Article Text

Abstract
Background C-type lectin-like molecule 1 (CLL-1) is highly expressed in acute myeloid leukemia (AML) but is absent in primitive hematopoietic progenitors, making it an attractive target for a chimeric antigen receptor (CAR) T-cell therapy. Here, we optimized our CLL-1 CAR for anti-leukemic activity in mouse xenograft models of aggressive AML.
Methods First, we optimized the CLL-1 CAR using different spacer, transmembrane and costimulatory sequences. We used a second retroviral vector to coexpress transgenic IL15. We measured the effects of each construct on T cell phenotype and sequential (recursive) co culture assays with tumor cell targets to determine the durability of the anti tumor activity by flow cytometry. We administered CAR T cells to mice engrafted with patient derived xenografts (PDX) and AML cell line and determined anti tumor activity by bioluminescence imaging and weekly bleeding, measured serum cytokines by multiplex analysis. After euthanasia, we examined formalin-fixed/paraffin embedded sections. Unpaired two-tailed Student’s t-tests were used and values of p<0.05 were considered significant. Survival was calculated using Mantel-Cox log-rank test.
Results In vitro, CLL-1 CAR T cells with interleukin-15 (IL15) were less terminally differentiated (p<0.0001) and had superior expansion compared with CD28z-CD8 CAR T cells without IL15 (p<0.001). In both AML PDX and AML cell line animal models, CLL-1 CAR T coexpressing transgenic IL15 initially expanded better than CD28z-CD8 CAR T without IL15 (p<0.0001), but produced severe acute toxicity associated with high level production of human tumor necrosis factor α (TNFα), IL15 and IL2. Histopathology showed marked inflammatory changes with tissue damage in lung and liver. This acute toxicity could be managed by two strategies, individually or in combination. The excessive TNF alpha secretion could be blocked with anti-TNF alpha antibody, while excessive T cell expansion could be arrested by activation of an inducible caspase nine safety switch by administration of dimerizing drug. Both strategies successfully prolonged tumor-free survival.
Conclusion Combinatorial treatment with a TNFα blocking antibody and subsequent activation of the caspase-9 control switch increased the expansion, survival and antileukemic potency of CLL-1 CAR T-cells expressing transgenic IL15 while avoiding the toxicities associated with excessive cytokine production and long-term accumulation of activated T-cells.
- adaptive immunity
- cytokines
- hematologic neoplasms
- immunotherapy
- adoptive
- receptors
- chimeric antigen
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
T cells genetically modified to express a chimeric antigen receptor (CAR) to specifically target a tumor cell surface antigen can successfully treat B cell malignancies, but have had less activity against acute myeloid leukemia (AML).1–3 The ideal CAR target antigen should be highly expressed on cancer cells and their clonal progenitors, but absent on normal cells and stem cells. Although no antigenic target fully meets these criteria for AML, a transmembrane glycoprotein (C-type lectin like molecule 1 (CLL-1); C-type lectin domain family 12 (CLEC12A); CD371), has emerged as a promising candidate.4 CLL-1 is present in leukemic cells in more than 87% patients with AML but is absent on early hematopoietic progenitor cells.5–7
Several studies have confirmed that CLL-1 can indeed be targeted by CAR T cells6 but their potency and durability have been suboptimal. TCR signaling and costimulation (signals 1 and 2) regulate expansion and persistence of T cells, and signal 3, which is provided mostly by cytokine stimulation, also influences activity.8 Previous studies demonstrated that cytokine exposure can enhance the anti tumor activity of CAR-T cells, with both interleukin 7 (IL7) and IL15 showing promise in mouse models of leukemia9 and glioblastoma.10 Here, we examine whether incorporating transgenic IL15 into CLL-1 CAR T cells can enhance efficacy for AML.
IL15 has pleiotropic effects on T cells that enhance their anti tumor activity, specifically influencing T cell migration and survival,11 12 sustaining central memory phenotype,13 and inhibiting activation-induced T cell death.14 15 Unfortunately, the concentrations of systemically administered IL15 required to achieve these benefits can be associated with uncontrolled T cell proliferation16 and severe in vivo toxicity.17 By contrast, gene-modified T cells or NK cells can produce high local but non-toxic systemic concentrations of IL15.7 8 16 CAR T cells coexpressing transgenic IL15 have superior anti tumor activity in several in vivo models of both lymphoid malignancies and solid tumors without significant toxicity. Furthermore, a clinical study has demonstrated the safety of cord blood NK cells engineered to express IL15 with a CD19 targeted CAR in chronic lymphocytic leukemia and lymphoma.8 9 18
Here, we tested T cells coexpressing a CLL-1 CAR and transgenic IL15 in xenografts of AML. We found transgenic IL15 significantly improved CLL-1 CAR-T cell expansion, persistence and anti tumor activity, allowing the combination product, but not the CLL-1 CAR alone, to eradicate disease. Unexpectedly, and in contrast to the safety of the CART- IL15 combination in other disease settings, in our models of AML, CLL-1-IL15 CART induced a severe and atypical form of cytokine release syndrome (CRS), associated with high levels of circulating tumor necrosis factor alpha (TNFα). Fortunately, we were able to predictably and consistently prevent these adverse effects while retaining anti tumor activity by sequencing the early administration of TNFα blocking antibody, followed by late termination of the ongoing anti tumor CAR-T response using an inducible safety switch. This combination approach allows CLL-1-IL15 CAR T cells to eliminate myeloid leukemias in the absence of discernible toxicity.
