Article Text

Abstract
Background Immunotherapies, such as immune checkpoint inhibitors and adoptive cell therapies, have revolutionized cancer treatment and resulted in complete and durable responses in some patients. Unfortunately, most immunotherapy treated patients still fail to respond. Absence of T cell infiltration to the tumor site is one of the major obstacles limiting immunotherapy efficacy against solid tumors. Thus, the development of strategies that enhance T cell infiltration and broaden the antitumor efficacy of immunotherapies is greatly needed.
Methods We used mouse tumor models, genetically deficient mice and vascular endothelial cells (VECs) to study the requirements for T cell infiltration into tumors.
Results A specific formulation of poly-IC, containing poly-lysine and carboxymethylcellulose (PICLC) facilitated the traffic and infiltration of effector CD8 T cells into the tumors that reduced tumor growth. Surprisingly, intratumoral injection of PICLC was significantly less effective in inducing tumor T cell infiltration and controlling growth of tumors as compared with systemic (intravenous or intramuscular) administration. Systemically administered PICLC, but not poly-IC stimulated tumor VECs via the double-stranded RNA cytoplasmic sensor MDA5, resulting in enhanced adhesion molecule expression and the production of type I interferon (IFN-I) and T cell recruiting chemokines. Expression of IFNαβ receptor in VECs was necessary to obtain the antitumor effects by PICLC and IFN-I was found to directly stimulate the secretion of T cell recruiting chemokines by VECs indicating that this cytokine-chemokine regulatory axis is crucial for recruiting effector T cells into the tumor parenchyma. Unexpectedly, these effects of PICLC were mostly observed in tumors and not in normal tissues.
Conclusions These findings have strong implications for the improvement of all types of T cell-based immunotherapies for solid cancers. We predict that systemic administration of PICLC will improve immune checkpoint inhibitor therapy, adoptive cell therapies and therapeutic cancer vaccines.
- adjuvants
- immunologic
- CD8-positive T-lymphocytes
- cytokines
- immunity
- cellular
- immunotherapy
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Over the past decade, there has been a remarkable resurgence in the field of cancer immunotherapy sparked by great clinical results obtained with immune checkpoint inhibitors (ICIs) and with T cell adoptive cell therapy (ACT).1 2 However, there remain several caveats that limit the applicability of these forms of cancer immunotherapy to most patients. The efficacy of the most promising ICI, programmed cell death 1 (PD-1)/programmed cell death ligand 1 (PD-L1), relies on an existing pool of tumor-reactive/tumor-infiltrating T cells (TILs), which is not a frequent occurrence. ACT requires either the isolation and expansion in tissue culture of TILs, which is only feasible and successful in rare occasions or in the generation of genetically modified T cells expressing T cell antigen receptors or chimeric antigen receptors a task that is technically challenging. The expansion of TILs and genetically modified T cells to large cell numbers is also laborious and expensive. Thus, there is an urgent need to develop alternative, cost-effective and broadly applicable cancer immunotherapies.
Our laboratory has worked for many years in the identification of CD8 and CD4 T cell epitopes from tumor antigens (TAgs) and the development of synthetic peptide-based vaccines comprising these epitopes.3 4 Using mouse tumor models, we developed peptide vaccination strategies capable of rapidly generating vast numbers of tumor-reactive CD8 T cells, similar to those observed during viral infections, where >10% of all CD8 T cells are specific for the immunogen.5 6 However, as with ICIs and TILs, these vaccines in many instances have limited success in eradicating large established tumors. Paradoxically, suboptimal therapeutic effects of these vaccines in advanced tumor models are observed even though high numbers of functional TAg-specific CD8 T cells are present in lymphoid tissues (spleen, bone marrow (BM), blood), suggesting that the lack of tumor control/rejection could be due to a lack of trafficking and infiltration of the T cells to the tumor parenchyma. In addition, various sources of immunosuppressive activities abound in the TME, which neutralize the effector function of the few T cells that manage to infiltrate the tumor parenchyma. Indeed, absence of T cell infiltration to the tumor site has been proposed as one of the major obstacles that limits ICI and TIL immunotherapy efficacy against solid tumors.7–10
T cell trafficking and infiltration to tissues where they are needed such as tumors is a complex multistep process, which involves the expression of adhesion molecules and corresponding ligands by vascular endothelial cells (VECs) and activated T cells and the production of T cell recruiting chemokines by the tissue cells in need of effector cells.11–14 Although this process readily occurs during most acute infections due to the interaction of pathogen-associated molecular patterns (PAMPs) with pattern recognition receptors (PRRs), clearly it is not effective in the case of cold tumors, where T cell infiltrates are absent and a relevant PAMP may not be present. We report here that systemic administration of a synthetic PAMP mimic consisting of polyinosinic:polycytidylic acid (poly-IC) formulated with poly-lysine and carboxymethylcellulose (PICLC), resulted in the enhancement of T cell infiltrates into mouse solid tumors, which correlated with a substantial therapeutic effect. In addition, we describe some of cellular and molecular mechanisms involved in the antitumor effects of PICLC.
Materials and methods
Mice and cell lines
Female C57BL/6 (B6) mice aged 6–8 weeks were from the National Cancer Institute/Charles River Program (Wilmington, Massachusetts, USA). MDA5 KO, TLR3 KO and mice expressing diphtheria toxin receptor (DTR) under CD11c promoter (CD11cDTR) were from Jackson Laboratory. Interferon (IFN)αβ receptor-deficient (IFNαβR KO) mice were originally provided by P. Marrack (National Jewish Health, Denver, Colorado, USA) and bred in our facility. Mice deficient of IFNαβR in endothelial cells were produced by crossing Tie2-Cre (Tek-Cre, Jackson Stock No: 004128) mice with IFNaR1 flox mice (Jackson Stock No: 028256). To generate BM chimeric mice, CD45.1 congenic B6 mice received one dose of total body irradiation (1000 rads) followed by an intravenous injection of ~1×107 BM cells from appropriate KO mice or CD11cDTR mice (all CD45.2) and 60 days later reconstitution efficiency was checked using flow cytometry. For CD11c+ cell depletion, mice reconstituted with CD11cDTR BM were injected intraperitoneally with 100 ng diphtheria toxin (DT)/mouse −2, 0 and +2 days of the PICLC injection. All animal care and experiments were conducted according to our institutional animal care and use committee (IACUC) guidelines. Lewis lung carcinoma (LLC) cells were the A9F1 subclone15 provided by L. Eisenbach (Weizmann Institute of Science, Rehovot, Israel). Mouse melanoma B16F10 cells were provided by Alan Houghton (Memorial Sloan Kettering Cancer Center, New York, New York, USA). Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza. The mouse endothelial cell lines from brain (bEnd.3) and heart (H5V) were from American Type Culture Collection.
Reagents and antibodies
PICLC (Hiltonol) is a clinical grade formulation of poly-IC stabilized with poly-L-lysine and carboxymethylcellulose manufactured by Oncovir. High molecular weight (hmw) poly-IC and poly-AU were purchased from InvivoGen. Poly-IC/PEI was prepared by mixing hmw poly-IC with in vivo-JetPEI (Polyplus) at a nucleotide:PEI ratio of 6 in a 5% glucose solution. Antimouse PD-L1 monoclonal antibody (mAb) (clone, 10F.9G2) was from BioXcell. Mouse recombinant Interferon-beta and human recombinant Interferon-α2 were purchased from BioLegend. CXCL10 ELISA kits were purchased from R&D Systems. EliSpot plates were from eBioSciences. Dispase I (neutral protease, grade I) was purchased from Sigma-Aldrich. Fluorescence-conjugated Abs for flow cytometry were from Biolegend or eBioscience.
