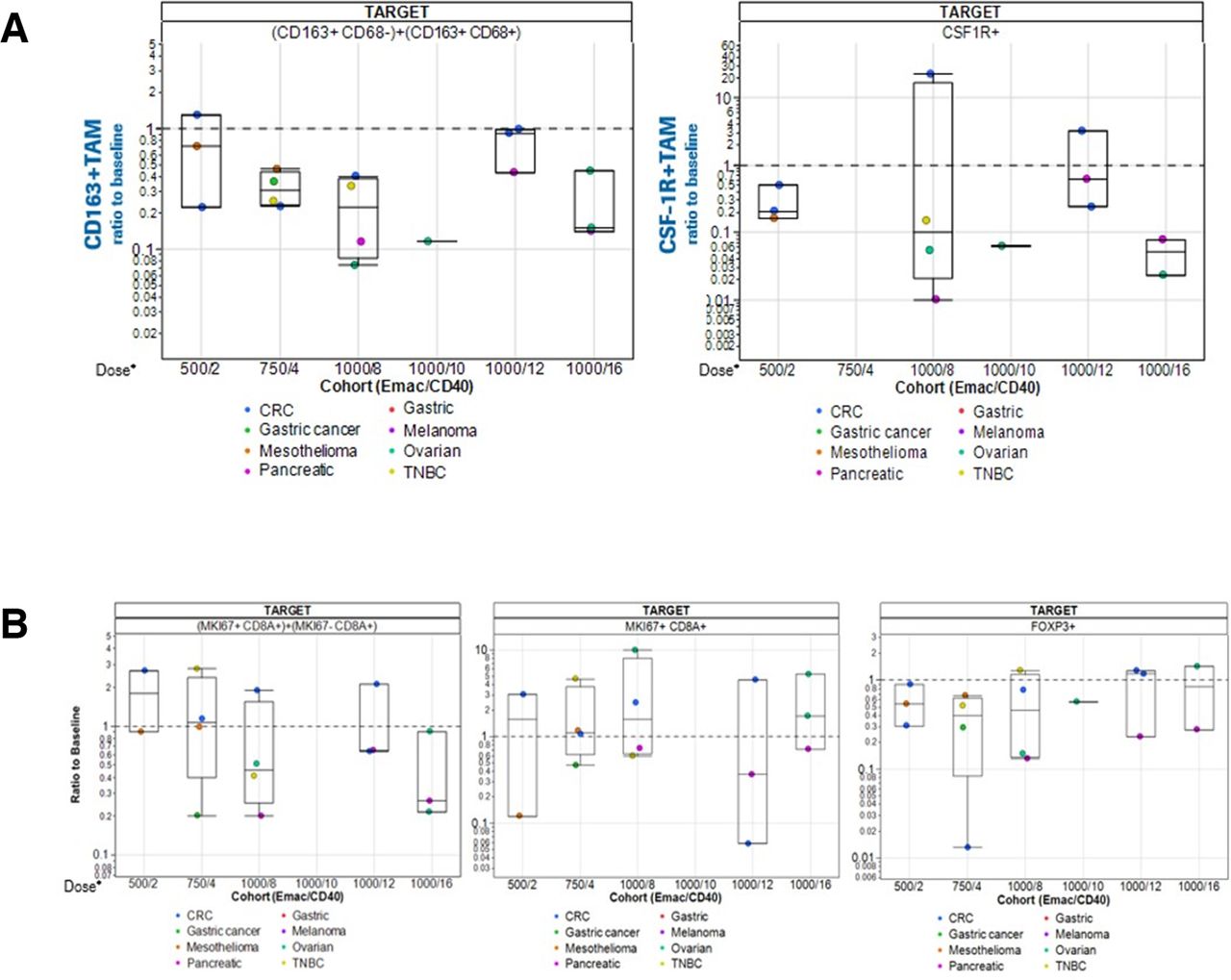
Paired tumor biopsies at screening and at cycle 2 day 1 were assessed for treatment-induced T-cell and TAM alterations in the TME. Overall, despite the limited number of patients in the dose escalation cohorts, the data demonstrated reductions of CD163+ and CSF-1R+ cells (figure 5A) as previously seen with emactuzumab monotherapy.25 The level of TAM reduction was comparable for increasing doses of selicrelumab with the exception of patients treated with 1000 mg emactuzumab and 12 mg selicrelumab. Here, data show the lowest TAM reduction compared with all other dose cohorts. Further increase of the selicrelumab dose to 16 mg resulted in a profound reduction of CD163+ and CSF-1R+ cells in the TME, comparable to the dose levels below 12 mg. As the overall hypothesis of the emactuzumab plus selicrelumab combination was to eventually generate a functional and effective CD8+ T-cell-driven anti-tumor immunity, the infiltration of overall CD8+ T cells as well as CD8+Ki67+ T cells in paired tumor biopsies was assessed (figure 5B).The data show a more pronounced reduction of CD8+ T-cell infiltrates at higher doses of emactuzumab and selicrelumab. Similar to the TAM reduction described above, for the 12 mg selicrelumab dose level, the lowest decrease of CD8+ T cells compared with the other doses tested was observed. In the TME, CD8+Ki67+ T-cell counts were stable across dose cohorts or at most showed a slight increase for higher doses once again with the exception of the 12 mg selicrelumab dose that showed a decrease of CD8+Ki67+ T cells. We could not generate any tumor T-cell data for the 1000 mg emactuzumab plus 10 mg selicrelumab cohort due to insufficient evaluable biopsy material. FOXP3+ Treg cells showed overall slight decreases over the different dose cohorts.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change from baseline of (a) CD163+ and CSF-1R+ TAMs in paired biopsies and (b) CD8+ T cells, Ki67+CD8+ TILs and FoxP3+ Tregs in paired biopsies. Doses (emactuzumab/selicrelumab) and tumor types are indicated. Please note: No data for T-cell analysis in situ for the 1000 mg emactuzumab plus 10 mg selicrelumab cohort were obtained due to insufficiently evaluable biopsy material. CRC, colorectal cancer; CSF-1R, anti-colony stimulating factor 1 receptor; TAM, tumor-associated macrophage; TNBC, triple-negative breast cancer.
Further, paired skin biopsies pre-treatment and at day 15 post treatment were performed to analyze dermal macrophage counts. Reductions of dermal macrophages could be seen across all dose cohorts (data not shown) similar to what has been seen for emactuzumab monotherapy.25
