Article Text

Abstract
Background Peptide-based vaccination is a rational option for immunotherapy of prostate cancer. In this first-in-man phase I/II study, we assessed the safety, tolerability and immunological impact of a synthetic long peptide vaccine targeting Ras homolog gene family member C (RhoC) in patients with prostate cancer. RhoC is a small GTPase overexpressed in advanced solid cancers, metastases and cancer stem cells.
Methods Twenty-two patients who had previously undergone radical prostatectomy received subcutaneous injections of 0.1 mg of a single RhoC-derived 20mer peptide emulsified in Montanide ISA-51 every 2 weeks for the first six times, then five times every 4 weeks for a total treatment time of 30 weeks. The drug safety and vaccine-specific immune responses were assessed during treatment and thereafter within a 13-month follow-up period. Serum level of prostate-specific antigen was measured up to 26 months postvaccination.
Results Most patients (18 of 21 evaluable) developed a strong CD4 T cell response against the vaccine, which lasted at least 10 months following the last vaccination. Three promiscuouslypresented HLA-class II epitopes were identified. Vaccine-specific CD4 T cells were polyfunctional and effector memory T cells that stably expressed PD-1 (CD279) and OX-40 (CD134), but not LAG-3 (CD223). One CD8 T cell response was detected in addition. The vaccine was well tolerated and no treatment-related adverse events of grade ≥3 were observed.
Conclusion Targeting of RhoC induced a potent and long-lasting T cell immunity in the majority of the patients. The study demonstrates an excellent safety and tolerability profile. Vaccination against RhoC could potentially delay or prevent tumor recurrence and metastasis formation.
Trial registration number NCT03199872.
- immunotherapy
- vaccination
- prostatic neoplasms
- T-Lymphocytes
- translational medical research
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Therapeutic antitumor vaccination may provide a safe and long-lasting immunotherapy treatment option for patients with cancer. Many trials are ongoing worldwide, with most recent developments favoring a patient-individual approach.1–3 It is acknowledged that vaccines should better be administered at an early stage of disease when the immune system of patients with cancer is not yet suppressed. For advanced patients, vaccines could also be applied in combination with for example, surgery, chemotherapy or checkpoint inhibitor therapy.1 2 In addition, cancer vaccines should not only be designed for the induction of cytotoxic T lymphocytes (CTLs), but also of effector CD4 T cells. CD4 T helper cells are crucial for CD8 T cell activation and expansion, as well as for the generation and maintenance of CD8 T cell memory.4–6 They also display a range of antitumoral effects, such as secretion of tumor necrosis factor (TNF) and interferon-γ (IFN-γ),7 8 activation of macrophages or natural killer cells and direct cytotoxicity, which together might be more powerful than the sole tumor killing by CTLs.9 10
To stimulate both CD4 and CD8 T cells, vaccines containing either a mix of known HLA-class I and -class II epitopes3 11 or (overlapping) synthetic long peptides (SLPs; 15–35 amino acids (aa))1 12 can be used. SLPs have been shown to be rapidly and more efficiently processed compared with the whole protein, and to activate CD4 T cells, but also CD8 T cells by cross-presentation.13 Since peptide processing takes place in vivo, prior knowledge of the precise T cell epitopes contained in the long peptides is not absolutely required, and such vaccines are generally applied to all patients, regardless of their HLA allotype.
The Ras homolog gene family member C (RhoC) belongs to the Rho GTPase family which comprises RhoA, RhoB and RhoC (85% sequence homology), all involved in the regulation of cytoskeleton organization.14 RhoC was shown to be an important player in tumor cell motility, invasion and metastasis formation.15 16 Since RhoC has a limited expression in normal cells but is highly expressed on advanced cancer cells and metastases,14 17 it could represents a suitable target for anticancer vaccination. Immunohistochemical analyzes of tumor samples from patients with prostate cancer (PCa) showed an increase in RhoC expression with advanced tumor stages and a strong correlation with the metastatic status. In addition, patients with RhoC expression have a significantly reduced overall-survival rate, indicating that RhoC could be used as a prognostic marker in PCa.18 Furthermore, reports have demonstrated RhoC expression in cancer stem cells,19 20 which are also found in PCa.21 In localized PCa, the presence of micrometastases has been associated with biochemical recurrence (BCR) after first-line treatment by radical prostatectomy.22 Targeting RhoC-expressing cancer cells and/or (micro) metastases by vaccination may therefore improve the clinical course of PCa patients and delay or prevent the onset of second-line therapies such as hormonal deprivation and/or chemotherapy. The immunogenicity of RhoC has been documented by our previous study, where CD8 T cells specific for a RhoC-derived 10mer anchor-modified peptide were found in the blood of melanoma patients. Cloned T cells could specifically kill HLA-A*03 and RhoC expressing tumor cell lines in vitro.23
In this clinical phase I/II study, we report the safety and immunogenicity of a 20mer SLP vaccine specifically targeting the RhoC protein in PCa patients.
Methods
Study design and patients
The study was an open-label, phase I/II trial. Patients previously treated with RP were identified, informed and followed at Copenhagen Prostate Cancer Center, Department of Urology, University of Copenhagen, Rigshospitalet. Vaccinations were administered at Zelo Phase I Unit, DanTrials ApS, Copenhagen, Denmark. All patients gave informed consent. Prior to study entry, patients underwent screening procedures including a full physical examination, and in case of BCR, a metastatic workup with computer tomography and bone scans. For inclusion and exclusion criteria, see online supplemental table S1. The primary endpoint of the study was the evaluation of the safety and tolerability of the vaccine. Treatment-emergent adverse events (TEAE) were analyzed in accordance with the common terminology criteria for adverse events (CTCAE), V.4.03. The secondary endpoint was the investigation of the immunological responses against the vaccine.