Therapeutic protocols and evaluation of antitumor effects
Mice were injected subcutaneously with 5×105 LLC, or 3×105 B16F10 cells in a shaved rear flank. Seven days (B16F10 tumor model), or 9 days (LLC tumor model) later, poly-IC, PICLC or JETPEI/poly-IC were administered intravenously, intramuscularly or intratumorally at 50 µg/dose. The administration of PICLC was repeated 3 times, 5 days apart. Anti-PD-L1 mAb (200 µg/dose) was administered intraperitoneally on days 1 and 3 after each PICLC administration. Tumor growth was monitored every 2–3 days in individually tagged mice by measuring two opposing diameters with a set of calipers. Mice were euthanized when the tumor area reached 400 mm2. Results are presented as the mean tumor size (area in mm2±SD for every treatment group at various time points until the termination of the experiment.
Tissue digestions and cell composition analysis
Established tumors and normal tissues (pancreas, lungs and hearts) were removed from mice, dissociated and treated with 1 mg/mL collagenase and DNase in phosphate-buffered saline (PBS) for 1 hour at 37°C. Cells were washed twice, and red blood cells were lysed using red blood cell lysis buffer, followed by filtration through a 40-μm strainer. For CD8 T cell detections, tumor/pancreas single suspensions were stained with anti-CD45 (Alexaflour700), anti-MHC-II (FITC) and anti-CD8 (Pacific Blue or PercpCy5.5). For myeloid-derived suppressor cells (MDSCs) detection, tumor single suspensions were stained with anti-CD45 (Alexaflour700), anti-MHC-II (FITC), anti-CD11b (PercpCy5.5) and anti-Gr1 (PE). For in vivo CD45 staining,16 tumor-bearing mice were injected with 5 µg APC-conjugated anti-CD45 (clone EM-05) in 200 µL PBS. Five minutes later, mice were euthanized, and blood and tumors were collected for in vitro CD45 staining using Alexaflour700-conjugated anti-CD45 (clone 30-F11). For measuring CXCL9 and vascular cell adhesion molecule 1 (VCAM-I) expression in tumor, heart/lung VECs B6 mice were inoculated subcutaneously with 5×105 B16F10 cells 10 days before intravenous administrations of PBS (No Tx), 50 μg poly-IC, or 50 μg PICLC on days 10 and 14. Tissues were harvested on day 15 and resuspended in Dispase-I, minced and dissociated in a gentleMACS (Miltenyi Biotec), at 37°C for 20 min. The cell digest was filtered through a 70 μm cell strainer, washed and stained with anit-CD45, anti-CD31 to specifically gate on VECs (CD45-negative, CD31-positive). Anti-VCAM-I was included to measure cell surface expression of this adhesion molecule. For chemokine detection, cells were incubated with 1 μL/mL GolgiPlug (BD Bioscience) at 37°C for 6 hours and followed by cell surface staining with anti-CD45 and anti-CD31 mAbs (for negative and positive gates, respectively) and with intracellular staining with anti-CXCL9 mAb. Cell florescence analyzes were performed in LSRII cytometers (BD Biosciences). Data analysis was performed using FlowJo software (V.8.5, TreeStar).
Immunofluorescence histology
Formalin-fixed paraffin-embedded tissues were sectioned at a 5 μm thickness. Sections were subsequently deparaffinized in xylene, rehydrated with decreasing concentrations of ethanol, and boiled in citrate buffer for antigen retrieval. Sections were assessed with a rat antimouse CD8α (Alexa Fluor 488) mAb, mounted in aqueous mounting media with 4′,6-diamidino-2-phenylindole (DAPI, ThermoFisher Scientific) and visualized under a phase contrast and fluorescence imaging by EVOS fluorescence microscope.
Human and murine IFN-I measurements
Primary human (HUVECs) or murine (bEnd.3 and H5V) vascular endothelial cell lines cells were stimulated with poly-IC, poly-AU or PICLC as indicated in the figure legends at 37°C in a humidified 5% CO2 incubator for 24 hours. Type I interferon (IFN-I) detection was performed using with HEK-Blue IFN-α/β cells (human) or B16-Blue IFN-α/β cells (murine) from InvivoGen according to manufacturer’s protocol using mouse IFN-β or human IFN-α2 for the standard curves.
Statistical analyses
Statistical significance to assess surface markers expression and cytokines production were performed using Student’s t-tests or one-way analysis of variance (ANOVA) as appropriate. Statistical significance to assess the antitumor efficacy was performed using two-way ANOVA. Results are presented as mean±SD. Statistical analyses were performed using GraphPad Prism (V.7).
Results
Systemic administration of poly-ICLC elicits antitumor effects accompanied by enhanced CD8 T cell tumor infiltrates
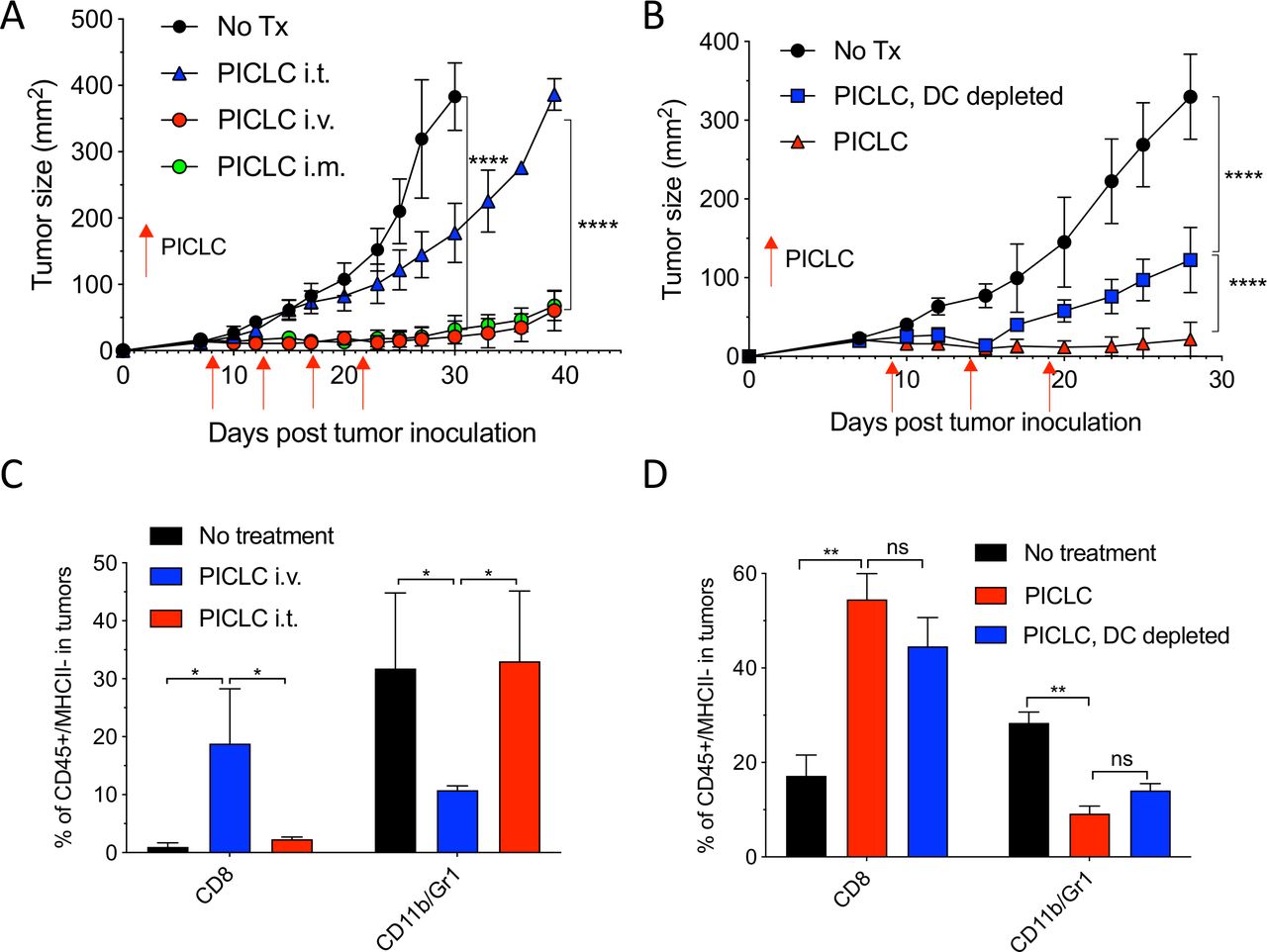