Discussion
The combination of aCD40 and anti-CSF-1R mAbs was associated with synergistic anti-tumor activity in three independent mouse models.26–28 Based on these data, we here report for the first time on the clinical translation of CD40 activation with simultaneous CSF1R blockade in advanced or metastatic solid tumor patients.
Co-administration of aCD40 selicrelumab and anti-CSF-1R emactuzumab was generally safe and tolerable and an MTD was not formally reached. Three DLTs were observed which were transient: all were grade ≥3 IRRs in the higher dose cohorts of selicrelumab, which is in concordance with safety observations reported for other aCD40 compounds.32 Selicrelumab was previously tested as monotherapy in phase I studies and revealed IRRs as the most prominent safety signal in up to 56% of patients.18 19 For weekly dosing of selicrelumab, the incidence of grade 3 IRRs was 11% at similar doses used in the present study.18 Similarly, intravenous administration of the aCD40 mAb SEA-CD40 led to IRRs in 70% of patients, with 11% being of grade 3.33 This is in line with the present data where the incidence of IRRs was 75.7% overall and 8.1% for grade ≥3 events, all considered related to selicrelumab. This suggests that the combination with emactuzumab did not increase the incidence and severity of selicrelumab-related IRRs. Importantly, there were no signs of liver toxicity shown in the present study, which is in line with monotherapy studies of either selicrelumab18 19 or emactuzumab25 in solid tumor patients and was also shown in a preclinical model of the combination treatment.34 Other frequent AEs for the combination treatment such as liver enzyme elevations and edema are likely caused by emactuzumab only, as shown previously to be a class effect of CSF-1R inhibitors.35 Interestingly, selicrelumab was administered subcutaneously in another study in combination with vanucizumab.36 Here, although injection-site reactions were seen for most patients (92%) and one grade 3 event was considered a DLT at 8 mg, doses up to 72 mg were administered and the incidence of IRRs was reduced. Similarly, an intratumorally injected aCD40 compound (ADC-1013) seemed to lead to improved tolerability, with only half of the patients experiencing IRRs.37 Hence, such administration routes may be able to increase the dose of selicrelumab and reduce AEs secondary to immune activation at the same time. Whether this results in better efficacy and PD activity remains to be shown.