Supplemental material
Twenty-four patients were screened and 22 included in the study (table 1). Median time for surgery to study entry was 1.2 years (range: 0.3–12.8). Patients were vaccinated subcutaneously alternating between the right and left upper arms with the vaccine consisting of 0.1 mg SLP RV001 (20aa C-terminal sequence of the RhoC protein: ATRAGLQVRKNKRRRGCPIL; the 16 last aa sequence is found only in RhoC, but not in RhoA/B, and is thus RhoC-specific) emulsified in Montanide ISA-51 (1 mL). Patients were vaccinated six times every 2 weeks, then five times every 4 weeks, resulting in a treatment duration of approximately 30 weeks (11 vaccinations in total). HLA-class I and II typing was performed before vaccination (visit 2, Tissue Typing Laboratory, Rigshospitalet, Copenhagen). For in vitro immunomonitoring, blood was taken before vaccination (visit 2), and after the 4th, 6th and 11th vaccinations (visits 6, 8 and 13). Follow-up samples were obtained every third month up to 13 months postvaccination (visits 14–17). The study started in March 2017 and was completed in March 2019. Serum prostate-specific antigen (PSA) levels were measured by routine clinical testing at every patient visit (median number of PSA measurements from study entry: 15 (range 13–17)). PSA doubling time was calculated using the MSK calculator available online.24 In men with measurable PSA at study entry, doubling time was calculated from the first measurable PSA until study entry, and from study entry to end of follow-up. Median follow-up time for serum PSA was 2.5 years (range 2.4–2.7 years). Of note, Patient 015 withdrew from treatment after seven injections, but completed all visits for blood collection, except visit 13. Study design is shown in figure 1 and patient’s characteristics, PSA levels and HLA-typing results are shown in online supplemental tables S2 and S3.
Main patient’s characteristics

RV001-specific T cells are induced after RhoC vaccination. (A) Vaccination and monitoring schedule. Patients were vaccinated 11 times. For immunoassays, blood was taken prevaccination, three times during the vaccination phase (vaccination) and four times postvaccination (follow-up 1 and follow-up 2) (blood drops). PBMCs were prestimulated with the RV001 and expanded for 12 days before IFN-γ ELISpot testing (0.2×106 cells/well, except for Patient 21 visits 2–13, and Patient 012 visits 16+17: 0.17×106 cells/well). (B) Exemplary result of an ELISpot (Patient 011). ddH2O and PHA were used as negative and positive control, respectively. (C–E) RV001-specific mean spot counts per analyzed time and normalized to 0.2×106 cells/well. Three independent ELISpot experiments were performed (indicated by the gaps). The sums of RV001-specific mean spot numbers (V6+8+13) are shown for strong- (C; n=7; ≥2500 spots), intermediate- (D; n=7; ≥1500–2500 spots), and weak/non- (E; n=7; 0–1500 spots) responders. (F) RV001-specific mean spot counts per patient and visit normalized to 0.2×106 cells/well. Light green indicates a statistical significance according to the DFR(2x) permutation test. n=21 patients. ddH2O, bidistilled water; IFN-γ, interferon-γ; na, cells not available; PHA, phytohemagglutinin; RhoC, Ras homolog gene family member C.
The following sections are prepared in accordance with the Minimal Information about T cell Assays (MIATA) guidelines.
Cell samples
Isolation of peripheral blood mononuclear cells
Blood samples (100 mL, heparinized) were processed within 8 hours after blood drawing (DanTrials ApS). Peripheral blood mononuclear cells (PBMCs) isolation was performed according to a standard, pre-established protocol, using prefilled 50 mL Leucosep tubes (Greiner Bio-One). Cells were counted with trypan blue and Tuerks solution (both Sigma-Aldrich). Six to 13×106 cells per cryovial (Nunc, Sigma-Aldrich) were frozen in 1 mL heat-inactivated (hi) foetal bovine serum (FBS) (ThermoFisher) containing 10% dimethylsulfoxide (DMSO, Sigma-Aldrich). Cells were kept in freezing containers (Nalgene Mr. Frosty, Sigma-Aldrich) at −70°C for 24–72 hours and transferred and stored in liquid nitrogen (−196°C). PBMCs were shipped on temperature-controlled dry ice to the Department of Immunology, Tübingen for immunological analyses. Cells were stored again at −196°C for 3–12 months before testing. PBMC samples from Patient 001 visit 2 and Patient 015 visit 13 were not available.
Immunological assessments
In vitro stimulation of antigen-specific T cells
PBMCs were thawed in IMDM (Gibco) containing 2.5% hi human serum (HS, Sigma-Aldrich), penicillin 100 units/mL and streptomycin 0.1 mg/mL (P/S, Sigma-Aldrich), and 50 µM β-mercaptoethanol (β-ME, Merck). After one washing step with serum-free medium (IMDM, P/S, 50 µM β-ME), cells were counted with trypan blue (Merck). The median live-cell yield after thawing was 63.5%. No cut-off was applied for further in vitro culture. On day 0, 1.0–3.5x106 or 3.5–6.5x106 PBMCs/well were seeded in T cell medium (TCM, IMDM with 10% hi HS, 1x P/S and 50 µM β-ME) in a 48-well or 24-well plate, respectively (Cellstar, Greiner bio-one), and further cultured at 37°C and 7.5% CO2. On day 1, cells were stimulated with 10 µg/mL RV001 peptide (purity ≥90%, PolyPeptide Laboratories France SAS) dissolved in 100% deionized water (ddH2O; LiChrosolv, Merck) plus 20 µg/mL Poly-ICLC (Hiltonol, Oncovir). Human interleukin (IL)-2 (R&D Systems) was added to the culture at 2 ng/mL on days 3, 5, 7 and 9. On day 12, cells were harvested, counted (median live-cell yield: 76.8%) and tested with enzyme-linked immune spot assay (ELISpot) and intracellular cytokine staining (ICS) assay.