While developing an intravenous peptide-based therapeutic vaccination strategy in mouse tumor models using poly-IC as the adjuvant and an ICI (anti-PD-L1 antibody), we observed a significant therapeutic effect in the control group receiving a non-TAg peptide, injected with poly-IC and ICI.5 Throughout these studies, we used a stabilized formulation of poly-IC (PICLC) containing poly-lysine and carboxymethylcellulose that protects this synthetic double-stranded RNA (dsRNA) from RNAse degradation.17 Subsequently, we reported that repeated intravenous administration of PICLC and anti-PD-L1 antibody alone (without any peptide) had a remarkable therapeutic effect mediated by CD8 T cells in several mouse tumor models.18 Moreover, in the case of LLC, which expresses low levels of PD-L1, repeated administration of PICLC alone was able to control tumor growth. Nevertheless, the mechanisms involved in the antitumor activity of the PICLC therapy remained unknown. We first hypothesized that PICLC could be functioning as an immune adjuvant potentiating CD8 T cell responses elicited by dendritic cells (DCs) presenting TAgs released by tumor cells. PICLC could also promote intratumoral CD8 T cell proliferation and survival. If these assumptions were correct, we predicted that intratumoral administration of PICLC should elicit superior antitumor effects as compared with systemic administrations (intravenous or intramuscular injections), which we routinely had used. To our surprise, intratumoral PICLC was significantly less effective in controlling growth of LLC tumors as compared with intravenous or intramuscular administrations (figure 1A). Also, DC depletions prior to intravenous PICLC therapy did not completely eliminate the antitumor efficacy (figure 1B). These results suggested that some of PICLC’s antitumor effects were taking place outside of the tumor parenchyma. Thus, we considered the possibility that PICLC could be facilitating the traffic and infiltration of effector CD8 T cells into the tumors. Mice bearing LLC tumors were treated twice with PICLC intravenous or intratumoral and tumors were resected the day after the second treatment and examined by flow cytometry for the presence of CD8 T cells. In agreement with our prediction, there was a substantial increase in the frequency of CD8 T cells in tumors from mice that received intravenous PICLC as compared with intratumoral PICLC or untreated control mice (figure 1C). In addition to these findings, the frequency of CD11b/Gr1-positive myeloid cells was significantly reduced in the tumors from mice intravenously treated with PICLC as compared with the other groups. The increase in tumor infiltrating CD8 T cells and decrease of CD11b/Gr1 cells induced by intravenous PICLC was also observed in DC-depleted mice (figure 1D).
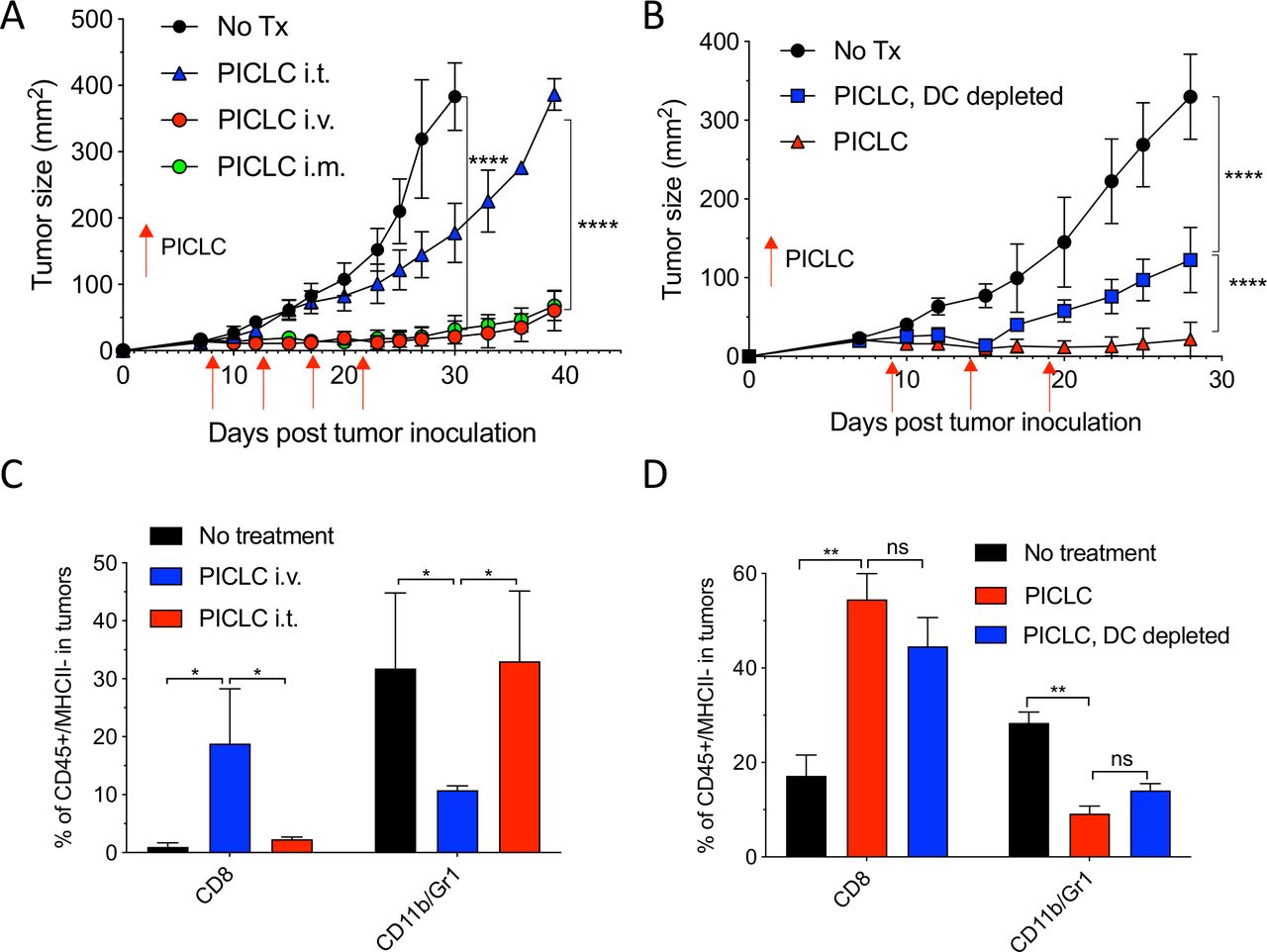
Systemic administration of poly-ICLC elicits antitumor effects accompanied with enhanced CD8 T cell tumor infiltrates. (A) Lewis lung carcinoma (LLC) tumor growth in mice after poly-IC stabilized with poly-lysine and carboxymethylcellulose (PICLC) treatment. Wild-type (WT) mice were subcutaneously inoculated on day 0 with 5×105 LLC cells, injected intravenously, intramuscularly or intratumorally with 50 µg PICLC on days 9, 14 and 19 and tumor growth was monitored. Data are shown as tumor average size±SD; n=5 mice per group. ****P<0.0001 (two-way analysis of variance (ANOVA)). (B) B16F10 growth in dendritic cell (DC)-depleted bone marrow chimeric mice. CD45.1 WT mice were irradiated and reconstituted with bone marrow cells from CD11cDTR mice. Two months later, mice were inoculated subcutaneously on day 0 with 3×105 B16F10 cells and treated intravenously with 50 μg PICLC on days 9, 14 and 19 plus 200 µg αPD-L1 monoclonal antibody (mAb) on days 10, 13 and 17 post-tumor inoculation. DCs were depleted in one group by injecting DT 100 ng/mouse on days −2 and 0 of each PICLC injection and the tumor growth was monitored. Data are shown as tumor size±SD; n=5 mice per group. ****P<0.0001 (two-way ANOVA). (C, D) CD8+ T cells and myeloid-derived suppressor cells (MDSCs) infiltration in the tumor after PICLC treatment. (C) Similar experiment to (A), except tumors were harvested 24 hours after the last PICLC injection, dissociated and the percentages of CD8+ T and CD11b+Gr1+ cells of the CD45+/MHC-II-negative populations were assessed in the single cell suspensions. Bars are shown as mean±SD; n=3 mice per group. *P<0.05 by Mann-Whitney unpaired test. (D) Similar experiment to (B), except that 24 hours after the last PICLC injection the percentages of CD8+ T and CD11b+Gr1+ cells were assessed in the single cell suspension. Bars are shown as mean±SD; n=3 mice per group. **P<0.01 by Mann-Whitney unpaired test. ns, not significant.
