Article Text

Abstract
Background Myeloid-derived suppressor cells (MDSC) are a functional myeloid cell subset that includes myeloid cells with immune suppressive properties. The presence of MDSC has been reported in the peripheral blood of patients with several malignant and non-malignant diseases. So far, direct comparison of MDSC across different diseases and Centers is hindered by technical pitfalls and a lack of standardized methodology. To overcome this issue, we formed a network through the COST Action Mye-EUNITER (www.mye-euniter.eu) with the goal to standardize and facilitate the comparative analysis of human circulating MDSC in cancer, inflammation and infection. In this manuscript, we present the results of the multicenter study Mye-EUNITER MDSC Monitoring Initiative, that involved 13 laboratories and compared circulating MDSC subsets across multiple diseases, using a common protocol for the isolation, identification and characterization of these cells.
Methods We developed, tested, executed and optimized a standard operating procedure for the isolation and immunophenotyping of MDSC using blood from healthy donors. We applied this procedure to the blood of almost 400 patients and controls with different solid tumors and non-malignant diseases. The latter included viral infections such as HIV and hepatitis B virus, but also psoriasis and cardiovascular disorders.
Results We observed that the frequency of MDSC in healthy donors varied substantially between centers and was influenced by technical aspects such as the anticoagulant and separation method used. Expansion of polymorphonuclear (PMN)-MDSC exceeded the expansion of monocytic MDSC (M-MDSC) in five out of six solid tumors. PMN-MDSC expansion was more pronounced in cancer compared with infection and inflammation. Programmed death-ligand 1 was primarily expressed in M-MDSC and e-MDSC and was not upregulated as a consequence of disease. LOX-1 expression was confined to PMN-MDSC.
Conclusions This study provides improved technical protocols and workflows for the multi-center analysis of circulating human MDSC subsets. Application of these workflows revealed a predominant expansion of PMN-MDSC in solid tumors that exceeds expansion in chronic infection and inflammation.
- biomarkers
- tumor
- immunity
- cellular
- immunoassay
- myeloid-derived suppressor cells
- head and neck neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Myeloid-derived suppressor cells (MDSCs) can expand in the peripheral blood of patients with several malignant and non-malignant diseases as a consequence of altered myelopoiesis. MDSCs represent a still relatively new functional myeloid cell subset and there are few methods available to compare this subset across different diseases.1 2 MDSCs are commonly subdivided into two main subtypes, based on their morphology, density and cell surface markers: polymorphonuclear MDSC (PMN-MDSC) and monocytic MDSC (M-MDSC). An additional subtype, which lacks macrophage and granulocyte markers, is called early-stage MDSC (e-MDSC) and it has been shown to accumulate in several disease settings.3 4 The main biological function attributed to MDSC is immune suppression of T cells through several mechanisms such as production of arginase 1, inducible nitric oxide synthase, indoleamine dioxygenase, cyclooxygenase and reactive oxygen species.5
Bronte et al published in 2016 an extensive paper containing recommendations for the study of human and mouse MDSC; the paper provided a useful list of markers, gating strategies and functional tests to be used for MDSC study.4 This was definitely one of the first solid milestones in MDSC study standardization. However, still there is the need to further refine the process by comparing MDSC frequencies and functions across multiple diseases. A significant problem in the field is the lack of robust and reproducible consensus protocols and markers that would allow the comparative analysis of MDSC in different disease settings and multicenter trials.
In order to overcome these limitations, the authors of this paper formed a study group within the COST Action Mye-EUNITER (www.mye-euniter.eu). The main goal of Mye-EUNITER was to further standardize and facilitate the study of human MDSC in multiple human diseases. Within this network, we developed an international research program (Mye-EUNITER MDSC Monitoring Initiative, Mye-MMI), which involved 13 European labs and performed the first pan-European cross comparative study on human MDSC, with almost 400 patients analyzed. This study uses a consensus standard operating procedure (SOP)-like protocol for the isolation and phenotyping of human circulating MDSC. Analysis of peripheral blood mononuclear cells (PBMC) from healthy donors showed that technical variables, such as the anticoagulant and separation method used, cause artifactual variabilities in the frequency of MDSC. When using an optimized and standardized protocol and comparing results to healthy control subjects, we observed a significant expansion of PMN-MDSC in solid cancers that was less pronounced in non-malignant chronic infectious and inflammatory disease settings. Expression of programmed death-ligand 1 (PD-L1) was mainly observed in M-MDSC and e-MDSC, while PMN-MDSC express Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1).
Methods
Human blood collection and description of patient cohorts
Lab 1 (Essen, Germany)
Peripheral blood was prospectively collected from treatment-naïve patients with primary head and neck cancer (HNC) and from healthy donors. Patients with synchronous carcinoma in another location or concomitant systemic infectious disease were excluded. All control samples are age matched to the patient group.
Lab 2 (Edinburgh, UK)
Peripheral blood (10 mL) was obtained from breast cancer patients from National Health Service, Edinburgh, Scotland, UK. The exclusion criteria for all patients with cancer at baseline included systemic metastatic disease, any inflammatory disorder and active infection or immunocompromised status not related to cancer. All the patients recruited were chemotherapy and radiotherapy naive before collection. All control samples are age matched and sex matched to the patient group.
Lab 3 (Nijmegen, The Netherlands)
Peripheral blood samples were collected from healthy individuals and glioma patients undergoing neurosurgical resection or biopsy for intracranial tumors at the Radboud University Medical Center (Radboudumc). Glioma patients receive dexamethasone as part of their treatment. All patients had histologically proven brain tumors diagnosed by neuropathologists of the Radboudumc. The tumors were classified according to WHO 2016 Classification of tumors of the Central Nervous System, and encompassed low-grade diffuse astrocytoma, isocitrate dehydrogenase (IDH)-mutant (WHO grade II), oligodendrogliomas, IDH-mutant and 1 p/19q-codeleted (WHO grade II, WHO grade III), and glioblastomas (grade IV). The healthy donors were anonymous and not age and sex matched.
Lab 4 (Heidelberg, Germany)
Peripheral blood samples (30 mL) were obtained from 21 melanoma patients of stage I–IV who were seen at the Skin Cancer Center (University Medical Center Mannheim, Germany) from February 2017 to March 2018. Tumor stages were determined according to the eighth edition of the American Joint Committee on Cancer (AJCC) classification. All the patients recruited were not treated within the last 6 months before blood collection. For control samples, mononuclear cells were isolated from peripheral blood obtained from the Institute of Transfusion Medicine and Immunology, Medical Faculty Mannheim, Heidelberg University, German Red Cross Blood Service Baden Württemberg–Hessen (Mannheim, Germany) after informed consent. All control samples are age matched to the patient group.