ELISpot
The IFN-γ ELISpot protocol has been described previously.25 If not otherwise stated, 0.2×106 cells were cultured per well in the presence of 50 µg/mL RV001 peptide for 26 hours at 37°C and 7.5% CO2 (triplicates). ddH2O (6 wells) and phytohemagglutinin-L (10 µg/mL, Sigma-Aldrich, 1 well) were used as negative and positive controls, respectively. Spots were counted with the ImmunoSpot series 6 ultra-V analyzer (CTL Europe) according to a standard protocol. Altogether, samples from n=21 patients were immunologically evaluated (results from Patient 002 were excluded because of inconsistent spot counts). For wells with counts above 2000 spots or stated as “too numerous to count”, TNTC), a count cut-off was set to 2000 spots. RV001-specific spot counts are defined as the mean spot counts in the RV001 stimulated wells minus the mean spot counts in the ddH2O wells. All spot counts are given in online supplemental table S4.
Multiparameter flow cytometry
PBMCs were analyzed by ICS either ex vivo or after culture. Thawed cells were rested in TCM for 4–6 hours prior to the ICS. Precultured cells were directly examined on day 12 or harvested from the ELISpot plate. Between 0.5 and 2×106 cells per well (96-well plate) were stimulated with 50 µg/mL RV001 or 10 µg/mL of single overlapping 15mers (RV001-derived: ATRAGLQVRKNKRRR (ATR15), AGLQVRKNKRRRGCP (AGL15), LQVRKNKRRRGCPIL (LQV15), all from JPT Peptide Technologies, ≥90% purity). ddH2O and Staphylococcus enterotoxin B (SEB, 10 µg/mL, Sigma-Aldrich) were added as negative and positive controls, respectively. The CD107a antibody (Ab) was added together with the stimulus; protein transport inhibitors Brefeldin A (10 µg/mL, Sigma-Aldrich) and Golgi Stop (BD) 1 hour thereafter. After 12 hours at 37°C and 7.5% CO2, cells were stained (Ab panel 1, online supplemental table S5). For ex vivo analysis of PD-1, OX-40, and LAG-3 expression (Ab panel 2, online supplemental table S5), fluorescence minus one controls were performed. After staining (for protocol, see online supplemental table S5), cells were acquired on the same day on a LSRFortessa SORP (BD) equipped with the DIVA Software (V.6). The analysis was performed with FlowJo (V.10.6.1), gating strategies are shown in online supplemental figure S1. All results were audited. Frequencies of RV001-specific T cells are defined as: % marker positive cells in the RV001-stimulated sample minus % of marker-positive cells in the ddH2O sample.
For the analysis of regulatory T cells (Tregs), 1.5×106 cells per test (96-well plate) from n=5 patients (four visits each) were stained. For the staining protocol, antibodies, and gating strategy, see online supplemental table S5 (Ab panel 3) and online supplemental figure S2. After staining, cells were washed twice, resuspended in FACS buffer, and acquired on a Spectral cell analyzer SP6800 (Sony) using the software V.2.0.2.14140. Analysis was done with FlowJo (V.10.6.2).
Identification of RV001-presenting allelic products
Potential RV001-derived HLA-class I and -class II epitopes were tested by using HLA-matched human lymphoblastic cell lines (LCLs) as peptide-presenting cells. C1R and its HLA-B*27:05 transfectant (C1R-B*27) were cultured in RPMI 1640 supplemented with 10% hi FBS, P/S, β-ME and G418 (Sigma-Aldrich) at 1 mg/mL. MGAR and H0301 were cultured in RPMI 1640 containing 20% hi FBS, P/S, β-ME (for HLA-typing results, see online supplemental table S3). Per condition, 8×106 LCL cells were loaded with 50 µg/mL RV001, 20 µg/mL RV001-derived single 15 mer peptides (ATR15, AGL15), or with the respective amount of ddH2O in 1.5 mL IMDM supplemented with 5% hi HS, P/S, and β-ME. After 7 hours at 37°C, 7.5% CO2, cells were washed three times. PBMCs stimulated with RV001 plus Poly-ICLC for 12 days were either incubated with the pre-loaded HLA-matched LCLs at a 2:1 effector to target ratio, with 50 µg/mL RV001, or with 10 µg/mL of each 15mer peptide (ATR15, AGL15, LQV15). ddH2O and 10 µg/mL SEB were used as controls. After 12 hours, an ICS was performed.
Immunological response definition
For each visit, a T cell response was defined by the DFR(2x) permutation resampling method.26 A patient was defined as an immunological responder if at least two out of three analyzed times were tested positive in the ELISpot. If a T cell reactivity against RV001 was already detected at baseline level, the patient was considered as a responder if the response was boosted during vaccination (specific spot counts after vaccination ≥2 x specific spot counts at baseline). If less than three vaccination samples were evaluable (n=1), T cell reactivity detected at one time was enough to consider the patient as an immunological responder. Patients were grouped according to the sum of RV001-specific mean spot numbers for visits 6+8+13: strong- (n=7; ≥2500 spots), intermediate- (n=7; ≥1500–2500 spots), and weak/non- (n=7 including three non-responders; 0–1500 spots) responders.
For the ICS, T cell reactivity was assessed within CD4 and CD8 T cell subsets. First, each activation marker (n=5) was defined as positive if the percentage of positive cells was ≥three fold for the peptide-stimulated cells compared with the ddH2O stimulated cells and, ≥20 marker-positive cells were counted. Second, at least three out of five markers must be positive.