Systemic PICLC elicits CD8 T cell infiltrates into the tumor parenchyma
Similar results of the effects of systemic administration (intravenous or intramuscular) of PICLC on T cell infiltration were obtained with mice bearing B16 melanoma tumors (figure 2A). However, in this experiment we assessed whether the CD8 T cells detected in the tumors actually reside in the tumor parenchyma or in the blood irrigating the tumors. This was done by injecting APC-labeled anti-CD45 antibodies intravenously 5 min prior to tumor resections. This procedure has been shown to label blood circulating lymphocytes but not TILs.16 An example of a PICLC intravenously treated mouse is presented in figure 2B showing that ~93% of the CD8 in tumors appear to reside in the parenchyma. The increased presence of CD8 T cells in tumor parenchyma of mice intravenously treated with PICLC as compared with intratumoral treatment was corroborated by immunohistology (figure 2C). CD8 T cells purified from B16 tumors of intravenous PICLC-treated mice recognized tumor cells in IFN-γ release EliSpot assays (online supplemental figure S1). Next, we compared the ability of PICLC to enhance T cell infiltration into tumors versus a normal organ. As shown in figure 2D, the T cell infiltration enhancement of intravenously administered PICLC was only significant in LLC tumors and not in a normal organ (pancreas). Similarly, the decrease of CD11b/Gr1 cells by PICLC was observed only at the tumor site.
Supplemental material
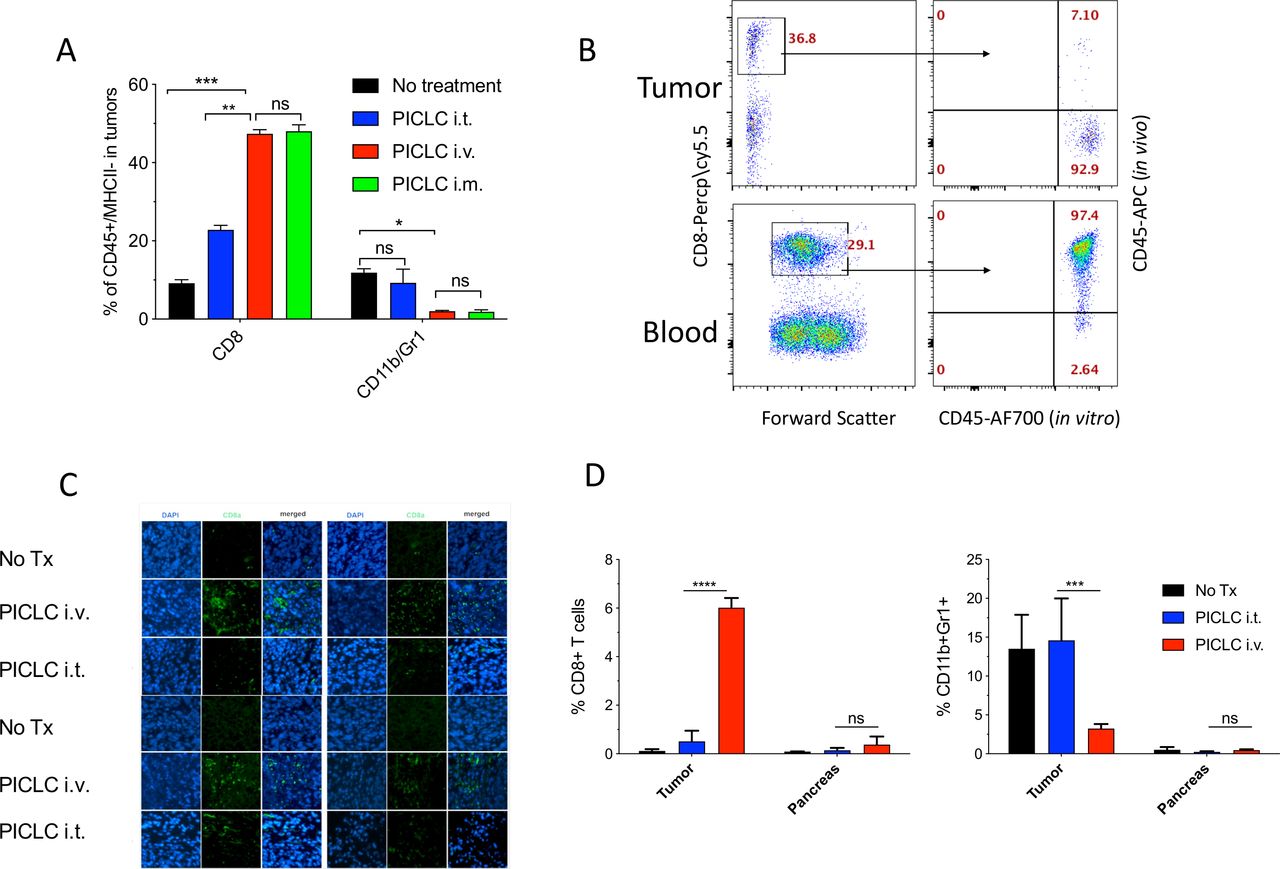
Systemic poly-IC stabilized with poly-lysine and carboxymethylcellulose (PICLC) elicits CD8 T cell infiltrates into the tumor parenchyma. (A) CD8+ T cell and myeloid-derived suppressor cell (MDSC) infiltration in B16F10 tumors after treatments. Wild-type (WT) mice were inoculated subcutaneously on day 0 with 3×105 B16F10 cells and injected intravenously, intramusculary or intratumorally with 50 µg PICLC on days 7, 12 and 17. On day 19, 5 min prior to tumor harvests the mice received 5 µg anti-CD45-APC intravenously and the % of CD8+ T and CD11b+Gr1+ cells were measured in the tumor single cell suspension. Bars are shown as mean±SD; n=3 mice per group. *P<0.05, **p<0.01 and ns, not significant by Mann-Whitney unpaired test. (B) In vivo CD45 staining as described in ‘Materials and methods’ section. A representative dot plot showing that the majority of the CD8 T cells harvested from tumor of an intravenous PICLC-treated mouse did not stain with the in vivo administered anti-CD45-APC, while most of the blood CD8 T cells did. (C) Immunofluorescence staining for the tumor tissue after PICLC treatment. Similar experiment to (A) except tumors were fixed in 2% formalin and stained with anti-CD8α monoclonal antibody (mAb) and DAPI and observed by fluorescence microscopy. (D) CD8+ T cells and MDSCs infiltration in tumors and the pancreas after PICLC treatment. WT mice were inoculated subcutaneously on day 0 with 5×105 Lewis lung carcinoma (LLC) cells and injected intravenously or intratumorally with 50 µg PICLC on days 9, 14 and 19, and the percentages of CD8+ T and CD11b+Gr1+ cells of all the live cells were measured in tumor and pancreas single cell suspensions 24 hours after the last PICLC injection. Bars are shown as mean±SD; n=2–3 mice per group. ****P<0.0001 ***p<0.001 **p<0.01 and ns, not significant by Mann-Whitney unpaired test.