Lab 5 (Belgrade, Serbia)
No blood samples from lab five are presented in this manuscript.
Lab 6 (Ankara, Turkey)
Peripheral blood samples (~10 mL) were obtained from healthy volunteers, and newly diagnosed, treatment-naïve colorectal cancer patients. All control samples are age matched and sex matched to the patient group.
Lab 7 (Warsaw, Poland)
For control samples, mononuclear cells were isolated from peripheral blood obtained from female healthy individuals from Military Institute of Medicine. Peripheral blood (10 mL) was obtained from ovarian cancer patients from Military Institute of Medicine (Warsaw, Poland). The exclusion criteria for all patients with cancer at baseline included systemic metastatic disease, any inflammatory disorder and active infection or immunocompromised status not related to cancer. All the patients recruited were chemotherapy and radiotherapy naive before collection. All control samples are age matched and sex matched to the patient group.
Lab 8 (Essen, Germany)
For control samples, mononuclear cells were isolated from the peripheral blood obtained from healthy volunteers at the Institute of Virology with their agreement to use the blood for the Mye-MMI study. Peripheral blood (10 mL) was obtained from non-treated patients with hepatitis B virus (HBV) infection from Clinic for Gastroenterology and Hepatology, University Hospital Essen. All control samples are age matched to the patient group.
Lab 9 (Paris, France)
For the study on HIV-1 infection, peripheral blood was collected on heparin from eleven antiretroviral treated, aviremic individuals living with HIV and from eleven healthy donors. The inclusion criteria for the people living with HIV-1 were as follows: plasma HIV RNA levels of <50 copies/mL for at least 24 months. Exclusion criteria were as follows: inflammatory diseases, infection with HIV-2, active co-infections, <200 CD4 cells, recent (<3 months) vaccinations. The healthy donors were anonymous and not age and sex matched.
Lab 10 (Lisbon, Portugal)
Peripheral blood (10 mL) was obtained from HIV-1 -infected subjects and healthy volunteer blood donors. The HIV-1-infected patients were studied immediately after diagnosis before starting antiretroviral therapy. All patients featured detectable plasma HIV RNA levels (802–8 61 828 copies/mL) and have broad CD4 T cell count distribution (10–866 cells/µL). The other exclusion criteria for HIV-1-infected patients included HIV-2 infection, no evidence of opportunistic infection or tumor, and no recent vaccinations (<3 months). All control samples are age matched to the patient group.
Lab 11 (Krakow, Poland)
For control samples, mononuclear cells were isolated from peripheral blood obtained from healthy individuals through blood donation center or research lab. Peripheral blood (10 mL) was obtained from psoriasis patients from Dept. of Dermatology, Zeromski Hospital, Krakow, Poland. The severity of the psoriatic skin lesions was assessed according to the Psoriasis Area Severity Index score. Patients on UV therapy, systemic or local corticosteroid treatment were excluded from the studies. Healthy control subjects had no clinical signs of dermatologic or inflammatory diseases. All control samples are age matched to the patient group.
Lab 12 (Stockholm, Sweden)
Healthy adults were intramuscularly vaccinated with one dose of the yellow fever vaccine (Stamaril, Sanofi) or the tick-borne encephalitis vaccine (Encepur, GlaxoSmithKline). Peripheral venous blood was collected at the day prior to vaccination and at day 2 after vaccination. Prevaccination controls and postvaccination samples from the same individuals were compared.
Lab 13 (Bergen, Norway)
Peripheral blood was collected from patients with acute chest pain admitted at Haukeland University Hospital (Bergen, Norway). The patients were included consecutively in two cohorts. We included subjects above 18 years of age who were hospitalized due to recent-onset chest pain of suspected cardiac origin and who received acetylsalicylic acid during ambulance transport or when arriving at the hospital, on suspicion of cardiovascular disease (CVD). Patients who were on immunomodulating medications, for example, corticosteroids, or with severe infections, were excluded from the study. Patients in whom CVD was confirmed were defined as acute chest pain patients with CVD. The patients where no CVD could be detected were defined as acute chest pain patients without CVD.
We initially planned to use and we strived for age and sex matched non-CVD donors for this study. However, due to time restriction and the available donors at the period of sampling, age and sex matching was not achieved for all patients.
Detailed clinical information for all patients involved in the study can be found in online supplementary table 1.
Supplemental material
Isolation of PBMC according to standardized protocol
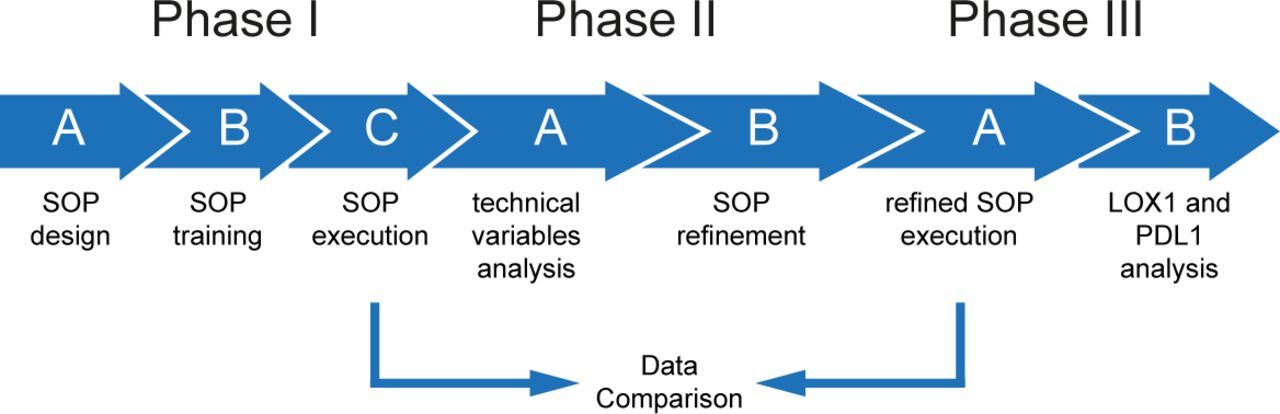
In phase I of this study (compare figure 1), participants used either heparin, trisodium citrate or Ethylenediaminetetraacetic acid (EDTA) anticoagulated blood. Blood was processed within 1 hour after blood sampling in all phases of this study. Before separation blood was admixed with the same volume of phosphate-buffered saline (PBS) and overlayed on 1.077 g/mL separation medium (Biocoll, Merck Millipore; Ficoll-Paque, GE healthcare; Pancoll, PAN-Biotech; Lymphoprep, Stemcell Technologies). Density centrifugation was performed at room temperature 400xg, 30 min without acceleration and brake. Afterward plasma was aspirated until 5 cm above PBMC fraction. PBMCs were collected using disposable Pasteur pipette into 50 mL centrifugations tubes (Falcon 352096). To remove thrombocytes, cells were washed by filling the tube up with PBS followed by centrifugation at 300xg for 8 min at room temperature until the supernatant was clear. After the last washing step, cells were resuspended in Roswell Park Memorial Institute (RPMI) 1640 culture medium supplemented with 100 IU/mL penicillin, 100 mg/mL streptomycin and 10% (v/v) heat-inactivated FCS for counting. To study the influence of the anticoagulants all participants received the same batch of heparin, trisodium citrate or EDTA collection tubes and Safety-Multifly-needle 21G (all Sarstedt, Nümbrecht, Germany) that were shipped from the core lab (lab 1) to the participating centers (phase IIA, figure 1).