Statistical comparisons
Statistical analyzes were performed with GraphPad Prism (V.6.01 and V.8.4.3). The Kolmogorov-Smirnov test with Dallal-Wilkinson-Lilliefors corrected p value was used to check Gaussian distribution. One-way and two-way analysis of variance (ANOVA) was performed for single-group and multiple-group comparisons, respectively. Correction for multiple comparisons was performed. Statistical differences were considered as significant for p≤0.05 (*), p≤0.01 (**), and p≤0.001 (***). The statistical test and the number of patients are given for each experiment.
General lab operation
All experiments were performed with standard reagents and according to laboratory-standard protocols for culture, assays and analysis. Protocols for ELISpot and ICS have been validated and the performance of the working group is regularly controlled by participation in external proficiency panels (CIP/CIMT and Immudex).
Results
Vaccination against RhoC induces potent and long-lasting RV001-specific T cell responses
The immunological response against the RV001 vaccine was assessed on PBMC samples obtained during the vaccination phase and at all follow-up times for 21/22 evaluable patients (figure 1A). An exemplary IFN-γ ELISpot result (Patient 011) is shown in figure 1B. Patients were grouped according to the strength of their T cell response into strong- (figure 1C), intermediate- (figure 1D) and weak/non-responders (figure 1E). In most cases, vaccine-reactive T cells were detected after 4 vaccinations (visit 6). In strong responders, T-cell frequencies reached a plateau at visit 13 (after 11 vaccinations), which lasted for 13 months postvaccination. Weak-responder patients (Patients 007, 015, 016, 017) showed a maximal response mainly at visit 15, 7 months postvaccination. Specific mean spot counts per patient and visit are displayed in figure 1F. In total, 18/21 (86%) of the patients mounted a T cell response during vaccination and 19/21 (90%) patients during the follow-up (light green). One spontaneous RV001-specific response was detected in Patient 012 PBMCs (visit 2), which was boosted by vaccination (approx. 20 fold) and lasted until the last follow-up visit (figure 1F). Interestingly, Patient 010, classified as non-responder during vaccination, developed a statistically significant response against the RV001 peptide at visit 15, which increased further at visits 16 and 17 (1.4 and 4 fold increase, respectively). Only one RV001-specific response was lost at the last follow-up visit (Patient 007). The high response rate among patients with various HLA allelic products indicates a broad immunogenicity of the vaccine. In addition, T cells were mostly detectable for at least 10 months (visit 16) postvaccination, suggesting the induction of a stable immunological memory.
Vaccine-specific T cells are mainly polyfunctional CD4 T cells
Multifunctional flow cytometry analysis was performed next to identify RV001-specific T cells in vaccine responders (n=18). Cells from one visit during vaccination were re-stimulated with the RV001 peptide for 12 hours and tested per ICS. A representative example for Patient 011 visit 13 is shown in online supplemental figure S1A. Seventeen out of 18 patients (94%) showed a CD4 T cell response against the RV001 (figure 2A). Patient 007, classified as weak-responder by ELISpot, did not reach the positivity threshold in the ICS. CD4 T cells of strong-responder patients expressed more activation markers (mean sum of CD107a, CD154, IL-2, TNF and/or IFN-γ: 21.7%, 95% CI 8.3% to 35.1%) on RV001 restimulation than CD4 T cells of intermediate-responders (mean sum: 9,0%, 95% CI 3.2% to 14.7%) and significantly more as CD4 T cells of weak-responders (mean sum: 2.4%, 95% CI −3.8% to 9.0%) (figure 2B). This supports our previous classification of patients in the three groups according to the ELISpot results. To assess multifunctionality, boolean gating was performed: 81% of the RV001-specific CD4 T cells expressed at least two activation markers and of these, almost half (43%) at least three markers simultaneously. When comparing the three patient groups, especially strong-responders showed a significantly higher frequency of RV001-specific T cells expressing two-three marker combinations (figure 2C). Among all patients, the three most frequent subsets of RV001-specific CD4 were those expressing CD154 and TNF only (mean: 2.8%, 95% CI 1.3% to 4.3%), CD154+TNF+CD107a (mean: 2.5%, 95% CI 0.8% to 4.3%), or CD154+TNF+CD107a+IFN-γ (mean 1.1%, 95% CI 0.1 to 2.1). On average, 0.49% (95% CI 0.19% to 0.80%) of the RV001-specific cells within the CD4 T cell population express all five markers simultaneously (figure 2D).