PD activity assessments of the combination of emactuzumab and selicrelumab were performed in peripheral blood as well as in paired tissue biopsies. We focused on PD markers that were identified for both compounds as monotherapy and confirm the previously reported dose-dependent transient decrease of peripheral B cells. Similar dose-dependent decreases were seen with single-agent selicrelumab,18 19 the combination of selicrelumab with vanucizumab36 and after intratumoral administration of the aCD40 ADC-1013.37 Although it was shown that peripheral B cells after aCD40 therapy showed signs of activation,18 19 37 it remains unclear whether B cells which marginated from peripheral blood are truly activated by aCD40 and act as additional APCs. As we did not observe similar B-cell-related PD effects (eg, increased expression of CD86 or CD54) for emactuzumab monotherapy (data not shown), this was likely induced by selicrelumab only. Further, we observed only a trend for a dose-dependent transient increase of peripheral activated CD8+ T cells which peaked 8 days after administration of the study drug combination. This is in contrast to Ruter et al who described a transient peripheral T-cell reduction for selicrelumab monotherapy.18 This reduction of peripheral T cells could be interpreted as an activation-induced margination effect of T cells and, due to the weekly selicrelumab administration in the study of Ruter et al, resulting in peripheral depletion and hyperacute T-cell activation. In the present study, the 3-weekly schedule prevented this hyperactivation-induced loss of T cells. On the other hand, the observed peripheral T-cell activation pattern in the present study is similar to the one described for a selicrelumab (administered subcutaneously q4w) and vanucizumab (administered IV q2w) combination therapy.36 Transient activation of CD8+ T cells had not been observed in emactuzumab monotherapy; hence, this effect seems to be driven by selicrelumab only. Preclinically, it has been shown that inhibition of CSF-1R signaling sensitizes TAMs to profound and rapid reprogramming in the presence of a murine aCD40 before their depletion. Despite the short-lived nature of macrophage hyperactivation, combined treatment with anti-CSF-1R and aCD40 mAbs was sufficient to create a proinflammatory tumor milieu that reinvigorates a pre-existing T-cell response in transplanted tumor.26 Clinically, emactuzumab reduced circulating CD14DimCD16bright monocytes in peripheral blood associated with a profound decrease of the TAM infiltrate in solid tumor patients.24 25 Whereas for emactuzumab monotherapy this monocyte subset showed a rapid and sustained depletion from peripheral blood at 1000 mg q3w,25 the addition of selicrelumab in the present study resulted in a delayed and less profound clearance of CD14DimCD16bright monocytes. This effect could be due to the selicrelumab-induced transient hyperactivation of CSF-1R+ monocytes that may have resulted in a temporary independency from CSF-1R-mediated survival signals. Notably, this peripheral non-classical monocyte subset most closely resembles the monocytic MDSC subset in the TME of murine tumors that was retained under CSF-1R blockade when an aCD40 was present.26 In agreement with published preclinical data, there was a similar depletion of CD163+ and CSF-1R+ cells/TAMs as compared with emactuzumab monotherapy.25 To determine a suitable on-treatment tumor biopsy time point to detect the hyperactivation of, for example, TAMs is challenging. In addition, the detection of soluble cytokines and chemokines that were released by hyperactivated TAMs in very limited tumor biopsy material is technically not feasible. However, we used the increased T-cell recruitment and activation in the TME as a surrogate for potential TAM reprogramming in the present study. Although there is a trend for increased activated CD8+ T-cell levels in paired tumor biopsies, the overall number of CD8+ T cells in the TME actually diminished with increasing doses of emactuzumab and selicrelumab. With the available data, it is difficult to identify the individual contribution of emactuzumab or selicrelumab to the observed PD TIL effects. However, emactuzumab single-agent treatment did not increase nor decrease the CD8+ infiltrates in a previous study.25 A direct comparison of this observation with other published reports is challenging due to differences in treatment intervals, the timing of the on-treatment biopsies, concomitant treatment regimen and tumor types treated. In a combination of an aCD40 agonist with aCTLA-A4, Bajor et al described a significant increase of overall CD8+ T cells in post treatment biopsies comparing eight pre-treatment and seven post treatment samples in melanoma patients; however, only two paired biopsies were available.20 Malmström et al reported a marked increase of a CD4+ T cell infiltrates of the TME combined with increased CD8+ T-cell infiltrates in bladder cancer patients after therapy with adenoviral vectors expressing CD40 ligand.38 Further, a recent publication by Kluger et al reported that the aCD40 mAb APX005M in combination with nivolumab resulted in an increase of TILs.39 Nevertheless, both reports describe combination therapies with only few biopsy samples, hence, conclusions on single agent contributions are difficult to draw.