Schematic representation of the Mye-MMI study. See manuscript text for details on the different phases and steps. The SOP-like protocol for MDSC immunophenotyping that was generated in the context of this study is available from the corresponding author on reasonable request. MDSC, Myeloid-derived suppressor cells; Mye-MMI, Mye-EUNITER MDSC Monitoring Initiative; PD-L1, programmed death-ligand 1; SOP, standard operating procedure.
In phase III of the study, the samples were collected in a harmonized fashion using trisodium citrate as anticoagulant and using Biocoll as the separation medium.
Immunophenotyping of MDSC subsets by flow cytometry
For characterization of MDSC subsets in phase I and II of the study, all participants used the same antibody clones with free choice of fluorochromes. In brief, 1×106 cells in a total volume of 50 µL were stained with CD15 clone HI98, CD14 clone MOP9, CD33 clone WM53, CD11b clone ICRF44, HLA-DR clone G46-6n and lineage cocktail including CD3 clone SK7, CD19 clone H1B19, CD20 clone 2H7 (not mandatory) and CD56 clone NCAM16.2 in the presence of FcR block in Fluorescence-activated cell sorting (FACS) buffer (PBS/2% bovie serum albumine (BSA)) for 20 min at 4°C. FMO control was used for CD11b and HLA-DR. Cells were washed with FACS buffer, centrifuged at 400xg for 5 min and resuspended in 200 µL FACS buffer for acquisition.
In phase III, participants included the functional marker PD-L1 clone 29E.2A3 and LOX-1 clone 15C4. Isotypes were used as negative controls. Isolation of PBMC, MDSC antibody labeling and data acquisition was locally performed in the participating centers. Because of technical constraints with multiparameter options at local flow cytometers and because of limited choices of commercially available fluorochromes for LOX-1, CD11b could not be included in the second step. Locally acquired data files were uploaded to a protected data server and centrally analyzed in the core lab (Essen, Germany) to ensure uniform conditions for gating for all samples from all centers.
T cell suppression assay
For validation of MDSC suppressive capacity, we used a previously published protocol of flow cytometry MDSC sorting and suppression of polyclonally stimulated autologous cancer patient-derived T cells.6 In brief, CD3 depleted PBMC from cancer patients were labeled with CD66b clone 80H3, CD33 and HLA-DR and sorted on lowest flow rate for HLA-DR-/CD33high (M-MDSC), HLA-DR-/CD33dim/CD66b+ (PMN-MDSC), HLA-DR-/CD33dim/CD66b- (e-MDSC).
The positively selected CD3 T Lymphocytes were labeled with 10 µM Proliferation Dye eFluor 450 (CPDye405) according to manufacturer instructions (eBioscience, Frankfurt am Main, Germany). T cells were stimulated with coated CD3 (1 µg/mL, clone OKT-3, eBioscience) and CD28 (2 µg/mL, clone CD28.2 Beckman coulter) in L-lysine and L-arginine free RPMI medium (Thermo Fisher Scientific) supplemented with 150 µM L-Arginine, 0.218 mM L-Lysine hydrochloride (both Sigma-Aldrich, Taufkirchen, Germany), 100 IU/mL penicillin, 100 mg/mL streptomycin and 10% (v/v) heat-inactivated FCS. Autologous MDSC-subsets were added in a T-cell: MDSC ratio of 2.5:1. CPDye405 intensity was analyzed by flow cytometry after 4 days of coculture and proliferation. Supernatants of the coculture were collected and IFN-γ measured by ELISA according to manufacturer’s protocol.
Patient and public involvement
Patients were not involved in the design of the study and the analysis and discussion of the data. The background of this study and the overall concept of Mye-EUNITER Initiative are shared with the general public by two publicly available videos. Videos are currently available at www.Mye-EUNITER.eu.
Analysis and statistics
Data from all participants were collected, uploaded to a protected data server and centrally analyzed in the core lab (Essen, Germany). GraphPad Prism Software (GraphPad Software, La Jolla, California, USA) was used for statistical analysis and significance was assessed with Mann-Whitney, Wilcoxon matched-pairs signed rank or Kruskal-Wallis test. Results were considered significant at *p≤0.05, **p≤0.01 and ***p≤0.001.
Results
Development of consensus protocols and execution of training schools
Within this network, we developed an international research program (Mye-MMI), which involved 13 European labs. The Mye-MMI study was divided into three phases: during phase I participants designed and agreed to use a SOP for the immunophenotyping and analysis of MDSC in the blood of human patients and controls (Phase IA, figure 1 ) The Mye-MMI network organized a training school, where scientists from all participating labs executed the SOP and trained together in the core lab (Essen, Germany) using the same samples; the outputs of the training school indicated that the SOP was reproducible and the variance among users minimal (phase IB, figure 1). The training school also provided all participants with practical and hands-on experience with the SOP. T cell suppression is a hallmark of MDSC function. We used a previously published protocol to assess the suppression of cancer patient-derived T cells by autologous circulating MDSC.6 This protocol was trained in the Essen core lab with selected participants and subsequently used for functional validation using patients with HNC and patients with melanoma. All these steps enabled the network to perform the study in different labs and geographical locations using a commonly agreed SOP for immunophenotyping and common assays validating MDSC function.