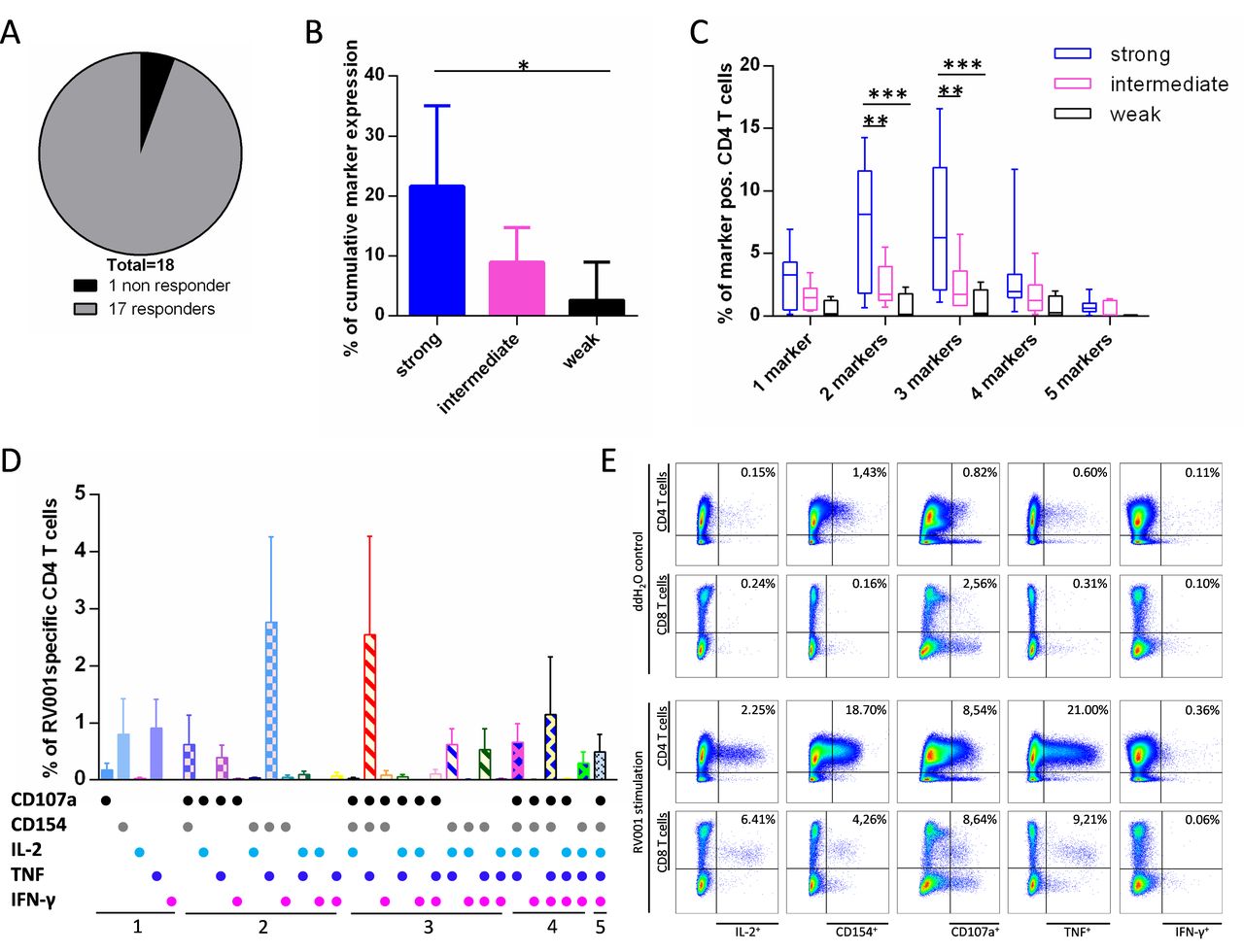
RV001-responding cells are multifunctional. ddH2O stimulated cells harvested from the ELISpot were restimulated with RV001 for 12 hours. Expression of CD107a, CD154, IL-2, TNF, and IFN-γ was examined by ICS on live CD4 and CD8 lymphocytes and the % of RV001-specific cells calculated for each of the 5 markers within CD4 or CD8 cell subsets. (A) Overview of CD4 T cell responses during vaccination (n=18 patients). (B) Mean+95% CI of cumulative marker expression on RV001-specific CD4 T cells for strong- (n=7), intermediate- (n=7) and weak- (n=4) responders. Kruskal-Wallis test with Dunn’s post-test. (C) Min to max percentages of RV001-specific CD4 T cells expressing one to five markers simultaneously, classified per strong- (n=7), intermediate- (n=7), and weak- (n=4) responders. Median values are indicated. Two-way ANOVA with Tukey’s post-test. (D) Mean+95% CI of RV001 specific CD4 T cells expressing each of the five activation markers or combinations thereof (all patients, n=18). (E) 12 day-cultured PBMCs from Patient 004 at visit 14 were restimulated with ddH2O (upper dot-plot panel) or RV001 (lower dot-plot panel). The activation marker expression was examined on living CD4 (upper rows) and CD8 (lower rows) lymphocytes. Percentages of marker+ cells within CD4 or CD8 cells are given. *P≤0.05, **p≤0.01, ***p≤0.001. Responder groups are defined based on the ELISpot results. ANOVA, analysis of variance; ddH2O, deionized water; IFN-γ, interferon-γ; IL-2, interleukin-2; TNF, tumor necrosis factor.
PBMCs obtained from n=10 patients (including the non-responder Patient 007) after the last vaccination (follow-up 1, visits 14 and 15) were also examined. Nine out of 10 patients still showed a CD4 T cell response against the RV001, while Patient 007 was still a non-responder (not shown). In addition to a CD4 T cell response, Patient 004 showed also a CD8 T cell response against the RV001 peptide at both visits (figure 2E). In summary, this multiparametric analysis shows that RV001-specific T cells are mainly polyfunctional CD4 effectors, and identifies in addition one CD8 T cell response.
Vaccination against RhoC induces memory effector CD4 T cells
A long-lasting antitumor immune response is mediated by T-cell memory formation. To address the phenotype of RV001-specific T cells, we examined the differentiation status (CD45RA/CCR7), as well as the expression of OX-40 (activation marker), PD-1 (activation/exhaustion marker), and LAG-3 (exhaustion marker) by ex vivo multiparametric flow cytometry. Gating strategy for Patient 018 is available in online supplemental figure S1B. Patients with a strong or intermediate IFN-γ response in the ELISpot were selected. RV001-specific T cells were identified by their TNF expression after stimulation (12 hours). This short culture-step does not modify the expression of the selected markers on T cells (data not shown). A representative overlay of CD4+TNF+ cells (black) on all CD45RA/CCR7 gated CD4 T cells (gray) is shown for Patient 009 in figure 3A. RV001-specific T cells were mostly effector memory T cells (CD45RA-CCR7-) and detectable already after the 4th vaccination at visit 6. The response peaked between visits 6 and 13 for all three patients tested (see also online supplemental figure S3). PD-1 and to a lesser extent OX-40 (but not LAG-3, data not shown) were expressed on RV001-specific cells. However, median fluorescence intensities did not appear to increase in the course of the vaccination as compared with those of the whole CD4 subset, suggesting that vaccine-specific cells did not especially differentiate towards an exhausted phenotype on repeated vaccination (figure 3B).