Materials and methods
Cell lines
AML cell lines THP-1, HL-60 and MOLM-13 were purchased from ATCC and maintained as described in the online supplemental methods appendix. All cell lines were transduced with a gammaretroviral vector encoding enhanced GFP-FFluc fusion protein as previous described.19 Patient-derived xenograft (PDX 401) cells were obtained from a multiply relapsed 16-month-old Caucasian boy diagnosed with AML FAB M5 (high-risk disease due to a KMT2A fusion partner) and were kindly provided by Alexandra Stevens.
Supplemental material
Plasmid construction and retrovirus production
A CLL-1 CAR previously described by our group19 was modified using in-Fusion Cloning (Takara/Clontech, California, USA) to generate a panel of CLL-1 CARs with altered spacer, transmembrane and costimulatory domains. Retroviral vector production and T cell transduction have been described previously.19 CAR T-cells were expanded in complete medium (45% RPMI 1640; Hyclone, Waltham, Massachusetts, USA), 45% Click’s media (Irvine Scientific, Santa Ana, California, USA), 2 mM L-glutamine (Gibco-BRL, San Francisco, California, USA) with added penicillin and streptomycin (Gibco-BRL) and 10% FBS (Hyclone) in the presence of 10 ng/mL IL-7 (Peprotech, Rocky Hill, New Jersey, USA) and 10 ng/mL 1 L-15 (Peprotech). We cotransduced CLL-1 CAR with a gammaretroviral vector SFG encoding the human IL15 gene linked via a 2A sequence8 with the inducible caspase-9 (iC9) suicide gene that induces apoptosis on specific binding with a small molecule chemical inducer of dimerization (CID) AP20187 (Takara Bio, California, USA).
Car expression and immunophenotoype of transduced cells
CLL-1 CAR was detected using AF647-conjugated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, Pennsylvania, USA). Anti-human CD45, CD4, CD8, CD3, CD45RA, CD19, CCR7, CD271, CD371 (Clec12A, CLL-1) and Mouse IgG2alpha isotype were obtained from BD Biosciences (San Jose, California, USA), Beckman Coulter (Indianapolis, Indiana, USA), Life Technologies (Carlsbad, California, USA) or Biolegend (San Diego, California, USA). Flow cytometric data were acquired on a Beckman Gallios (Beckman Coulter, Indianapolis, Indiana, USA) or BD FACS Canto II (BD Biosciences) and analyzed using FlowJo 10 (BD).
Coculture, sequential killing assays and detection of cytokine secretion
The details of coculture and sequential killing assays are included in the online supplemental methods appendix. For in vivo experiments, mice were bled at the specified time points and the sera snap frozen. Human TNF-α, IL −15, interferon (IFN)-g, IL-2, IL-1a, IL-6, IL-10 were measured using a Luminex xMAP Multiplex Assay with Millipore Sigma antibodies (Burlington, Massachusetts, USA). For in vitro experiments, we cultured HL-60 and blast cells obtained from the PDX model with CAR-T cells, and supernatants were collected at the time points described. Cytokine production by T-cells was measured by flow cytometry using Intracellular Cytokine Staining Kit (BD Biosciences).
Xenogeneic and PDX AML mouse models
NOD.SCID IL-2Rg-/- (NSG-SGM3) mice were were bred in the Baylor College of Medicine Transgenic Mouse Facility using breeder pairs obtained from Jackson Laboratories. Tumor burden in mice engrafted with FFluc-modified cells was measured using In Vivo Imaging System (IVIS) imaging as described before.19 Disease progression in PDX models was measured in peripheral blood of mice by flow cytometry at specified time points. Mouse experiments were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Activation of suicide gene and administration of TNFα antibody in vivo
We activated the iC9 suicide gene in adoptively transferred T-cells by i.p injection of AP20187 (Takara Bio). Human TNFα-specific antibody with IgG2a mouse Fc region (Absolute Antibody, Boston, Massachusetts, USA) was injected intravenously at the stated times.
Pathological analysis
Formalin-fixed tissues post mortem were embedded in paraffin blocks, cut into sections, H&E stained and examined macroscopically and microscopically for evaluation of disease or tissue toxicity by a trained pathologist.
Statistical analysis
Unpaired two-tailed Student’s t-tests were used for group comparisons and values of p<0.05 were considered significant. Survival was calculated using Mantel-Cox log-rank test (Graph Pad Prism).