Expansion of circulating MDSC subsets in cancer exceeds expansion in infectious and inflammatory diseases
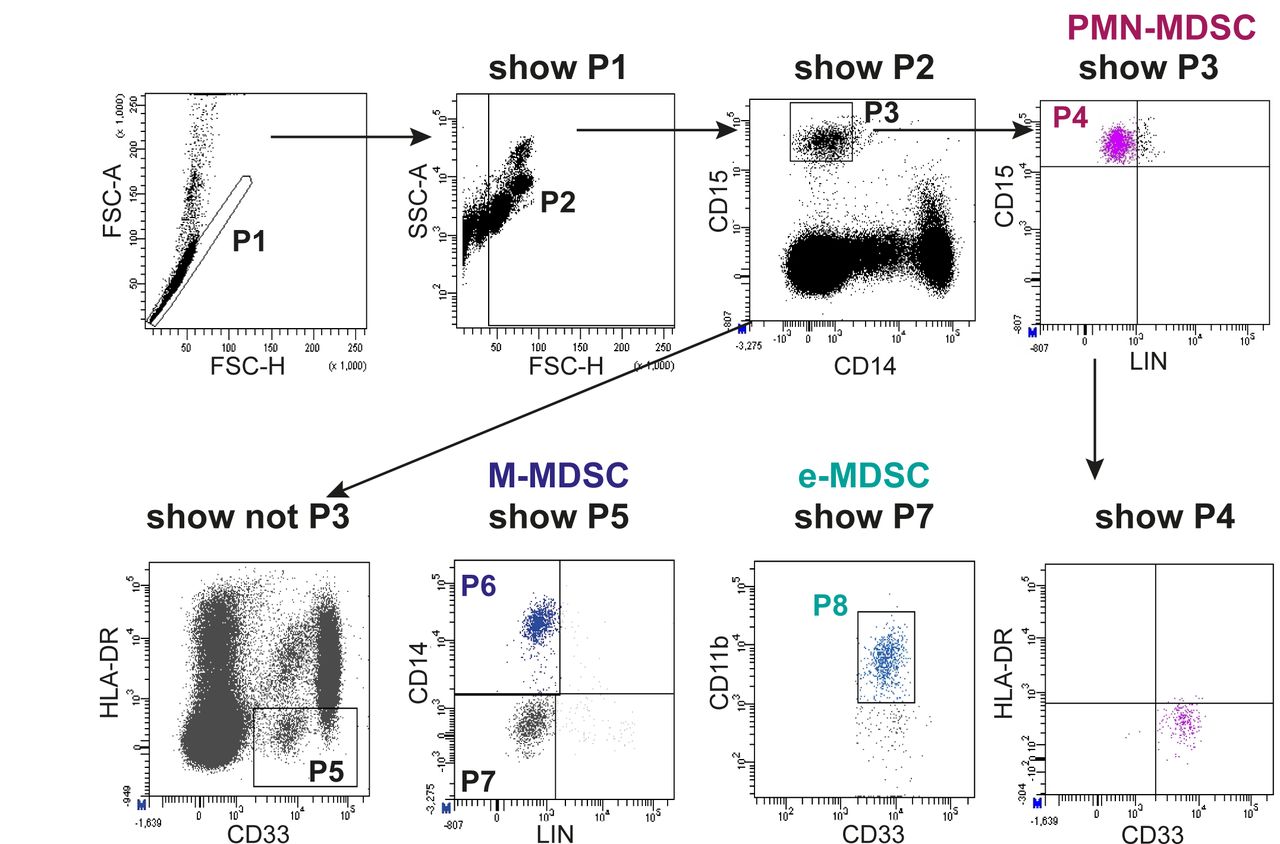
After development of an initial consensus protocol for the immunophenotyping of circulating MDSC and execution of a training school, the trained researchers performed MDSC immunophenotyping in their own Centers (phase IC, figure 1). In each center, a particular cancer type or infection or inflammatory disease was investigated. Data files (.fcs format) from all patients and control subjects were uploaded to a protected server at the coordinating Center (Essen) and centrally analyzed. Figure 2 shows an example of the employed gating strategy. Based on previous experience in the participating centers, we derived a sequential gating strategy that identifies the three major human circulating MDSC subsets with no overlap between subsets. In a first step, we determined the frequency of PMN-MDSC, M-MDSC and e-MDSC in six different types of cancer, major viral infections, psoriasis, a mixed cohort of patients with inflammatory CVDs and in patients that received the yellow fever or the tick-borne encephalitis vaccine. In each center, an independent group healthy donor controls was analyzed (see online supplementary table S1 for patient and healthy donor characteristics). At this stage, the anticoagulant and the blood separation medium were free of choice to the lab.

Flow cytometry gating of the three major subsets of human circulating MDSC. PBMCs were isolated from the peripheral blood of patients and healthy donor controls by density gradient centrifugation in 12 different participating centers and according to a harmonized protocol. Use of predefined clones was mandatory. In a centralized analysis, MDSC subsets were classified as CD15+/CD14-/CD33dim/HLA-DRneg=PMN-MDSC (P4), CD15-/CD14+/CD33pos/HLA-DRneg=M-MDSC (P6) and CD15-/CD14-/CD11b+/CD33dim/HLA-DRneg as e-MDSC (P8).4 An example for the gating strategy is shown (head and neck cancer patient). Note that all MDSC subsets are negative for the lymphocyte lineage markers CD3, CD19 and CD56 (CD20 not mandatory). MDSC, myeloid-derived suppressor cells; PBMCs, peripheral blood mononuclear cells; PMN, polymorphonuclear.
In most cancer types, we observed a substantial induction of PMN-MDSC frequency over healthy controls (figure 3A). No statistically significant increase of PMN-MDSC was observed in patients with infectious and inflammatory diseases, although a tendency for higher frequency of PMN-MDSC was observed in some diseases, in agreement with previous reports.7 In patients with cancer, the induction of the PMN-MDSC subset exceeded expansion of other MDSC subsets (figure 3A). When compared with healthy donors the PMN-MDSC frequency was induced in 5/6 cancer types (figure 3B). In contrast, M-MDSC frequency was only significantly induced in glioma or was even reduced in breast cancer. A particular induction of M-MDSC was observed in patients with inflammatory CVD and after vaccination confirming previous findings,8 but did not change in any other conditions. With the exception of glioma and viremic HIV-1, e-MDSC frequencies were not significantly altered between healthy donors and patients.

Frequency of the three MDSC subsets in malignant and non-malignant disease. For each center the frequency of the three MDSC subsets was determined for patients and the respective local healthy controls. Staining procedure and gating were performed according to figure 1. (A) Median frequency of MDSC compared with healthy donor’s controls (set as value 1) and (B) total frequency within the PBMC are shown. Mean values with SD are shown. Mann-Whitney U test was used for statistical analysis. Results were considered significant at *p≤0.05, **p≤0.001 and ***p≤0.0001. CVD, cardiovascular disease; HBV, hepatitis B virus; MDSC, Myeloid-derived suppressor cells; PBMCs, peripheral blood mononuclear cells.