MDA5 and IFN-I signaling are essential for the antitumor effects of PICLC
Poly-IC and PICLC function as agonists for the endosomal TLR3, but both can also stimulate the RIG-I-like dsRNA sensor MDA5 in circumstances when these polynucleotides are able to leak from endosomes into the cytoplasm. Activation of TLR3 and MDA5 results in the production of immunomodulatory cytokines such as IFN-I.19 To assess the roles of TLR3, MDA5 and IFN-I in the antitumor effects of intravenously administered PICLC, an experiment was performed in genetically deficient mice bearing LLC tumors. As shown in figure 3A, PICLC did not elicit antitumor responses in MDA5 and IFN-I receptor (IFNαβR)-deficient mice. In contrast, PICLC reduced tumor growth in TLR3-deficient mice to the same extent as with wild-type (WT) mice. These findings correlated directly with the levels of CD8 T cell tumor infiltrates, and inversely correlated with the frequency of CD11b/Gr1 myeloid cells (figure 3B). These results indicate that PICLC promotes CD8 T cell tumor infiltration and growth control via the production of IFN-I by stimulating MDA5. PICLC contains poly-lysine, a poly-cation that binds to negative charges in the polynucleotide’s backbone, which has been used as a transfection reagent to deliver DNA (and RNA) into the cytoplasm. Polycations such as poly-lysine and polyethyleneimine (PEI) facilitate cytoplasmic delivery of DNA and RNA via the proton pump effect, which ruptures endosomes.20 21 Thus, PICLC should exhibit higher antitumor effects as compared with poly-IC, and PEI should increase the antitumor effects of poly-IC. Indeed, as shown in figure 3C, both PICLC and poly-IC/PEI elicited significantly higher antitumor effects as compared with poly-IC. These results correlated with the increases of infiltrating CD8 T cells and decreases of CD11b/Gr1 cells (figure 3D).

MDA5 and type I interferon (IFN-I) signaling are essential for the antitumor effects of poly-IC stabilized with poly-lysine and carboxymethylcellulose (PICLC). (A) The role of MDA5, TLR3 and IFNαβR on the antitumor effect of PICLC. Wild-type (WT), MDA5 KO, TLR3 KO and IFNαβR KO mice were inoculated subcutaneously on day 0 with 5×105 Lewis lung carcinoma (LLC) cells, injected intravenously on days 9, 14 and 19 with PICLC and tumor growth were monitored. Data are shown as tumor size ±SD; n=4–5 mice per group. ****P<0.0001, ns, not significant by two-way analysis of variance (ANOVA). (B) The role of MDA5, TLR3 and IFNαβR CD8+ T cells and myeloid-derived suppressor cells (MDSCs) infiltration. Wild-type (WT), MDA5 KO, TLR3 KO and IFNαβR KO mice were inoculated subcutaneously on day 0 with 3×105 B16F10 cells, injected intravenously on days 7, 12 and 17 with PICLC and the percentages of CD8+ T cells and CD11b+Gr1+ cells were assessed in the tumor single cell suspensions on day 19. Bars are shown as mean±SD; n=3 mice per group. ***P<0.001, **p<0.01 and ns, not significant by Mann-Whitney unpaired test. (C) The antitumor effect of poly-IC/PEI. WT mice were inoculated subcutaneously on day 0 with 5×105 LLC cells, injected intravenously on days 9, 14 and 19 with 50 µg of poly-IC, PICLC or poly-IC/PEI and tumor growth was monitored. Data are shown as tumor size±SD; n=5 mice per group. ****P<0.0001 by two-way ANOVA. (D) CD8+ T cells and MDSCs infiltration after poly-IC/PEI treatment. WT mice were inoculated subcutaneously on day 0 with 3×105 B16F10 cells and injected intravenously on days 7, 12 and 17 with 50 µg of poly-IC, PICLCor poly-IC/PEI and the percentages of CD8+ T cells and CD11b+Gr1+ cells were assessed in the tumor single cell suspensions on day 19. Bars are shown as mean±SD; n=3 mice per group. ***P<0.001, *p<0.05 and ns, not significant by Mann-Whitney unpaired test.
MDA5 and IFNαβR on stromal cells is required for the antitumor effects and T cell tumor infiltration of PICLC therapy
Next, we assessed which cells responding to PICLC promote CD8 T cell tumor infiltration and antitumor effects. One possibility is that BM-derived cells capable of capturing PICLC such as DCs, monocytes and macrophages would be the main participants in this event. The results in figure 1B,D indicate that DCs may play some role but are not solely responsible in this process. There is evidence in the literature that stromal cells are also capable of responding to poly-IC and produce IFN-I via MDA5 stimulation.19 Thus, we generated several BM chimeric mice to evaluate the roles of MDA5 and IFNαβR expression on either BM derived or stromal cells in the antitumor effects and immune cell tumor infiltration of PICLC therapy. As shown in figure 4A, the most effective and significant antitumor effect of PICLC was observed when BM of MDA5-deficient mice was implanted into WT mice. BM of WT into either MDA5-deficient or IFNαβR-deficient hosts resulted in reduced, but still significant therapeutic antitumor responses by PICLC. These results correlated with tumor infiltrations of CD8 T cells (figure 4B). These findings indicate that both, immune cells derived from the BM and non-BM-derived stromal cells respond to PICLC and participate in the antitumor effects.