Results
CD28z-CD8 CLL-1 CAR T cells lack sustained disease control in an aggressive in vivo model of AML
We previously reported that T cells expressing a CLL-1 CAR with 4-1BB and CD3 zeta endodomains, CD8 transmembrane and CD8 hinge (4-1BBz-CD8) could control AML in xenograft models with low leukemia burden.19 To determine if this CAR construct could eradicate bulkier disease, or whether such functionality required a construct able to produce more rapid expansion of CAR-T cells, we chose a CLL-1 CAR from the panel of CLL-1 CAR constructs described in online supplemental figure 1, based on its expansion and anti tumor effector function. At day 20, T cells expressing the CLL-1 CAR with CD28 transmembrane and endodomain and CD8 hinge (CD28z-CD8) had a two log greater expansion in culture compared with 4-1BBz-CD8 CAR (p<0.001) (figure 1A,B). As shown in figure 1C, this increased expansion was in part due to their lower death rate compared with 4-1BBz-CD8 CAR T cells. As previously described, the tonic CAR-derived 4-1BB signaling may be associated with continuous TRAF2-dependent activation of NF-kB pathway and augmented Fas-dependent cell death, impairing T cell expansion and diminishing their anti tumor activity particularly when it is expressed from a gamma retroviral vector.20

CLL-1.CD28z-CD8 CAR T cells lack sustained disease control in vivo. (A) Schematic representation of CLL-1 CAR constructs. (B) Expansion of CAR-expressing T-cells in culture after gammaretroviral transduction. P value was determined by unpaired Student’s t-test (n=3). (C) Viability of different CLL-1 CAR T cells through days in culture (n=3). P value was determined by unpaired Student’s t-test. (D) The schematic figure of HL 60 xenograft mouse model comparing CLL-1.CD28z.CD8 (n=11) vs CLL-1.4-1BBz.CD8 (n=10) CAR vs control (n=7) T cells. Mice aged 10–16 weeks old were irradiated with 1.16 Gy followed by injection of FFLuc-labeled HL-60 cells (1.5×106 cells per mouse). CLL-1 CAR T cells (2×106 cells per mouse) were injected intravenously 3 days later. (E) Representative images showing leukemia progression in groups from week 1 to week 7. (F) Kaplan-Meier curve showing the survival of mice in each experimental group. P value was determined by log-rank Mantel-Cox test. CAR, chimeric antigen receptor; CLL, C-type lectin-like molecule 1; NTR, non-transduced.
We next tested the ability of these CART cells to control established AML in vivo. NSG-SGM3 mice were sublethally irradiated (1.16 Gy) and injected with FFLuc-modified HL-60 AML cell line (1.5×10ˆ6 cells per mouse). Three days later, we injected 2×10ˆ6 CLL-1 CART cells with the chosen CD28z-CD8 backbone and an identical dose of T cells expressing the same CLL-1 scFv but with the 4-1BBz-CD8 backbone19 (figure 1D). Both CARs decreased tumor growth compared with controls (figure 1E), but survival was significantly prolonged in the group receiving CD28z-CD8 CAR T cells (>50 days vs 36 days for 41BB construct, p=0.002) (figure 1F). This outcome suggests either that initial tumor cell depletion was deeper in the recipients of CLL-1 CD28 CAR T, consistent with their more rapid expansion compared with 41BB constructs, or that anti tumor activity was sustained for longer. In a tumor-free mouse, CLL-1 CAR-expressing T cells persisted in peripheral blood to week 7 after CD28z-CD8 CAR T cell injection (online supplemental figure 2A,B). Even after receiving CD28z-CD8 CAR T cells, however, we observed relapses at day 10, 21 and 30. Two of these mice were sacrificed at week 7 after CD28z-CD8 CAR T cell injection and we observed CLL-1 antigen-positive disease in bone marrow and spleen. No CAR T cells were detected within these tissues in the mice (online supplemental figure 2C). Therefore, we hypothesized that despite the superior initial performance of CD28z-CD8 CLL-1 CART, expansion and persistence remain insufficient for sustained disease control. To augment their antileukemic function, we, thus, modified the CD28z-CD8 CLL-1 CAR T cells to incorporate IL15 as a signal 3.