In the present study, we cannot conclude that the observed PD activity on TILs supports our preclinical hypothesis. Single-agent control arms or less frequent dosing intervals with emactuzumab were not pursued. The prolongation of emactuzumab-free treatment intervals may allow the macrophages to repopulate the tumor and to repeat the reprogramming with the subsequent aCD40/anti-CSF1R combination therapy.
Despite the proposed biological synergism, there was no evidence that the observed PD effects translated into objective tumor responses. The best clinical response was SD achieved in 40.5% of patients and therefore did not confer higher activity than what would have been expected with emactuzumab or selicrelumab alone. In fact, selicrelumab monotherapy in 29 solid tumor patients showed an objective response rate of 13.8%,19 although in this study, PRs were unconfirmed and seen in melanoma patients only. Remarkably, one of the responding patients continued treatment on a 2-monthly schedule and remained in complete remission more than 5 years later.16 However, in another monotherapy study with selicrelumab, no objective responses could be shown in a 27-patient set.18 Monotherapy with SEA-CD40 in 34 solid tumor patients showed one PR (2.9%) in a basal cell carcinoma patient.33 Emactuzumab monotherapy has no overt clinical activity in solid tumor patients as shown recently25 and the combination presented here did not provide any additional activity. Underlying reasons may be: (1) The 3-weekly and concomitant schedule of selicrelumab and emactuzumab administration used in the present study may not be suitable to generate hyperactivated human TAMs accompanied by a pronounced T-cell activation as preclinically described in murine models. While in preclinical models, CD40 activation relies on FcR-mediated trimerization of the receptor, the human IgG2 aCD40 selicrelumab functions independent of FcR engagement.19 (2) Certain tumors express high levels of CD40 and direct activation of CD40 results in growth inhibition and sensitization to cytotoxic agents.40 However, CD40 expression levels in tumors were not evaluated in this study and might have been too low to contribute to any anti-tumor effects. (3) The tumor types enrolled in the present study was based on the prognostic relevance of macrophage infiltration. However, responding patients for selicrelumab monotherapy were exclusively melanoma patients,19 in contrast to only one melanoma patient enrolled in this study. Preclinical data for this combination therapy suggested that ongoing T-cell response against the tumor is a prerequisite for the combination to result in tumor shrinkage. Hence, patients with immunogenic tumors like melanoma, microsatellite instability-high tumors or renal cell carcinoma who failed immune checkpoint blockade may derive better clinical benefit from this combination therapy. (4) The dose of selicrelumab for IV administration was limited to 16 mg due to IRRs. Higher and possibly more efficacious doses may be administered for different routes such as subcutaneously or intratumorally36 37 but were not pursued in this study.
In summary, combination treatment with aCD40 selicrelumab and anti-CSF-1R emactuzumab was tolerable and triggered B-cell margination, CD8+ T-cell increase and monocyte decrease in the periphery and a decrease of TAMs in the tumor; however, despite the suggested biological rationale, this did not translate into objective clinical responses in the current study.