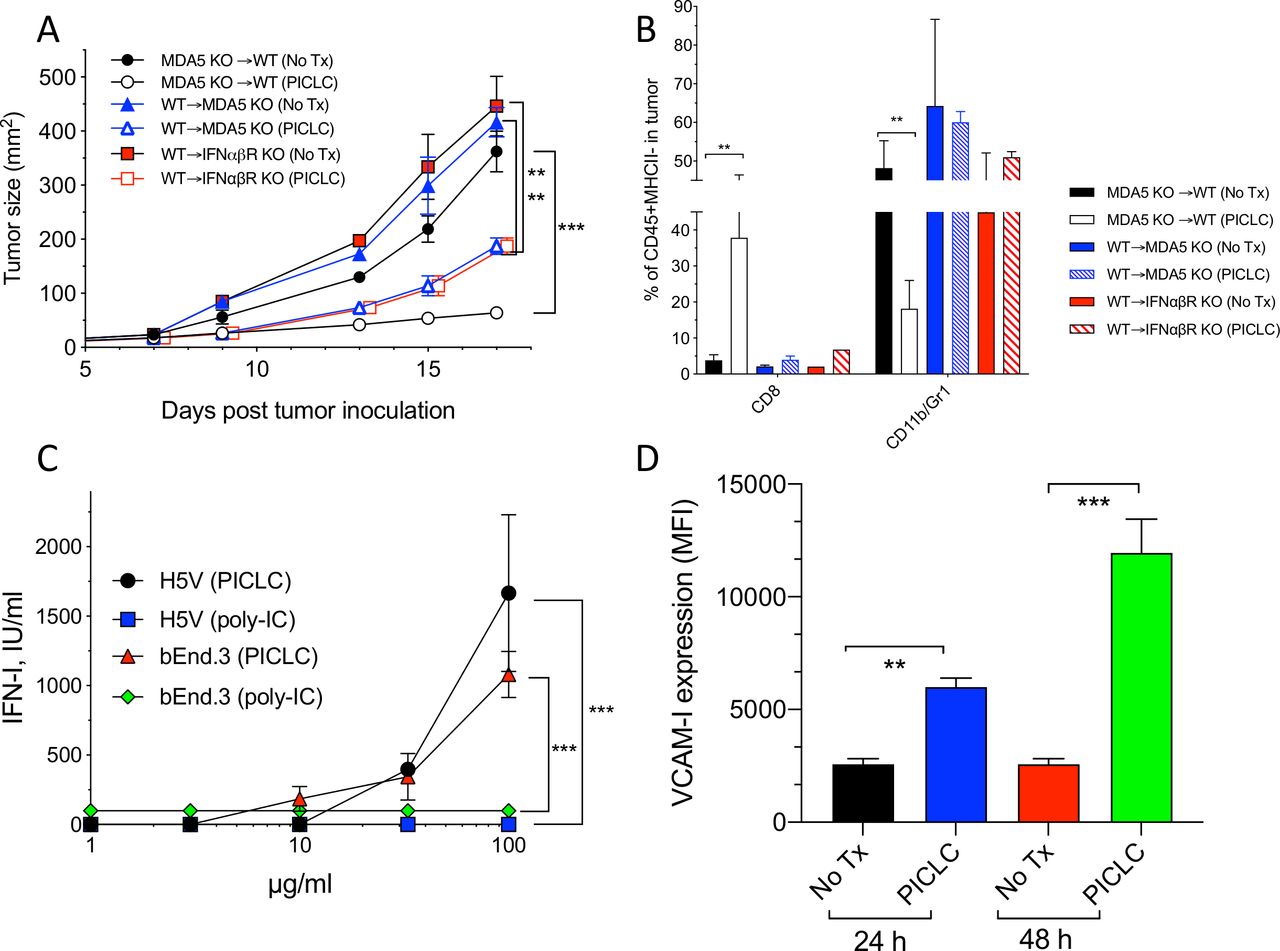
MDA5 and IFNαβR on stromal cells is required for the antitumor effects and T cell tumor infiltration of poly-IC stabilized with poly-lysine and carboxymethylcellulose (PICLC) therapy. (A, B) The role of MDA5 on stromal cells in the antitumor effect and infiltration. (A) Bone marrow chimeric mice were generated as described in ‘Materials and methods’ section and were inoculated subcutaneously on day 0 with 3×105 B16F10 cells and treated with 50 μg PICLC (intravenously) in combination with αPD-L1 monoclonal antibody (mAb) as described in figure 1. (A) Tumor growth was monitored (data are shown as tumor size±SD; n=2–3 mice per group; ***P<0.001, **p<0.01 by two-way analysis of variance) and (B) the percentages of CD8 T cells and CD11b+/Gr1+ cells were assessed on day 19. Bars are shown as mean±SD; n=2–3 mice per group. **P<0.01 by Mann-Whitney unpaired test. (C) IFN-I production by endothelial cells after poly-IC or PICLC treatment. H5V or bEnd.3 (1×105) mouse endothelial cells were stimulated with PICLC or poly-IC and 24 hours later and the production of IFN-I was assessed in the supernatants. (D) VCAM-I expression in endothelial cells after poly-IC or PICLC treatment. 1×105 bEnd.3 cells in triplicates were stimulated with 50 µg PICLC and the VCAM-I expression was assessed by flow cytometry 24 and 48 hours later. Error bars represent the SD of the mean fluorescence intensity (MFI). **P<0.01 and ***p<0.001 by Mann-Whitney unpaired test.
As to the nature of the stromal cells capable of responding to PICLC, we proposed that vascular endothelial cells (VECs) would be the most likely candidates that could promote the extravasation of T cells into the tumor parenchyma, since this is their function during inflammatory responses such as with infections. In addition, VECs are known to express various types of scavenger receptors capable of binding poly-anionic and poly-cationic compounds such as polynucleotides and poly-lysine, which would enable their capture and endocytosis. In view of this, we tested the ability of PICLC and poly-IC to stimulate the production of IFN-I in two mouse VEC lines (H5V and bEnd.3). Both VEC lines produced significant amounts of IFN-I when stimulated with PICLC but not with poly-IC (figure 4C). Stimulation of one of these cell lines (bEnd.3) with PICLC enhanced the expression of the adhesion molecule VCAM-I (figure 4D). Notably, incubation of these VECs with poly-IC or IFN-I (IFN-β) did not enhance the expression of VCAM-I (data not shown). Because these cells express TLR3 (data not shown) and did not respond to poly-IC, we believe that the production of IFN-I and enhancement expression of VCAM-I is likely mediated via MDA5.
PICLC induces the adhesion molecules and chemokine expression in vascular endothelial cells
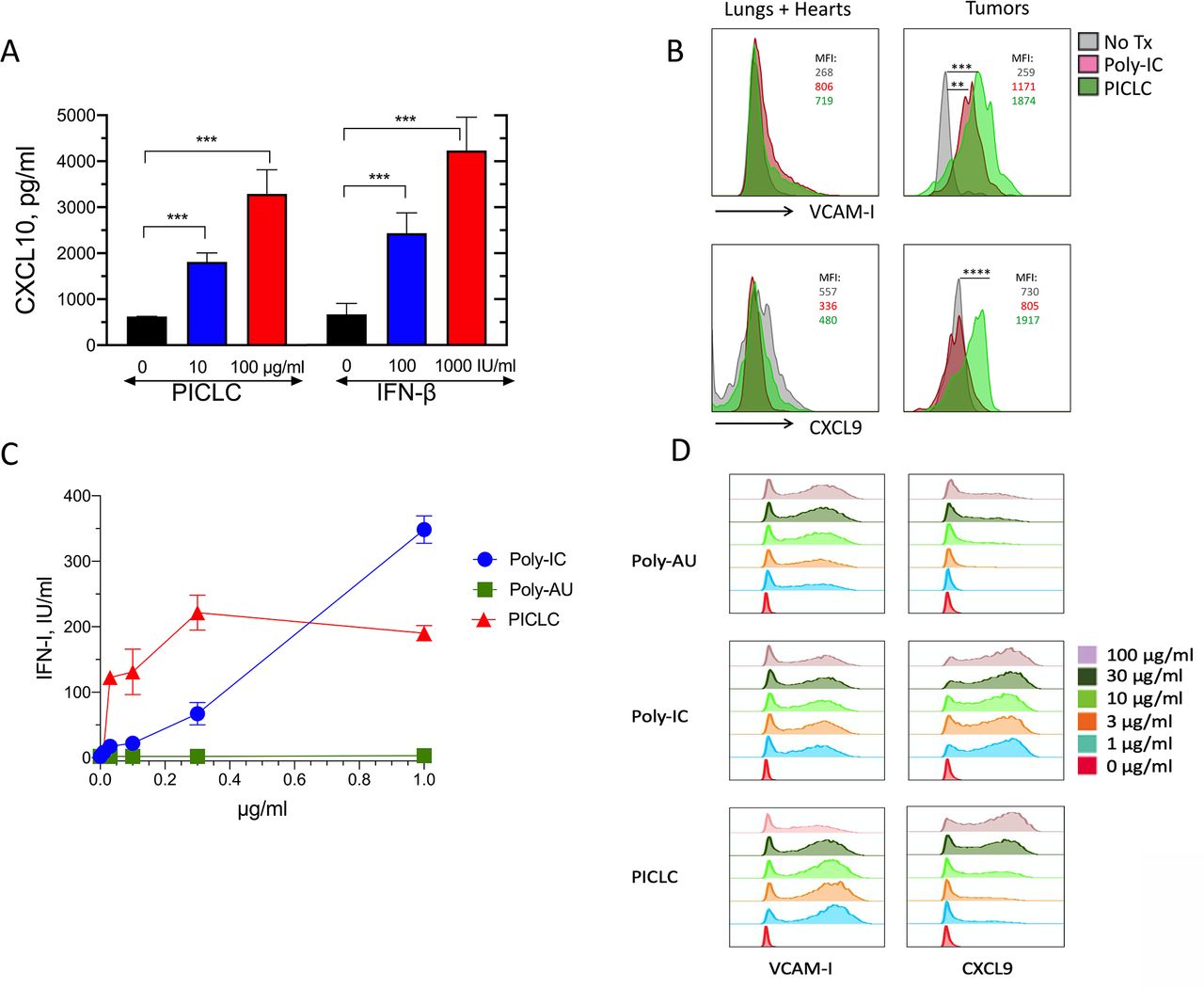
In addition to the role of adhesion molecules, chemokines such as CXCL9 and CXCL10 participate in the recruitment of CD8 T cells into tissues and solid tumors.22–26 PICLC was effective in stimulating the production of CXCL10 in the bEnd.3 VEC line (figure 5A). Because INF-β also elicited the production of CXCL10 to roughly the same levels, we assume that the production of CXCL10 by PICLC in VECs is mediated through IFN-I. Since PICLC systemic administration enhanced T cell infiltration into tumors, but not into normal tissues, we isolated VECs from tumors and normal organs (lungs and hearts) after intravenous PICLC or poly-IC injections and measured the expression of VCAM-I and CXCL9 by flow cytometry. Both PICLC and poly-IC enhanced cell surface expression of VCAM-I in tumor VECs to a much higher extent as compared with VECs from normal tissues (figure 5B). Intracellular expression of CXCL9 was enhanced by PICLC and not by poly-IC in tumor VECs and no changes were observed in normal organ VECs. Both PICLC and poly-IC enhanced the expression of ICAM-I in tumor and normal organ VECs (data not shown).