Transgenic IL15 augments CAR T cell expansion and persistence in vitro
In order to increase expansion, prolong survival and augment the anti tumor activity of CD28z-CD8 CLL-1 CAR T cells, we cotransduced them with a gammaretroviral vector encoding human IL15 gene. Since high concentrations of IL15 have been associated with toxicity, we also included in the construct an iC9 safety switch21 22 (iC9-IL15, figure 2A). Most of the T cells were doubly transduced with CAR/iC9-IL15 (figure 2B). The double transduced T cells expanded at the same rate as control non-transduced cells, approximately twice as much as unmodified CLL-1 CART (p<0.001). These results, together with our published data,9 10 support the functional activity of the transgenic IL15 (figure 2C). The CD4:CD8 ratio was unaltered by double transduction, although the proportion of naïve like cells (CD45RA+, CCR7+) was higher in both the CD4+ and CD8+ subpopulations of CLL-1 CAR and iC9-IL15 double transduced cells than in CLL-1 CAR T cells (21.7% vs 9.5%, p<0.0001; 26% vs 14%, p<0.0001) (figure 2D). In short-term killing assays, we found no difference in CLL-1 CAR T-cell anti tumor activity with or without iC9-IL15 cotransduction, irrespective of whether the target cells expressed high, intermediate, or low levels of the CLL-1 target antigen (figure 2E). The benefits of transgenic IL15, however, became apparent when we measured T cell proliferation and cytotoxic function in sequential (recursive) killing assays against the THP-1 cell line, which expresses high levels of CLL-1 (figure 2F). In comparison to CLL-1 CAR T cells cultured alone, proliferation was promoted and antileukemic activity was more sustained when CLL-1 CAR +iC9-IL15 CAR T cells were co cultured with the THP-1 cell line.

Transgenic IL-15 improves CLL-1. CAR T cell function in vitro. (A) Schematic figures of CLL-1.CD28z-CD8 CAR (CLL-1 CAR) and IL15 constructs. (B) The cotransduction of CLL-1 CAR and IL15 construct shown in flow plot at day 3 after transduction. (C) Expansion of NTR, CLL-1 CAR and CLL-1 CAR+ IL15 T cells in culture shown as fold increase in total cells. P value was determined by unpaired Student’s t-test (n=3). (D) The distribution of CD4 T cells (top) and CD8 T cells (bottom) at day 7 after transduction. P value was determined by unpaired Student’s t-test (n=3). (E) The MFI ratios of cell lines THP-1 (left), HL60 (middle) and MOLM-13 (right), the residual counts of tumor cells in THP-1 (left), HL-60 (middle) and MOLM-13 (right) after coculture at 72 hours with CLL-1 CAR and CLL-1 CAR+ IL15 E:T (1:4). (F) Residual live tumor (top) and T cell counts (bottom) at sequential killing assay with THP-1 E:T (1:4). (n=3). CAR, chimeric antigen receptor; CLL, C-type lectin-like molecule 1; IL-15, interleukin 15; Iso Ctrl: isotype control; NTR, non-transduced.
Transgenic IL15 increases both CLL-1 CAR T expansion and toxicity in PDX and xenograft models of AML
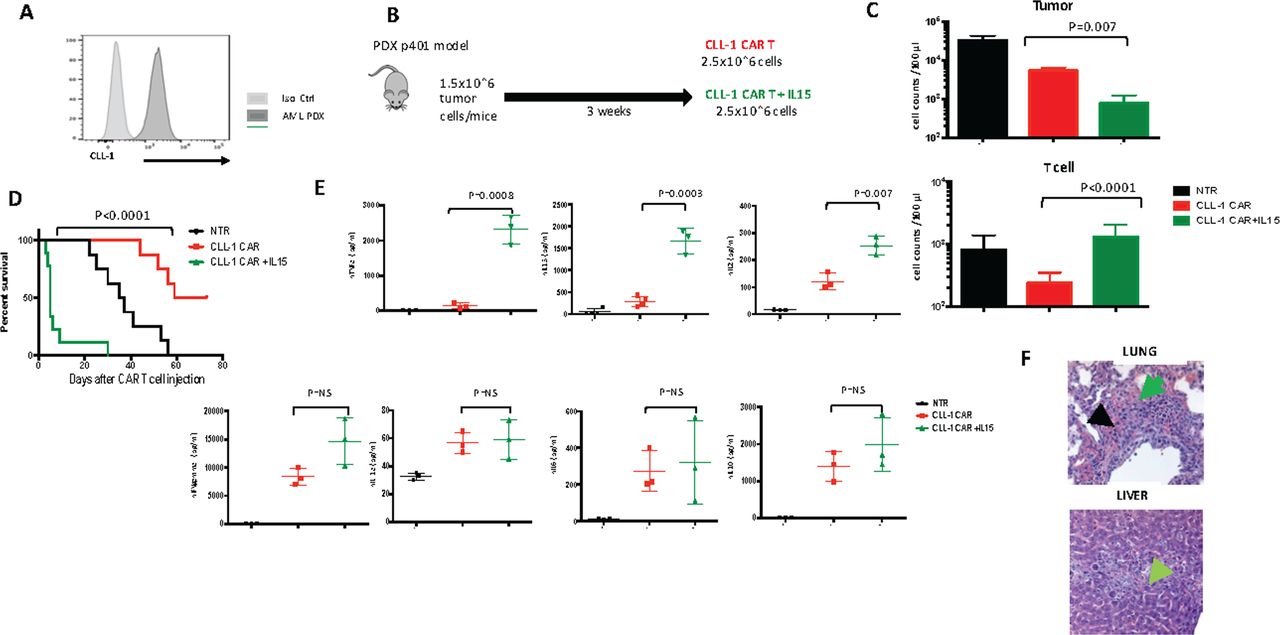
To evaluate the in vivo influence of transgenic IL15 on the persistence and anti tumor activity of CLL-1 CART, we engrafted NOD.SCID IL-2Rg-/- (NSG-SGM3) mice with a AML (PDX) with high CLL-1 expression (figure 3A). We repeated the same experiment with an intermediate CLL-1 expressor cell line, HL-60, AML xenograft model. As described below, both models showed a similar pattern of response.