RV001-specific T cells are effector memory T cells. PBMCs from Patient 005 (visits 6–15) and Patients 009 and 018 (visits 6–17) were thawed, rested and stimulated either with RV001 or ddH2O for 12 hours. Live RV001-specific CD4 lymphocytes were identified by TNF expression and examined for the expression of CD45RA, CCR7, PD-1, OX-40, and LAG-3. (A) Exemplary results (Patient 009): CD4+TNF+ cells (black) are overlaid on the whole CD4 cell population (gray). Numbers indicate CD4+TNF+ cell counts. (B) Expression profile of PD-1 (upper row) and OX-40 (lower row) for n=3 patients at visit 6 and visit 15. Numbers indicate MFI ratios of the two receptors between CD4+TNF+CD45RA-CCR7- cells (dark gray) and CD4+TNF-CD45RA-CCR7- cells (light gray). Histograms show event counts normalized to mode. (C–E) Assessment of Tregs (CD3+CD8-CD4+CD127-CD25+Foxp3+) from 5 patients (4 visits). (C) Mean+95% CI of total CD4 cells (left-axis, black curves) and Tregs (right axis, blue curves) within lymphocytes. (D) Kinetics of Treg percentages within CD4 T lymphocytes for each of the 4 patients. (E) Exemplary dot-plots for Patient 003, percentages of CD3+CD8-CD4+CD127-CD25+Foxp3+ cells are indicated. ddH2O, deionized water; MFI, median fluorescence intensity; TNF, tumor necrosis factor.
We further examined the proportion of regulatory T cells (Tregs defined as CD3+CD8-CD4+CD25+CD127-FoxP3+) in two strong- (Patients 003/011), one medium- (Patient 009), one low- (Patient 016) and one non- (Patient 024) responders before (visit 2), during (visit 6 or 8), and after vaccination (visits 14 and 17). In mean, total CD4 T cells increased from 19.3% (visit 2) to 27.3% (visits 6/8; 1.41 fold) and Tregs from 0.42% (visit 2) to 0.53% (visits 6/8, 1.26 fold) during vaccination. Thirteen months after the last vaccination (visit 17), both subsets had returned to prevaccination levels (figure 3C). Examination of the patients according to their responder groups (figure 3D) revealed that Tregs decreased approximately 2-fold during vaccination in strong-responder patients (one representative example is shown in figure 3E), were stable for the medium responder patient, and increased slightly (~1.3-fold) for the low-responder and non-responder patients.
Altogether, these results indicate the induction of functional effector memory cells, but not of Tregs, on vaccination.
Several epitopes are recognized by T cells within the RV001 sequence
The 20 aa long RV001 peptide might serve as a CD4 T cell epitope itself, but might also contain several shorter CD4 T cell epitopes. To identify such sequences, we expanded RV001-specific T cells from selected PBMC samples (n=7, two follow-up visits each). After 12 days, cells were restimulated with RV001, or with single RV001-derived 15mer peptides (ATR15, AGL15, LQV15; 15aa overlap) for 12 hours, followed by ICS staining. Representative results for CD4 T cells of n=3 patients from visit 16 samples are shown in figure 4A and in online supplemental figure S4. We found that all three peptides could be recognized, although at various rates. Peptide AGL15 was recognized by cells from all three patients, peptide LQV15 by cells from Patients 001 and 003, and ATR15 by cells from Patient 019 only.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The RV001 sequence comprises several promiscuous HLA-class II epitopes and one HLA-B*27:05 restricted epitope. The expression of CD107a, CD154, IL-2, TNF and IFN-γ was examined on live CD4 or CD8 cells. (A) Cells were restimulated with RV001 or with RV001-derived 15mer peptides (ATR15, AGL15, LQV15) for 12 hours. Shown are the percentage or mean+SD percentage (n=2 repeated measurements) of peptide-specific CD4 cells expressing each activation marker for three patients at visit 16. (B) LCLs were preloaded with RV001, ATR15, or AGL15 and incubated with HLA-matched patient cells at 1:2 ratio for 12 hours. Shown are the specific percentages of CD4 cells expressing the indicated activation markers. (C) Cells were restimulated either with the RV001 peptide alone or with RV001-preloaded C1R or C1R-HLA-B*27:05 cells for 12 hours in the ICS. Shown are the percentages of specific marker expression on CD8 cells. IFN-γ, interferon-γ; IL-2, interleukin 2; LCLs, lymphoblastic cell lines; TNF, tumor necrosis factor.
Next, we used two human LCLs MGAR and H0301 to identify the presenting HLA-class II allele/s for Patients 001, 003 and 019. According to the four-digit HLA-typing, the HLA-DRB1*13:02 allele was expressed by Patient 001, Patient 003, and by H0301, whereas HLA-DRB1*15:01, -DQB1*06:02, -DPB1*04:01 alleles were shared between Patient 019 and MGAR (online supplemental table S3). RV001 prestimulated PBMCs were mixed with preloaded (RV001, ATR15, or AGL15) LCLs for 12 hours. Both 15mers were recognized by CD4 cells of Patients 001 and 003. For Patient 019, only a response against RV001 was detected, while coincubation with ATR15 or AGL15 preloaded LCLs led to an increase in IFN-γ, CD154 and IL-2, but did not reach the predefined positivity threshold (figure 4B). Based on these findings, we concluded that several epitopes derived from the RV001 sequence are presented by HLA-DRB1*13:02, and possibly by DRB1*15:01, and/or -DQB1*06:02, and/or -DPB1*04:01, three alleles co-expressed in 80% of the HLA-DRB1*15:01+ patients in the cohort.