Poly-IC stabilized with poly-lysine and carboxymethylcellulose (PICLC) induces the adhesion molecules and chemokine expression in vascular endothelial cells (VECs). (A) In vitro induction of CXCL10 in mouse vascular endothelial cells. Mouse VECs (bEnd.3) were stimulated with PICLC or interferon-β (IFN-β), and 24 hours later, CXCL10 levels in the supernatants were assessed by ELISA. (B) VCAM-I and CXCL9 expression in tumor and normal (lungs and hearts) VECs. Wild-type (WT) mice were inoculated subcutaneously with 5X105 B16F10 cells and received intravenous administration of phosphate-buffered saline (PBS) (No Tx), 50 μg of Poly-IC or 50 μg of PICLC on days 10 and 12. On day 13, the levels of VCAM-I and CXCL9 in VECs from normal tissues (lungs and hearts) and tumors were analyzed by flow cytometry in the CD45−, CD31+ population. Mean fluorescence intensity (MFI) values for each condition are shown. Data are pooled from three mice per group. Statistical differences between treatment groups were calculated using a univariate χ2 test within the FlowJo software. **P=0.000151; ***p=0.000231; ****p=8.38X10-8. (C, D) IFN-I production, VCAM-I and CXCL9 expression in human endothelial cells. Human vascular endothelial cells (HUVECs) were stimulated Poly-AU, Poly-IC or PICLC for 24 hours and levels of secreted IFN-I (C) and expression of VCAM-I and CXCL9 were measured by flow cytometry (D).
We also assessed whether human VECs (HUVECs) could respond to PICLC stimulation. Primary HUVECs were stimulated with various concentrations of PICLC, poly-IC or poly-AU (a TLR3 agonist that does not stimulate MDA5). The results showed that PICLC was substantially more effective in inducing IFN-I secretion as compared with poly-IC, and that poly-AU did not stimulate the HUVECs (figure 5C). Both PICLC and poly-IC and to some extent poly-AU (but at much higher concentrations) induced the production of CXCL9 on HUVECs while all three compounds were able to increase the expression of VCAM-I (figure 5D). These results provide support to the conclusion that systemically administered PICLC enhances CD8 T cell-mediated antitumor responses by augmenting tumor T cell infiltration via MDA5 stimulation in VECs resulting in the production of IFN-I and T cell recruiting chemokines.
Conditional deletion of IFNαβR in endothelial cells impairs the antitumor efficacy of PICLC
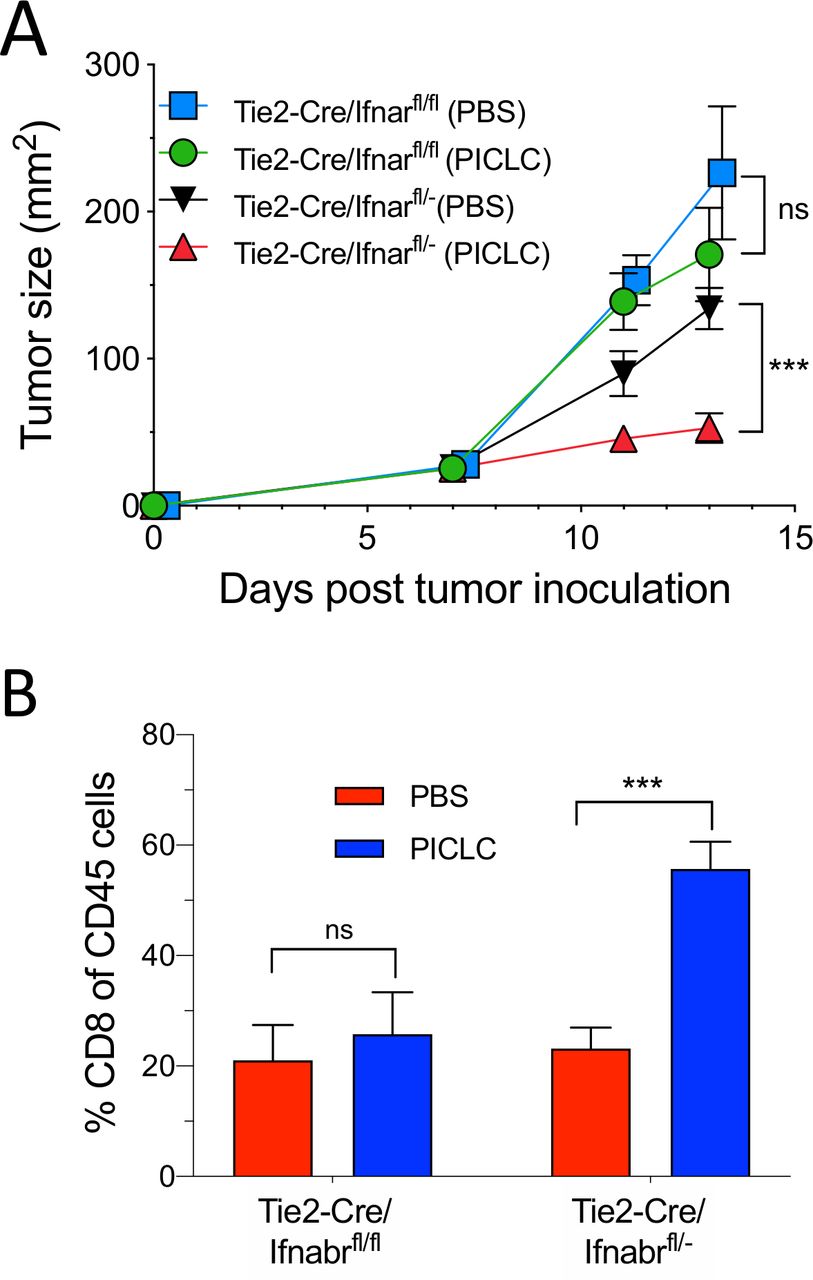
Since the role of IFN-I on VECs appears to be essential for the capacity of PICLC to promote tumor T cell infiltration, we examined the antitumor effects of PICLC and the presence of tumor T cell infiltrates in conditional IFNαβR-deficient mice. Using Tie2-Cre mice and Ifnar1fl mice we generated mice deficient in the expression of IFNαβR in endothelial cells. As shown in figure 6A, intravenous administration of PICLC in Tie2-Cre/Ifnar1fl/fl mice failed to reduce the tumor growth as compared with the mice injected with PBS. On the other hand, PICLC injected into Tie2-Cre/Ifnar1fl/− control mice significantly decreased the tumor growth as compared with mice injected with PBS. Simultaneously, PICLC therapy resulted in increased CD8 T cell tumor infiltrates in the Tie2-Cre/Ifnar1fl/− control mouse group but not in Tie2-Cre/Ifnar1fl/fl mice (figure 6B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Conditional deletion of IFNαβR in endothelial cells impairs the antitumor efficacy of poly-IC stabilized with poly-lysine and carboxymethylcellulose (PICLC). (A, B) IFNαβR in endothelial cells is important in the antitumor effect of PICLC. Tie2-Cre/Ifnar1fl/fl or Tie2-Cre/Ifnar1fl/− control mice were inoculated with 5×105 B16F10 cells and received intravenous administration of 50 µg PICLC on days 7 and 12. αPD-L1 mAb (200 µg) was administered on days 8, 10 and 13. (A) Tumor growth (data are shown as tumor size ±SD; n=3 mice per group; ***p<0.001, and ns, non-significant two-way analysis of variance) and (B) percentages of CD8+ T cells in the tumor CD45+/MHC-II-negative single cell suspensions, 2 days after the last PICLC injection. Bars are shown as mean±SD; n=3 mice per group. ***P<0.001 by Mann-Whitney unpaired test.