Transgenic IL-15 in in vivo model. (A) CLL-1 expression of AML PDX model. (B) Schematic figure of AML PDX model. in the PDX model, mice received PDX cells intravenously (1.5×106 cells per mouse) followed 18–21 days later by a single injection of CLL-1 CAR T cells (2.5×106 cells per mouse). (C) Residual tumor cells (top) and number of T cells (bottom) at day 5 after CLL-1 CAR (n=8) CLL-1 CAR+ IL15 (n=5) T cell infusion. P value was determined by unpaired Student’s t-test. (D) Kaplan-Meier curve showing the survival of mice in each experimental group. (E) Serum human cytokine levels (mean±SEM) (TNF alpha, IL15, IL-2, IFN-gamma, IL-1a, IL-6, IL-10) of representative mice at day 7 after CLL-1 CAR (n=3) and CLL-1 +IL15 CAR (n=3) T cell infusion. P value was determined by unpaired Student’s t-test. (F) Histopathological examination of lung (top) and liver (bottom) tissues (H&E, ×100) in a mouse died at day 4 after CLL-1+ IL15 CAR infusion. Hemorrhage, edema is marked with green and apoptosis is marked with black in lung. The necrosis of liver is marked with green. P values were determined by log-rank Mantel-Cox test. CAR, chimeric antigen receptor; CLL, C-type lectin-like molecule 1; IFN, interferon; IL-15, interleukin 15; Iso Ctrl, isotype control; NS, not significant; NTR, non-transduced; PDX, patient derived xenografts; TNF, tumor necrosis factor.
After PDX engraftment, we injected CLL-1 CAR T cells or CLL-1 CAR+iC9-IL15 (figure 3B). In both models, all animals receiving the iC9-IL15 coexpressing CART developed a rapidly progressive shock-like syndrome with hypothermia, tachypnea, tachycardia and death. In the PDX model, this syndrome was associated with rapid tumor destruction, since tumor burden was more than one log lower in mice receiving CLL-1 CAR +iC9-IL15 CAR T cells compared with recipients of CLL-1 CAR T cells (p=0.007 at day 5) (figure 3C). This initial reduction in tumor burden was associated with greater CAR-T expansion in peripheral blood (p<0.0001) (figure 3C). But since the rapid tumor destruction and T cell expansion were associated with acute toxicity, recipients of CLL-1 CAR T cells without IL15 survived longer, although with higher tumor burden (median survival; 66 days vs 5 days, p<0.0001) (figure 3D).
To understand the cause of this syndrome, we measured human cytokine levels in the serum of these NSG-SGM3 mice. At day 7 after CAR T cell injection, we detected increased levels of human TNFα, IL15, and IL2 in CLL-1 CAR+iC9-IL15 CAR T compared with control CLL-1 CAR T cells (mean, pg/mL; 2314 vs 135, p=0.0008; 1667 vs 289,p=0.0003; 253 vs 121, p=0.007). Other human cytokines, in particular IL1 and IL6, were low and not significantly different between groups (figure 3E), perhaps reflecting the lack of human cell subsets responsible for initiating and sustaining production of these proteins during an inflammatory response.23 The elevated levels of hIL15 and hTNFα we observed in recipients of CLL-1 CAR +iC9-IL15 CAR T were associated with tissue damage, since we observed ×10 normal levels of serum transaminases and LDH and severe lung and liver injury on histopathological examination (figure 3F).
We observed an identical pattern of toxicities in a second xenograft model, although with delayed kinetics. Mice with HL-60 tumors who received CLL-1 CAR T cells survived longer than CLL-1 CAR +iC9-IL15 CAR T cells recipient (median survival, undefined >60 days vs 32 days, p=0.04) (online supplemental figure 3A–C).
Increased TNFα secretion is less evident in vitro with CLL-1 CAR+iC9-IL15 CAR T cells
We did not anticipate these results based on our preclinical experiments. For example, there was no difference in TNFα release between constructs with or without IL15 during 9 days of in vitro coculture of CLL-1 CAR T cells with HL 60 leukemic targets. Even repeated addition of fresh leukemic targets failed to replicate what we had observed in vivo (online supplemental figure 4A). More sensitive analyzes using intracellular staining for cytokines detected higher TNFα in the T cell, but not the myeloid, compartment when CLL-1 CAR +iC9-IL15 CAR CD4 T cells were co cultured with HL60 leukemic targets compared with control CLL-1 CAR CD4 T cells (online supplemental figure 4B,C). This effect was more obvious when CLL-1 CAR +iC9-IL15 CAR CD4 T cells were co cultured with primary blasts from the PDX model. This effect was greatest when CLL-1 CAR +iC9-IL15 CAR CD4 and CLL-1 CAR +iC9-IL15 CAR CD8 cells were cultured at high tumor to CAR T ratio (online supplemental figure 4D–G).