Although our study was focused on the immunomonitoring and immunophenotpying of MDSC in different diseases, we sought to test and confirm the T cell suppressive activity of the MDSC subsets under investigation. Employing previously published protocols6 we tested the immunosuppressive capacity of MDSC isolated from patients with HNC and melanoma using identical protocols after execution of a training school. In line with earlier findings,6 and in both tumor entities tested, PMN-MDSC were superior to M-MDSC and e-MDSC in suppressing the function of polyclonally stimulated autologous T cells (figure 4).
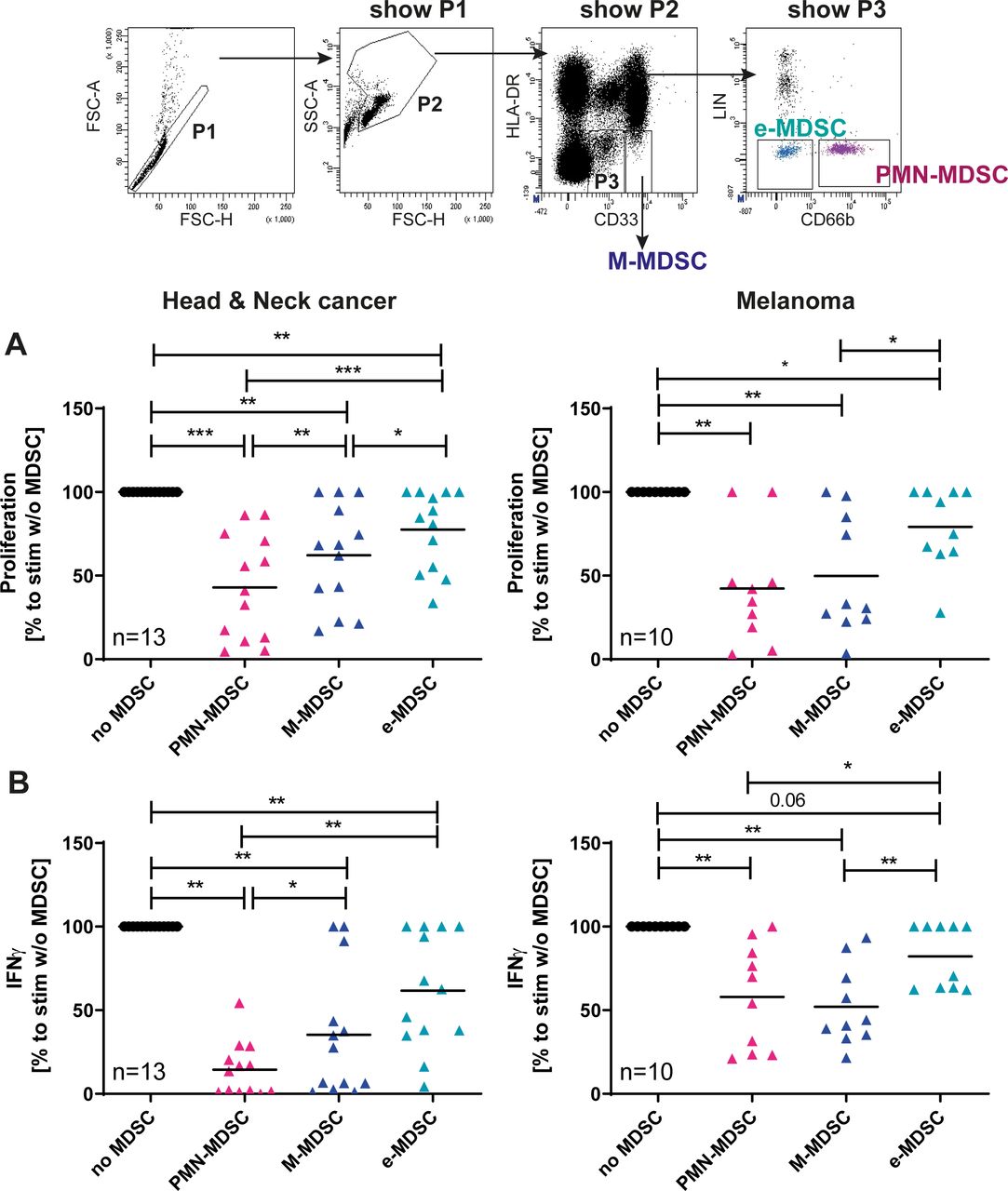
Suppressive activity of MDSC subsets. Suppressive activity of the MDSC subsets was confirmed in two independent laboratories using a shared and previously published protocol.6 (A) Cell proliferation dye labeled Responder T cells from patients were stimulated with plate-bound CD3 and CD28 mAb in the presence or absence of autologous MDSC (T cell: MDSC ratio of 2.5:1). Lymphocyte proliferation was measured at day 4 and analyzed centrally. Relative proliferation to stimulated T cell without MDSC (set as 100%) and mean values are shown. (B) Levels of IFNγ were determined in supernatants of cocultures consisting of T cells activated by plate-bound CD3/CD28 with and without additions of MDSC. IFNγ was determined after 4 days by ELISA. Relative release was calculated to stimulated T cells without MDSC. Mean values are shown. Wilcoxon signed-rank test was used for statistical analysis. Results were considered significant at *p≤0.05, **p≤0.001 and ***p≤0.0001. Gating strategy data are from a patient with head and neck cancer. IFNγ, interferon-γ; M-MDSC, monocytic myeloid-derived suppressor cells; PMN, polymorphonuclear.
Identification of technical and experimental variables that influence immunomonitoring of MDSC
Intercenter analysis of data presented in figure 3 revealed an interesting variability in the frequency of PMN-MDSC in healthy donors (figure 3B, upper panels). Frequency of this MDSC subset in healthy donors varied between 0.08% (lab 1) and 1,8% (lab 3) in the cancer Centers and between 0.3% (lab 11) and 1.9% (lab 12) in the infection & inflammation Centers. We considered it unlikely that this difference is based on biological differences between healthy donors in the respective Centers. A reanalysis of the underlying data and protocols further revealed that, despite standardized immunolabelling and analysis, the participating centers used different anticoagulants and separation media for isolation of MDSC from venous blood of healthy donors and patients. In order to test for a potential effect of the anticoagulant (sodium citrate, EDTA, heparin), we purchased the three types of blood collection tubes at the coordinating Center (lab 1) and shipped them to eight participating Centers for further use (phase IIA, figure 1). Centers obtained venous blood from healthy donors into three different collection tubes and used their preferred separation medium for MDSC isolation. MDSC labeling was performed according to the standardized consensus protocol and data files were sent to the coordination Center for centralized analysis. Analysis of MDSC frequencies showed that the type of anticoagulant had little influence on the frequency of M-MDSC and e-MDSC. In contrast, a clear increase in PMN-MDSC frequency was observed when heparin was compared with sodium citrate (figure 5A). This increase was observed for all types of separation medium. In order to test for a potential effect of the separation medium, we reanalyzed the data of healthy donors with the separation medium being the variable factor (figure 5B). Results revealed an increased frequency of PMN-MDSC when Ficoll was used as the separation medium (figure 5B, left column). Samples prepared via Biocoll showed the lowest frequency of M-MDSC in healthy donors. For PMN-MDSC Biocoll and Lymphoprep showed the lowest frequencies. Based on these data we concluded that a combination of Biocoll and sodium citrate would provide lowest frequencies of MDSC in healthy donor blood samples; these data were, therefore, used to refine the SOP by including precise indications on the separation medium and anticoagulant to be used (phase IIB, figure 1).