The next step was to identify the presenting allele of the RV001-derived HLA-class I epitope that was recognized by CD8 T cells from Patient 004 (HLA-A*02/A*30+, -B*18/B*27+), (figure 2E). PBMCs from Patient 004 visit 15 were prestimulated with RV001, then incubated with either RV001 or with C1R or C1R-HLA-B*27 LCLs preloaded with RV001. A CD8 T cell response (CD107a, CD154, TNF and IL-2 expression) was observed on stimulation with the RV001 peptide or with the RV001 preloaded HLA-B*27 LCL, but not with the non-transfected LCL (figure 4C). These findings clearly show that a RV001-derived sequence is presented to CD8 cells by the HLA-B*27:05 allele.
In summary, the RV001 peptide contains at least three different HLA-class II peptides promiscuously presented on various HLA-class II allelic products, as well as one HLA-class I peptide presented by the HLA-B*27:05 allele.
Vaccine safety and PSA levels
No adverse events led to discontinuation of the treatment in any patient. Most frequent treatment-related events were fatigue and injection site reactions of grades 1 or 2. All patients experienced at least one TEAE of injection site reaction that was considered related to the RV001 vaccine. Four patients (18%) experienced at least 1 grade 2 TEAE of injection site reaction. One patient had a TEAE of fatigue (grade 1) and one patient a TEAE of hot flush (grade 1), both events probably related to the RV001 vaccine. No treatment-related side effects of grade 3 or higher occurred (table 2).
Treatment-related side effects
Clinical response was not a primary endpoint of this phase I/II study, but patients were routinely monitored for PSA serum levels (online supplemental table S2). PSA doubling time is regarded as a surrogate parameter for PCa progression, and can predict the risk of mortality in men with localized PCa.27 Three patients progressed biochemically during follow-up (Patients 006, 015, 018), from whom two had BCR at study entry (Patient 006: followed for 24 months, 12 PSA measurements; Patient 018: followed for 52 months, 9 PSA measurements). Patient 006 had a PSA increase from 0.5 to 1.1 µg/L 29 months after study entry, and Patient 018 from 1.1 to 1.5 µg/mL 24 months after study entry. Patient 015, who received only seven vaccinations and presented with a pT2c R+ Gleason score 7 (4+3) PCa, developed BCR with a PSA doubling time of 1.2 years and a final PSA level of 0.28 µg/L 26 months after last vaccination. When comparing the prestudy PSA doubling time to that on study, we observed an increase from 1.3 to 2.1 years for Patient 006 and from 1.95 to 3.8 years for Patient 018. The PSA levels and IFN-γ spot counts are shown for both patients in the online supplemental figure S5. Interestingly, Patient 018 showed stable PSA and IFN-γ spot counts during the observation period, whereas raising PSA at the end of the treatment in Patient 006 was concomitant to a reduction of vaccine-specific cells. Thirteen months postvaccination, Patients 015 and 006 were in BCR (visit 17), however, PSA level in Patient 006 declined the last months on follow-up, and from a clinical perspective, the patient was considered as biochemically stable. No patient developed clinical recurrence.
Discussion
Recent cancer vaccine studies targeting the tumor mutanome have demonstrated a high rate of immunological responses as well as encouraging clinical courses.1 2 Such studies are per se individualized, since they target mutations that mostly occur in individual patients. Other vaccine strategies that are suitable for tumors with low level of mutational events focus on non-mutated, tumor-specific or tumor-associated antigens.3 11 28 29 Prostate tumors generally harbor only few mutations and respond poorly to checkpoint Ab therapy. Clinical studies have demonstrated that vaccination against prostate-associated or overexpressed antigens is safe, immunogenic and can impact clinical course. Most of these trials have been conducted at advanced stages of the disease (castration-resistant metastatic PCa), but few at earlier times, for example, at biochemical relapse.28–31 Immune intervention at BCR, when tumor load is limited and immunosuppression absent or limited, might lead to more favorable clinical outcome. In this context, targeting of antigens associated with cancer stem cells and metastases, like the RhoC protein, could improve tumor control.
The SLP vaccination against RhoC was safe for all patients over the complete treatment period. Side effects were predominantly injection site reactions and fatigue (grade 1/2). These events are most likely related to the adjuvant and carrier Montanide ISA-5132 and did not necessitate specific medical intervention.
Immunogenicity of the vaccine was assessed in n=21 patients. Most of these (94%) had developed T cells specific for the RhoC peptide after the 4th vaccination. After a short presensitization, we frequently observed high IFN-γ ELISpot counts (>1000 spots/200.000 cells), indicating excellent immunogenicity of the vaccine in vivo and/or a robust in vitro proliferation capacity. T cell responses were overall stable and were detected almost 1 year after the last vaccination. Vaccine-specific cells were also detectable by ex vivo ELISpot in strong-responder patients (n=4 tested, data not shown). This long-lasting functional immune response indicates the establishment of an immunological memory, which is essential for immunosurveillance of recurrent tumors and/or metastases. We found that antivaccine T cells belonged predominantly to the effector memory CD4 subset and were polyfunctional cells (>80% of RV001 specific CD4 T cells expressed at least two of the activation markers/cytokines tested). They often expressed the cell-surface degranulation marker CD107a, suggesting that at least a fraction of those were cytotoxic effectors. Moreover, ex vivo phenotyping (PD-1, LAG-3) strongly suggests that RV001-specific cells did not shift towards an exhausted phenotype after multiple vaccine applications. During vaccination, Tregs appeared to decrease in the two strong-responder patients, while they increase slightly in the two low/non-responder patients. Altogether, essentially functional T helper cells were induced by the vaccine.