Because IFN-I signals could play a role in the function and survival of CD8 T cells we examined the role of the IFNαβR on the CD8 T cells by generating T cell receptor OT-I (ovalbumin specific) transgenic mice on an IFNαβR background. Purified OT-I IFNαβR-deficient cells (CD45.1+/CD45.2+) were mixed at a 1:1 ratio with WT OT-I cells (CD45.1+) and were transferred into mice bearing subcutaneous B16 tumors expressing ovalbumin. After treating the mice with intravenous PICLC the tumors were examined for the presence of OT-I T cells. PICLC enhanced OT-I infiltrates into the tumor but no significant differences were observed between the percentages of OT-I IFNαβR-deficient cells and OT-I WT cells (online supplemental figure S2). Also, no significant differences in the ratio of these T cells were observed in spleens. These results indicate that the enhancement of CD8 T cell tumor infiltration by PICLC treatment is not influenced by IFN-I signals on the T cells.
Discussion
Many of the successful cancer immunotherapies require the participation of tumor-reactive T lymphocytes. Various vaccination modalities such as the use of synthetic peptides, DNA, mRNA and proteins aim at eliciting T cell responses capable of eliminating established tumors, or at least, limiting their growth and metastatic activity. Since most vaccines so far used in the clinic fail to elicit huge T cell responses, which in our view are necessary to eliminate tumors,6 many groups have opted to use adoptive T cell therapies that so far appear to be more promising than vaccines.27 28 Another promising mode of cancer immunotherapy is the use of ICIs, which augment the function of already existing antitumor T lymphocytes.1 29 30 Nevertheless, we believe that one of the main barriers of all these T cell-based cancer immunotherapies, which limits their antitumor efficacy, is due to a suboptimal traffic and infiltration of the effector T cells into the tumor parenchyma. The results presented herein reveal that systemic administration of PICLC, a particular formulation of the synthetic dsRNA mimic poly-IC, promoted CD8 T cell infiltrates into the tumor parenchyma, resulting in significant reduction of tumor growth in mice.
It was reported that peptide vaccines administered subcutaneously simultaneously with intramuscular PICLC had a therapeutic benefit in mice with intracranial glioblastomas.31 The antitumor effect required IFN-I and IFN-γ and was mediated by CXCL10, which increased T cell tumor infiltrates. Although these studies demonstrated that systemically administered PICLC enhanced tumor T cell infiltration, the roles of the 2 PRRs for PICLC, TLR3 and MDA5, and the cells responding to this synthetic PAMP mimic were not studied. Our results show that MDA5 and not TLR3 mediated the IFN-I response that enhanced tumor T cell infiltration induced by systemically administered PICLC. The present findings indicate that both BM-derived cells (presumably DCs) and stromal cells responding to PICLC via MDA5 were necessary to obtain optimal antitumor effects and tumor T cell infiltration. We previously reported that intravenous injections of PICLC into mice resulted in more than 10-fold higher production levels of IFN-I as compared with poly-IC, which was mediated by MDA5 and not TLR3.32 Several experiments presented here provide evidence that the stromal cells responding to PICLC are VECs, which would make sense since these cells play a critical role in allowing T cell infiltration into tissues where they are needed. It is well-known that VECs express numerous scavenger receptors allowing them to capture molecules such as lipids and nucleic acids. Indeed, VECs were shown here to produce IFN-I when stimulated with PICLC but not with poly-IC, which could be explained by the induction of the proton sponge effect by poly-lysine component of PICLC allowing the endosomal escape of the synthetic dsRNA into the cytoplasm to stimulate MDA5. In vitro studies with DCs using a proton sponge inhibitor bafilomycin A1 have shown that IFN-I production by PICLC requires endosomal escape via this mechanism.32 VECs stimulated with PICLC upregulated VCAM-I, CXCL10 and CXCL9, which play an important role in T cell tissue infiltration.11 12 14 24 Interestingly, systemic administration of PICLC promoted T cell infiltration into tumors while not affecting much T cell infiltration into other organs. This difference appears to be due to the preferential enhancement of VCAM-I and CXCL9 expression induced by PICLC systemic administration in the tumor vasculature as compared with normal tissues. Mouse and human VECs responded somewhat differently to poly-IC and PICLC. The two mouse VEC lines tested here were substantially less responsive (required higher concentrations) to stimulation by PICLC as compared with primary HUVECs (figure 4C vs figure 5C), suggesting the possibility that long-term cultured VECs may have diminished capacity to capture and internalize polynucleotides. The ability of HUVECs to respond better to poly-IC and PICLC as compared with poly-AU indicated that increases of IFN-I and CXCL9 expression are likely mediated by MDA5 stimulation and not so much by TLR3 activation. On the other hand, enhanced VCAM-I expression appeared to be mediated via TLR3 stimulation. Our results also indicate that poly-IC without the poly-lysine component may be able to stimulate MDA5 and generate an antitumor effect but not as effective as the one elicited by PICLC. Nonetheless, it should be noted that in vivo administration of PICLC to human cancer patients will also be more successful than using poly-IC because PICLC is formulated to be resistant to RNAse degradation. We observed in most experiments that the increases of CD8 T cell tumor infiltration by systemic administration of PICLC was accompanied by reductions in the CD11b/Gr1+ (presumably MDSCs) population. At present time, we do not know whether this is due to a reduction in the rate of infiltration of these cells or whether they are simply diluted by the increase of CD8 T cells at the tumor site. Further studies are warranted to determine what mechanisms are involved in these changes.
In summary, the present findings have strong implications for the improvement of T cell-based immunotherapies for cancer. We predict that systemic administration of PICLC will improve ICI therapy, therapeutic vaccines capable of eliciting substantial tumor-reactive T cell responses and adoptive cell therapies using either TILs or genetically modified T cells (expressing tumor-reactive T cell receptors or chimeric antigen receptors) since all of these modalities require that the T cells traffic and infiltrate the tumor parenchyma. This prediction is somewhat confirmed by recent findings in an autovaccination pilot clinical study combining sequential intratumoral (in situ vaccine) and intramuscular PICLC where immunologically cold tumors were converted into hot tumors, with marked increases in the activation marker PD-L1 and infiltration with CD4 and CD8 T cells, and a corresponding clinical response.33 34 These findings suggest that after priming of CD8 T cells by the initial intratumoral PICLC injections, T cell infiltration into the tumors may have been driven by the subsequent intramuscular PICLC administrations.
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
All animal studies were approved by the Institutional Animal Care and Use Committee of Augusta University and carried out in accordance with guidelines of the National Institutes of Health on animal care.
References
Footnotes
X @estebancelis
HS and JW contributed equally.
Correction notice This article has been corrected since it was first published online. Figure 5 and the associated figure legend have been updated.
Contributors EC conceived and designed the experiments. HS, JW, VIF, AEF and DA performed the experiments. Data interpretation and analysis was performed by HS, JW, AMS and EC. AMS produced and provided the poly-ICLC used in this study. HS, JW, EC and AMS contributed with important intellectual input. Writing, reviewing and manuscript editing was done by HS, JW, AMS and EC.
Funding This work was supported by start-up funds from Augusta University, Georgia Cancer Center and the Georgia Research Alliance (GRA).
Competing interests EC is a paid consultant for Oncovir, Inc. AMS is President and CEO of Oncovir, Inc. which is developing poly-ICLC (Hiltonol®) for the clinic.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.