Acknowledgments
The authors would like to thank the patients and their families for their participation in this study, and the staff at the study sites.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JPM, CGR, JMM, JPD, PC and DR contributed to the study design, data collection, analyses and interpretation. JPM, CGR, JMM, DZ, TM, GC, LE, JPD and PC served as clinical investigators at the study sites and conducted the clinical study. WJ, A-MJ, FM, RC, CW, GB, KK, IK, PCT, SH, CR, GML, MW and DR contributed to collection, analyses and interpretation of the data. The primary data were made available to the investigators for independent central review and analyses. The first draft of the manuscript was written by WJ, MW, DR, MC and JPM with review and revision by the other coauthors. All authors had full access to all data in the study, made the decision to submit these data for publication, were involved in writing the report, and agreed upon final content of the paper.
Funding This study was funded by F. Hoffmann–La Roche Ltd.
Competing interests Jean-Pascal Machiels: Advisory board consulting for Pfizer, Roche, AstraZeneca, Bayer, Innate, Merck Serono, Boerhinger, BMS, Novartis, Janssen, Incyte, Cue Biopharma and ALX Oncology; travel expenses from Amgen, BMS, Pfizer, MSD; data safety monitoring board support for Debio, Nanobiotix and PsiOxus. Carlos Gomez-Roca: Consultancy for AstraZeneca and BMS; travel grant from Boehringer Ingelheim, BMS, Pierre Fabre, Roche and Sanofi Aventis; honoraria from BMS, Pierre Fabre and Roche; Jean-Marie Michot: Consultancy from Celgene, Bristol Myers Squibb, AstraZeneca and Janssen; travel grant and non-financial support from AstraZeneca, Roche, Novartis, Gilead, Celgene and Bristol Myers Squibb; Dmitriy Zamarin: Consultancy fees from Merck, Synlogic Therapeutics, Biomed Valley Discoveries, Trieza Therapeutics, Tesaro, and Agenus; Tara Mitchell: Advisory board consulting for Merck, BMS and Array; Gaetan Catala: Travel grants from Roche, Pharmamar, MSD and AstraZeneca; advisory role for MSD. Lauriane Eberst: None. Wolfgang Jacob: Sponsor employee and sponsor stock ownership. Anna-Maria Jegg: Former sponsor employee and has patent issued in the use of emactuzumab. Michael A Cannarile: Sponsor employee and sponsor stock ownership. Carl Watson: Sponsor consultant. Galina Babitzki: Sponsor employee. Konstanty Korski: Sponsor employee. Irina Klaman: Sponsor employee. Priscila C Teixeira: Sponsor employee. Sabine Hoves: Sponsor employee, sponsor stock ownership and has patent issued in the use of emactuzumab. Carola Ries: Former sponsor employee and has patent issued in the use of emactuzumab. Georgina Meneses-Lorente: Sponsor employee. Francesca Michielin: Sponsor employee. Randolph Christen: Sponsor employee and sponsor stock ownership. Dominik Rüttinger: Sponsor employee, sponsor stock ownership and has patent issued in the use of emactuzumab. Martin Weisser: Sponsor employee and sponsor stock ownership. Jean-Pierre Delord: Consulting or advisory role for Novartis, Roche/Genentech, Bristol Myers Squibb, MSD Oncology; research funding from Genentech, Bristol Myers Squibb, MSD Oncology. Philippe Cassier: Honoraria from Novartis, Roche/Genentech, Blueprint Medicines, Amgen and AstraZeneca; research funding from Novartis, Roche/Genentech, Eli Lilly, Blueprint Medicines, Bayer, AstraZeneca, Celgene, Plexxikon, AbbVie, Bristol Myers Squibb, Merck Serono, Merck Sharp & Dohme, Taiho Pharmaceuticals, Toray Industries, Transgene, Loxo, GlaxoSmithKline, Innatre Pharma and Janssen; travel grants from Roche, Amgen, Novartis, Bristol Myers Squibb, Merck Sharp & Dohme and Netris Pharma.
Patient consent for publication Not required.
Ethics approval Local ethics committee approval was obtained in six centers in Belgium, France and the USA and all patients provided written informed consent.
Provenance and peer review Not commissioned; externally peer-reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Qualified researchers can request access to individual patient-level data through the clinical study data request platform. For more on Roche’s criteria for eligible studies, see www.clinicalstudydatarequest.com. For more on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.