Early administration of CID resolves toxicity of CLL-1 CAR+iC9-IL15 CAR T but diminishes long-term anti tumor activity
To determine whether depleting CAR +iC9-IL15 CAR T cells by activating the embedded suicide switch could ameliorate toxicity while sparing anti tumor activity, we initially activated the iC9 using a small molecule dimerizing drug (AP20187) at a 10 nM concentration.9 After establishing HL60 leukemia in NSG-SGM3 mice, we injected CLL-1-CAR T cells (2.5×10ˆ6/mouse) or CLL-1 CAR +iC9-IL15 CAR T cells (2.5×10ˆ6/mouse) (figure 4A). Based on the timing of the severe toxicity described above, we administered CID on day 6 and 12 by intraperitoneal injection. We monitored iC9-IL15 CAR T depletion on day 14. In four of six mice, iC9-IL 15 CAR T cell expansion was reduced by >2 logs. In two mice, iC9-IL 15 CAR T cell expansion was less well controlled, and these animals were reinjected with CID at day 14, whereupon iC9-IL15 CAR T cells were reduced by 2 logs. In all mice, residual CLL-1 CAR +iC9-IL15 CAR T cells began to re-expand 6 days after depletion (figure 4B). The level and duration of CAR-IL15 T cell depletion was sufficient to prolong the survival of CLL-1 CAR +iC9-IL15 CAR treated mice (median survival, 53 days vs 17 days for CLL-1.CAR.IL15 T cell treated mice; p=0.002) (figure 4C). In these mice, serum levels of TNFα and IL15 were reduced by a mean of 50% following CID administration (figure 4D), and autopsy showed no macroscopic or microscopic organ damage (figure 4E). By contrast, mice in the CLL-1 CAR +iC9-IL15 CAR T cell group that did not receive CID had high levels of TNFα and died early with the same severe organ damage described previously. However, while early administration of CID controlled the CRS and prevented early death, CAR T activity was insufficient for disease eradication and the mice subsequently succumbed to AML (figure 4C,F).
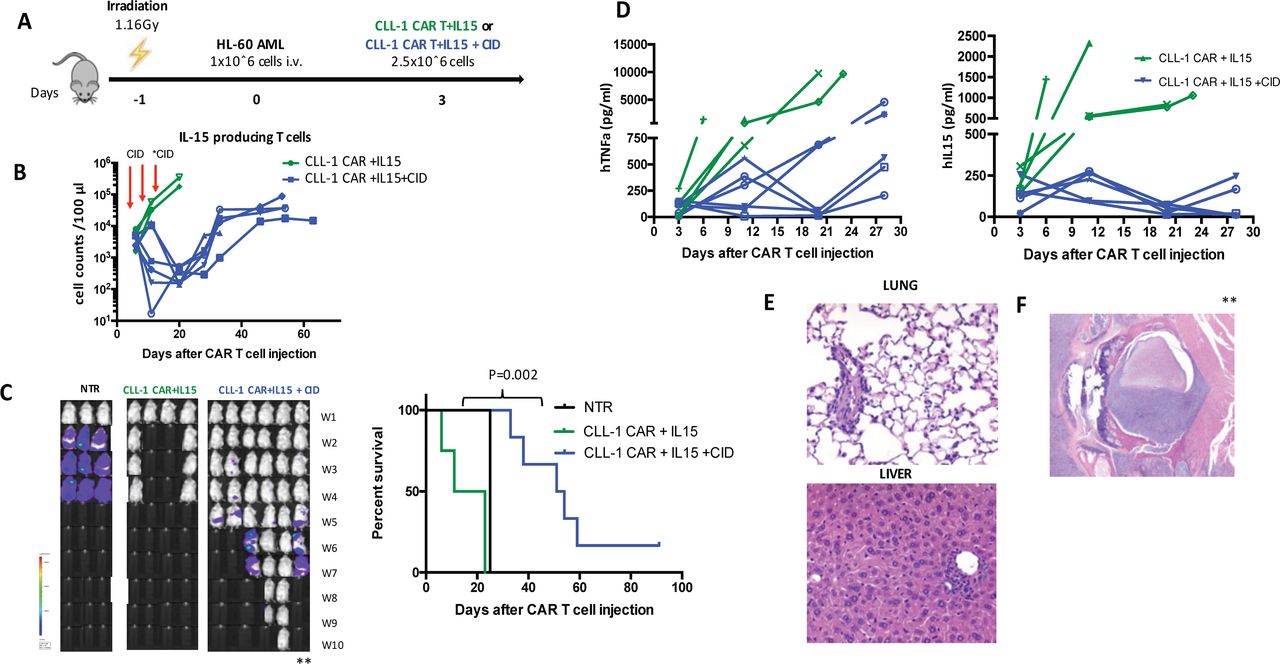
CID eliminates IL15-producing T cells in vivo. (A) Schematic figure of HL-60 tumor model comparing CLL-1 CAR+IL15±CID (50 ug/mouse on day 6, 12 for all mice and on day 14 for three mice). (B) Total T cell and IL15 producing T cell counts in peripheral blood in each experimental group through the experiment. red arrows represent the CID administration