Identification of further technical variables in MDSC immunomonitoring. Participating centers stained for MDSC subsets in healthy donor controls. Beforehand every participant received the identical batch of blood collection tubes. From the same donor sodium citrate-, EDTA and heparin blood was collected and PBMC were stained for MDSC subsets in PBMC. Lymphocyte separation medium was free of choice. MDSC frequency was determined in the core lab as described for figure 2. Influence of anticoagulants (A) and lymphocyte separation medium (B) on total frequency of putative ‘MDSC subsets’ in healthy individuals with mean and SD is shown. Kruskal-Wallis was used for statistical analysis. Results were considered significant at *p≤0.05, **p≤0.001 and ***p≤0.0001. M-MDSC, monocytic myeloid-derived suppressor cell; PBMC, peripheral blood mononuclear cell; PMN, polymorphonuclear.
Validation of the refined SOP and comparison with previous dataset
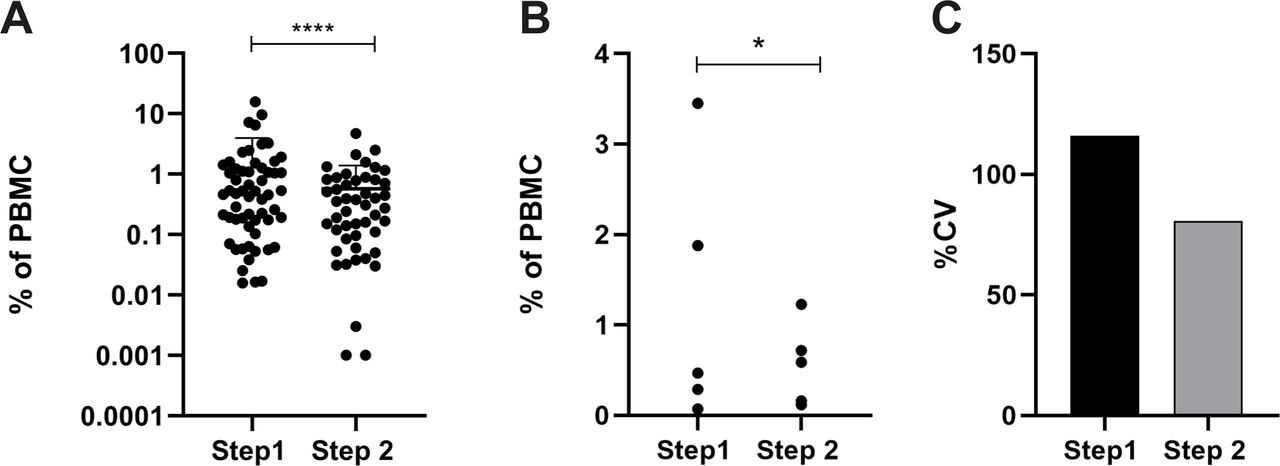
In order to confirm the utility of standardization of the isolation procedure, five participating Centers volunteered to repeat the analysis shown in figure 3 using the refined SOP (phase IIIA, figure 1). Figure 6 shows the level of variance in healthy donors for the original data (figure 3, phase IC) and the variance of data with the harmonized isolation procedure (phase IIIA). Data show that choice and standardization of blood collection tubes and separation medium reduced PMN-MDSC frequency in healthy donors, further improved intercenter comparability of results and reduced differences in MDSC counts between healthy control cohorts from different Centers.

Standardization of anticoagulant and separation medium reduces intercenter variability of PMN-MDSC frequencies. All participants used sodium citrate as anticoagulant and Biocoll as separation medium and determined the frequency of PMN-MDSC in healthy donor controls. Data were compared with data obtained for figure 2. (A) Frequency of PMN-MDSC in Step1 (figure 2, free choice of anticoagulant and separation medium) and step 2 (sodium citrate and Biocoll) from all healthy blood donors in the five centers is shown (plus mean and SD). (B) The mean frequency was determined for each of the five participating centers and (C) % cv was calculated. In all panels the comparison to step 1 (data from figure 2) is shown. F test was used for statistical analysis. Results were considered significant at *p≤0.05 and ****p≤0.0001. PBMC; peripheral blood mononuclear cell.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of molecules associated with T cell suppression expression of PD-L1 and LOX-1 on MDSC subsets in patients and healthy donor controls was determined using the harmonized flow cytometry labeling protocol, Biocoll as standard separation medium and sodium citrate as standard anticoagulant. Flow cytometry analysis was performed in the core lab to ensure standardized gating. Staining intensity of PD-L1 (A) and LOX-1 (B) on MDSC-subsets was determined in five different disease settings. Delta median (median signal intensity of antibody minus median signal intensity of isotype control) is shown. Data are depicted as mean and SD is shown. CVD, cardiovascular disease; M-MDSC, monocytic myeloid-derived suppressor cells; PD-L1, programmed death-ligand 1; PMN, polymorphonuclear.
PD-L1 is primarily expressed in mononuclear MDSC and is not upregulated as a consequence of disease
LOX-1 has been suggested as a marker associated with suppressive activity of PMN-MDSC.9 PD-L1 is expressed on myeloid cells and considered as an important biomarker and functional target in current immunotherapies.10 In the final step of our study (phase IIIB, figure 1), we tested the expression of both markers in five participating centers.
Quantitative analysis of expression levels showed that PD-L1 is primarily expressed on M-MDSC and e-MDSC (figure 7 and online supplementary figure S1). In contrast, PD-L1 is absent or expressed at very low levels on PMN-MDSC. As expected, LOX-1 is expressed on PMN-MDSC and absent on M-MDSC and e-MDSC (figure 7B, online supplementary figure S2). It is important to note that in all tested diseases and conditions both markers were not upregulated in patients over healthy controls.