Although the vaccine contained a single 20mer peptide, we could identify three HLA-class II 15mer RV001-derived epitopes. The high response rate in our patient cohort indicates that these, and possibly further as yet unidentified class-II epitopes, are presented promiscuously on several HLA-class II alleles (including DRB1*13:02). Hence, the RhoC vaccine can be applied broadly, independently of the patient’s HLA allotype. In addition, a CD8 response restricted to HLA-B*27:05 could be observed in one patient. The exact epitope is under characterization, and further HLA-B*27+ patients will be assessed. Interestingly, RV001-specific CD8 T cells in that patient were polyfunctional, with high levels of TNF and/or CD107a, IL-2, CD154. CD154+ CD8 T cells (also called CD8 helper cells) have been shown to support their own expansion and differentiation and to activate dendritic cells to promote antitumor immunity.33 Although the RhoC 20mer contains an embedded HLA-A*03 binding peptide, we did not observe any CD8 T cell response (one single time point tested) in the two patients carrying the HLA-A*03 allele.23 Since epitope spreading was shown to be associated with a better clinical outcome,34 we also tested CD8 T cell reactivities against epitopes derived from PCa-associated antigens (PSA, PSMA, prostein, TRPP8 and survivin) in five HLA-A*02 patients.28 Two individuals responded to 2–3 epitopes before (visit 2), during (visits 6/8) and after vaccination (visits 14/17), and this was increased in one patient after vaccination (data not shown). Such analyzes will be pursued in the ongoing phase II trial.
CD4 T cells are now widely recognized as key players in antitumor immunity. Their role in dendritic cell and CD8 T activation, and in memory formation are well described.6 35 36 CD4 T cells were also shown to kill HLA-class II positive tumors via granzyme/perforin release or Fas/Fas-L interaction,9 37 and there is also preclinical evidence that CD4 T cells can reject tumors better than CD8 T cells.10 We could not test the HLA-class II expression of PCa tissues in our cohort, but HLA-class II expression has been detected on primary PCa cells and on PCa cell lines, or could be induced by IFN-γ.38 39 Even in the absence of HLA-class II expression on tumor cells, indirect tumor killing or tumor senescence can be mediated by CD4 T cells, in particular via IFN-γ and TNF,7 8 or by the recruitment of nitric oxide producing macrophages within the tumor.40 Altogether, our phenotyping and functional data indicate that the profile of vaccine induced, RV001-specific CD4 T cells is in line with that of antitumor effectors.
Vaccine-based studies in mice and patients have started to unravel the contribution of CD4 T cells in tumor control.11 41 42 High rates of polyfunctional CD4 cells, together with CD8 T cells, were detected in melanoma patients vaccinated with a personalized, neoantigen-based SLP vaccine containing up to 20 aa long mutated peptides. Four out of six patients had no recurrence 25 months postvaccination, while the two patients with tumor recurrence achieved tumor regression when treated with an anti-PD-1 Ab.1 Case reports document tumor regressions after adoptive transfer of antitumor CD4 cells.43 44 In mouse models, there is also evidence that CD4 chimeric antigen receptor (CAR) T cells are more potent than CD8 CAR T cells, as they can kill tumors, and in addition, exhibit long-lasting effector function.45 46
The finding of an increase in PSA doubling time in two patients following vaccination shall be interpreted with caution, as PSA levels were altogether low, and kinetics are influenced by the period over which the PSA values are measured (limited time span in our study) and the number of tests drawn.47 It is estimated that approximately one in four patients undergoing RP will eventually experience BCR. The risk of BCR varies according to preoperative PSA and histopathological findings.48 49 The individual risk of BCR in the first year following enrolment in this study ranged from 2% to 19% and did not change substantially in the period of follow-up.50
In summary, vaccination against RhoC is a well-tolerated treatment option which induces a long-lasting immune response in the large majority of patients. Vaccine-specific cells are polyfunctional and equipped for an antitumor response. A correlation between the induction of immune responses to RhoC on vaccination and clinical outcome is at this stage premature, but will be examined in a recruiting double-blind, placebo-controlled, phase II trial for PCa patients in BCR (NCT04114825). RhoC vaccination to impair tumor spreading might also synergize with many tumor vaccines (such as those targeting patient-individual tumor antigens) or other therapies, and become a valuable approach for PCa and for further tumor entities.
Acknowledgments
We thank all patients for participating in this study, A. Ljungqvist for support throughout the study, L. Yakes for proofreading of the manuscript, R. Klein for help with HLA-typing and N. Mitrakis for technical help.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conception and design: CG, PTS, JuS and JeS. Development of methodology: JuS and CG. Acquisition, analysis, reagents and interpretation of data: JuS, TB, JRR, CS, SH, JoS, KB, MAR, PTS and CG. Writing: JuS, CG and all authors. Study supervision: CG, PTS and H-GR.
Funding The study was sponsored by RhoVac ApS, Agern Alle 24, DK-2970 Hoersholm Denmark. CG, JuS and H-GR were supported by a research grant from RhoVac ApS.
Competing interests CG and H-GR receive a research grant from RhoVac ApS. PTS is consultant at RhoVac ApS. CS is employee of DanTrials. TB is owner and CEO of DanTrials.
Patient consent for publication Not required.
Ethics approval The trial was approved by the local ethics committee (H-1604701) and the European Union regulatory authorities (EuDRACT number: 2016-004189-24), and was conducted according to the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data relevant to the study are included in the article or uploaded as online supplemental information. Additional information is available on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.