on day 6 and 12 to each mouse, on day 14 CID administered to two mice marked with (*). (C) Representative images showing leukemia progression in groups from week 1 to week 10. Kaplan-Meier curve showing the survival of mice in each experimental group. P values were determined by log-rank Mantel-Cox test. (D) Serum human TNF-alpha and IL-15 levels in each representative mouse. (E) No tissue damage was shown in histopathological examination of lung (top) and liver (bottom) tissues (H&E, ×100) in a representative mouse which received CID. (F) In histopathological exam of spinal cord showed the compression with tumor cells in a mouse marked in C with (**) from CID group that relapsed and showed hind and limb paralysis (H&E, ×100). AML, acute myeloid leukemia; CAR, chimeric antigen receptor; CLL, C-type lectin-like molecule 1; CID, chemical inducer of dimerization; IL-15, interleukin 15; TNF, tumor necrosis factor.
TNFα blocking antibody reduces TNFα levels without impeding CLL-1 CART anti-tumor activity
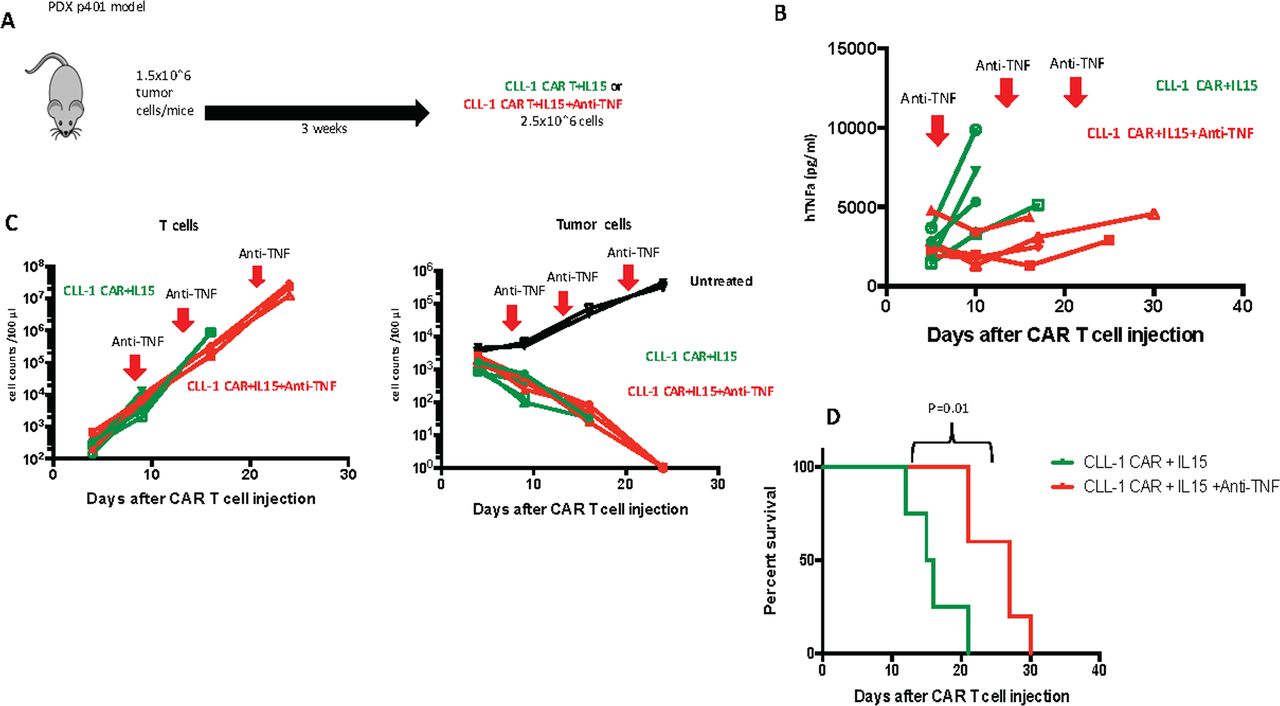
To discover whether the excessive production of TNFα was indeed the primary driver of fatal CRS in these models, we used anti-TNFα as an alternative mitigating strategy to iC9-IL15 CAR T cell destruction with CID. We used a chimeric antibody containing infliximab-derived TNFα-specific variable regions fused with murine Fc region for additional stability in mice. Three weeks after intravenous inoculation of 1.5×10ˆ6 p401 PDX tumor cells/mouse, we injected CLL-1 CAR +iC9-IL15 CAR T cells (2.5×10ˆ6/mouse) T cells (figure 5A), followed by intravenous injections of anti-TNFα.24 25 The TNFα levels were significantly reduced in mice treated with the first dose of anti-TNFα (figure 5B) but were not well controlled by subsequent injections. This treatment had no adverse effect on T cell expansion or tumor control (figure 5C). The anti-TNFα treated mice did not develop early CRS and had a survival advantage compared with untreated mice (median survival, 27 days vs 16 days; p=0.01) (figure 5D). However, CRS was only deferred, since T cell expansion was minimally impeded by cytokine blockage; once T cell numbers reached >10ˆ8, even the mice with TNFα blockade died with acutely progressive and fatal shock-like syndrome.