Supplemental material
Supplemental material
Discussion
Since their initial identification, MDSCs have received substantial consideration from immunologists, who performed several studies to elucidate their immunopathological role in cancer, inflammation and infectious diseases.11–14 In addition, MDSCs are considered as major cellular mediators of resistance to cancer immunotherapy and may serve as potential future biomarkers to predict response to conventional and immune-based cancer therapy.15 Thus, precise and reliable immunomonitoring of MDSC, even in intercenter analyses, is of utmost importance.
Human circulating MDSCs are normally isolated from whole blood after density gradient centrifugation and subdivided into at least three different subsets (granulocytic PMN-MDSC, monocytic M-MDSC and myeloid precursor-like e-MDSC) using flow cytometry.6
The main limitation of the current studies on human MDSC lies in the extreme variability in the protocols used to extract, identify and phenotype these cells. Variable isolation procedures, antibody panels and gating strategies are used to isolate and characterize human MDSC. Previous reports already highlighted the need for marker and gating harmonization4 16 and outlined technical variables that could affect MDSC immunomonitoring.17 A recommendation paper was published in order to suggest minimal characterization standards when researching MDSC.4 However, also in those reports no consensus strategy for MDSC preparation, isolation and immunophenotyping is provided and an accepted gating strategy for enumeration of non-overlapping human MDSC subsets does not exist until now. These obstacles make it nearly impossible to directly compare MDSC data sets between published studies.
In this study, we developed a consensus protocol for the isolation, identification and immunophenotypic characterization and analysis of human MDSC. After centralized hands-on training, this protocol was executed in the participating Centers, followed by a centralized flow cytometry analysis that utilized a uniform gating strategy. Our initial analysis on malignant and non-malignant infectious/inflammatory disease patients revealed that PMN-MDSC are significantly expanded in malignant disease with 5/6 cancer types (ie, Glioma as well as head and neck, breast, colorectal and ovarian cancer) showing statistically significant upregulation even with our relatively small sample size per cancer type, and consistent to what is already reported in the literature.6 18–22 No statistically significant induction of PMN-MDSC frequency was found in melanoma. Interestingly, and in contrast to cancer, this induction of PMN-MDSC was not observed in patients with infection and inflammation. For M-MDSC and e-MDSC we did not find such significant changes in most disease entities. We acknowledge that the sample size could be responsible for the lack of statistically significant expansion of MDSC in some types of disease. Nevertheless, the comparative pan-disease analysis in our study unequivocally shows that PMN-MDSC expansion in solid cancers exceeds expansion of this MDSC subset in infectious and inflammatory diseases and also exceeds the relative expansion of M-MDSC in cancer. These data partially challenge previously published data that reported robust MDSC expansion in infections and non-malignant inflammatory diseases such as psoriasis, HIV, HBV and CVDs.23–27
In this context, it is interesting to note the very divergent levels of MDSC expansion reported in the literature. The use of different methodologies and techniques to isolate MDSC could potentially lead to technical, rather than biologically caused differences among labs. It is a particular strength of our study that, by using a SOP-like protocol, harmonized reagents and antibodies, core Center analysis and a uniform gating strategy, we were able to facilitate a comparative analysis of human MDSC expansion across many different disease types.
Our study also revealed the importance of further technical variables during blood collection and separation. These findings were triggered by initial observations that showed substantial differences in the frequency of MDSC in healthy donors across the different Centers (see PMN-MDSC frequencies in healthy donors in figure 3B). These differences suggested that additional technical variables, not standardized in the original SOP, could affect the results. We then decided to analyze the anticoagulant and gradient separation medium variables, by performing additional experiments; results indicated that different anticoagulants and separation media can alter the baseline frequency of MDSC in healthy donors. In particular the combination of sodium citrate as anticoagulant and Biocoll as stratification solution allowed for the lowest MDSC frequencies in healthy donors.
Anticoagulants can affect the frequency of MDSC in human blood as already reported by Apodaca et al; the authors compared the total number of M-MDSC in blood of healthy donors collected using EDTA or Heparin and observed a significant difference between the two anticoagulants.17
We included these two additional standardized variables in the SOP and repeated the experiments in a selected number of centers, showing a reduced degree of variance among healthy donors across centers. It is worth noting that by this approach we obtained PMN-MDSC frequencies well below 1% in four out of five Centers. Again, a great variability in MDSC, and in particular PMN-MDSC frequency is reported in the literature.4 17 28
In the final phase of our study, we also evaluated the expression of PDL1 and LOX-1, two surrogate suppressive functional markers associated with M-MDSC and PMN-MDSC, respectively. Our data confirmed the restricted expression of LOX-1 on PMN-MDSC as previously reported for circulating PMN-MDSC9 and PMN-MDSC in cancer tissues.29 On a per cell basis the expression level of LOX1 was not different between PMN-MDSC extracted from healthy donor controls and cancer patients. It is, however, important to note that LOX1-positive PMN-MDSC were substantially expanded in cancer patients over healthy controls. This is also in line with previous studies9 that showed an expansion of this LOX-1-positive subset in the unseparated blood of cancer patients. In contrast, strong expression of PD-L1 was restricted to M-MDSC and e-MDSC and similar to LOX-1, on a per cell basis, the expression was not substantially induced in patients with cancer.
PD-L1 is a promising target for checkpoint blockade immunotherapy. From our data we may assume that this type of immunotherapy will primarily co-target M-MDSC in cancer patients. Most likely, additional and distinct therapeutic interventions will be required to target human PMN-MDSC. In the context of these considerations, we are convinced that the use of standardized protocols for immunomonitoring of MDSC subsets and their associated functional molecules will aid future patient selection and stratification for treatment.
Conclusion
In summary, our optimized study design allows us to conclude that expansion of PMN-MDSC exceeds expansion of other MDSC subsets in cancer and is more pronounced in solid tumors as opposed to various infectious and inflammatory diseases. In addition, we report several technical and analytical aspects that will guide the future analysis of MDSC, an important potential cellular resistance mechanism in current cancer immunotherapies.
Acknowledgments
Lab1: We would like to thank Delia Cosgrove for her enthusiastic organizational input and for support of this project. We also thank the staff of the ORL department for their support during blood sample collection. Lab2: The work was supported extensively by the Edinburgh Breast Unit team and particularly by Lorna Renshaw and Jane Keys in this unit. We would like to thank the CIR blood resource (AMREC #15-HV-013) for the recruitment of blood from normal controls and the CIR flow facility (Shonna Johnston, Will Ramsay and Mari Pattinson). Lab3: We would like to thank Sandra Bossmann en/of Fleur Brienen for excellent support. Lab4: We thank the staff of the Core Facility Live Cell Imaging Mannheim and S. Uhlig from FlowCore Mannheim for help with the cell sorting. Lab6: We would like to thank all patients and nurses who contributed to the study, especially Nuraydın Sahin for collecting blood samples. Lab9: We would like to thank Beatrice Jacquelin for her contribution in supervising the PhD student and obtention of financial support. We would like to thank all the members of the Kremlin-Bicêtre hospital who helped for this study. Lab10: We would like to thank the Infectious Diseases Department of Hospital de Santa Maria/CHULN/Lisboa Portugal and all the patients for their collaboration, as well as the team members of Flow Cytometry Unit at Instituto de Medicina Molecular Joao Lobo Antunes (Lisboa, Portugal) for technical assistance. Lab11: We would like to thank lab members that enrolled in this study as healthy donors. Lab12: We would like to thank Tyler Sandberg for the help in the recruitment of the healthy volunteers. Lab13: We would like to thank all patients who contributed to the study. We would like to thank Aud Valle Hansen at the Western Norway University of Applied Sciences for collecting blood samples and Professor Øystein Bruserud at Department of Clinical Science, University of Bergen, for kindly letting us use the BD FACS Verse flow cytometer at the Leukemia Research Laboratory. Participants of this study also thank all members of the Mye-EUNITER network for their scientific input and for fruitful discussions.
References
Footnotes
Twitter @lbodnar
Contributors Lab1 (SB, KB, BH and SL) coordinated the study, designed the SOP, participated to Phase I, II and III of the study and performed centralized data analysis. Lab2 (LC, MD and JWP) coordinated the study, designed the SOP and participated to phase I, II and III of the study. Lab3 (GA, KS, EK-R and MtL) designed the SOP and participated to phase I and II of the study. Lab4 (VU and XH) designed the SOP and participated to phase I and II of the study. Lab5 (SB, MG and JFS) contributed to phase I of the study.Lab6 (GE and UH) participated to phase I of the study. Lab7 (BT, LB, EG and MK) participated to phase I of the study. Lab8 (GZ, OEA and UD) participated to Phase I and II of the study. Lab9 (TG-T, B and MM-T) participated to phase I of the study. Lab 10 (AG-S, RB and AEdS) participated in Phase I and II of the study. Lab11 (JC, JS-M, OO and MK-M) participated to phase I, II and III of the study. Lab12 (AL and KL) participated to phase I, II and III of the study. Lab13 (IMR, AOK and EE) participated to phase I, II and III of the study.
Funding Lab1: This research was supported by European Cooperation in Science and Technology (COST) Action Mye-EUNITER (BM1404) and by a grant from the Deutsche Forschungsgemeinschaft to SB (DFG, BR 2278/6–1). Lab2: This research was supported by Wellcome Trust (101067/Z/13/Z), MRC Centre\ grant MR/N022556/1 to JWP. Lab3: This work was supported by grants from the STOPbraintumors Foundation, COST (European Union) and grants from the Dutch Cancer Society awarded to GA and CBüll (KUN2015-7604) and GA, KS and P Wesseling (KWF11266). Lab4: The work was supported by the grant from German Research Council RTG2099/2 (JSU, VU). Lab5: Our work is supported by a grant from the Serbian Ministry of Education, Science and Technological Development (451-03-68/2020-14/200015)'. Lab6: This work was partially supported by The Scientific and Technological Research Council of Turkey, (TÜBİTAK; project no. 115S636) and covered under the European Cooperation in Science and Technology (COST-EU) Action BM1404 (Mye-EUNITER). Lab7: This result is part of a project that has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programmeprogram (Grant agreement No. 715048). Lab9: TG-T was recipient of a fellowship from the Pasteur Paris University International PhD program and and Institut Carnot Microbes et Santé. This work was supported by Sidaction. Lab10: This work was funded by the following grants: PTDC/MED-IMU/30474/2017 – project cofunded by FEDER LISBOA-01–0145-FEDER-030474, through Programa Operacional Regional de Lisboa, do PORTUGAL 2020, and Fundação para a Ciencia e a Tecnologia and by Gilead Genesis to AES. Lab 11: This work was supported by grants from Polish National Science Center UMO-2011/02/A/NZ5/00337 and UMO-2017/25/B/NZ6/01003 (to JC). Lab12: This study was funded by the Swedish Research Council (2019–01036). Lab13: This work was supported by grants given by Faculty of Engineering and Natural Sciences, Western Norway University of Applied Sciences, Norway.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Lab1: The collection and use of peripheral blood cells was approved by the ethics committee of the medical faculty of the University of Duisburg–Essen. Lab2: All study protocols were approved by The University of Edinburgh (Edinburgh, UK) ethics committees as appropriate. Lab3: Experiments were performed in accordance to the Helsinki Declaration and approved by the local Medical Ethics Committee of the Radboudumc (registration number 2011/307). Lab4: The study protocol was approved by the Institution's ethics committee (2010–318N-MA). Lab6: The study was approved by Hacettepe University and University of Health Sciences - Diskapi Yildirim Beyazit Research and Training Hospital local ethics committees and conducted in agreement with guiding principles of the declaration of Helsinki and the good clinical practice. Lab7: All study protocols were approved by Military Institute of Medicine (Warsaw, Poland) ethics committees as appropriate. Lab8: All study protocols were approved by University Hospital Essen's Ethics Committee (Approval Number 15–6495-BO) as appropriate. Lab9: Peripheral blood samples from non-HIV-infected blood donors were obtained from the French blood bank (Etablissement Français du Sang) as part of an agreement with the Institut Pasteur (C CPSL UNT, number 15/EFS/023). Lab10: All study protocols were approved by the Faculdade de Medicina da Universidade de Lisboa and Centro Hospitalar Universitário Lisboa Norte (Lisbon, Portugal) ethics committees. Lab11: All study protocols were approved by the Jagiellonian University Institutional Bioethics Committee. Lab12: The study was approved by the Stockholm Local Ethical Committee and was performed according to the Declaration of Helsinki principles. Lab13: Both clinical protocols (phase I and II) were approved by The Norwegian Regional Committee for Medical and Health Research Ethics (REK Vest 2017/49).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Experimental data and protocols are available on reasonable request via the corresponding author. Reuse requires citation of this manuscript. Identifiable patient information and data are not available, as patient material was used in an anonymous manner. The SOP-like protocol for MDSC immunomonitoring generated in the context of this study is available from the corresponding author on reasonable request.