TNFα antibody (infliximab) reduces TNFα levels in vivo. (A) Schematic figure of AML PDX model comparing CLL-1 CAR+IL 15±Anti TNFα (1 mg/kg on day 7, 14 and 22). (B) TNFα levels throughout the experiment. red arrows show the days that anti-TNFα was administered. (C) Total T cell counts (left) and residual tumor cell counts (right) in peripheral blood of mice. (D) Kaplan-Meier curve showing the survival of mice in each experimental group. P values were determined by log-rank Mantel-Cox test (n=4). CAR, chimeric antigen receptor; CLL, C-type lectin-like molecule 1; IL-15, interleukin 15; TNFα, tumor necrosis factor α.
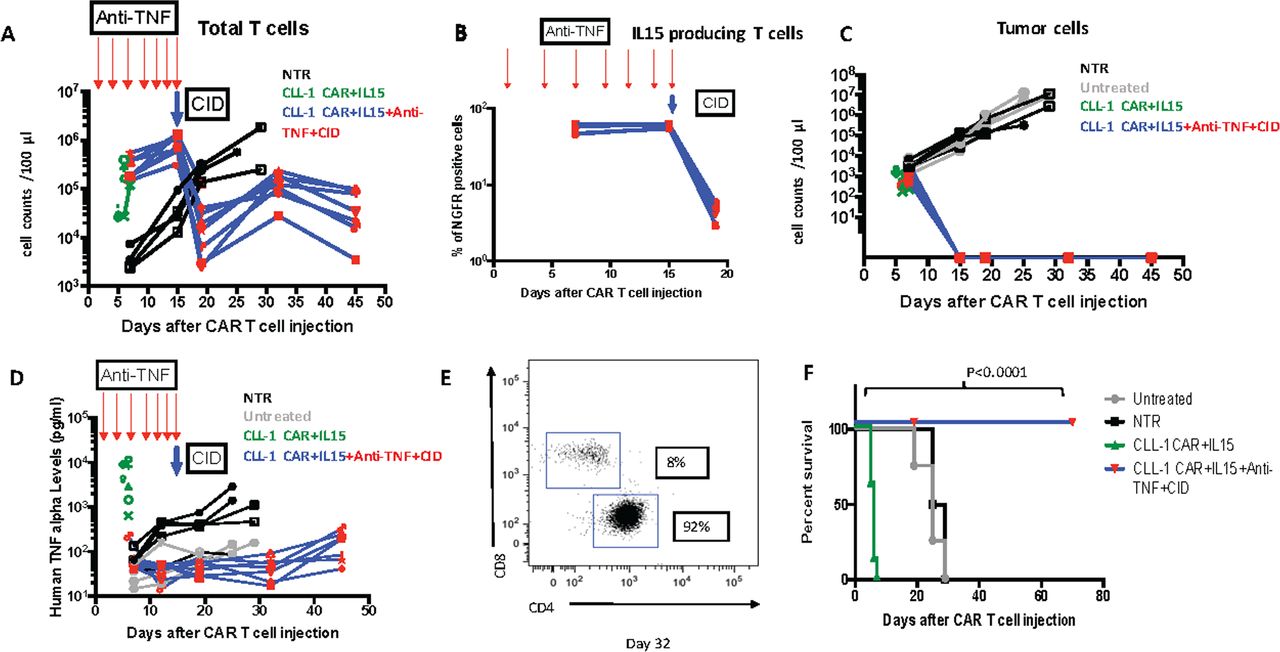
Sequential Anti-TNFα and CID fully mitigate CRS and allow both sustained tumor control and long-term survival
To assess whether TNFα antibody and iC9-induced cell death had complementary benefit, we injected CLL-1 CAR +iC9-IL15 CAR T cells (2.5×10ˆ6/mouse) 3 weeks after establishing a PDX model. We then injected a sequential combination of TNFα antibody,25 followed by later administration of CID. We observed initial expansion of CLL-1 CAR +IL15 CAR T cells and associated tumor control in the absence of toxic concentrations of TNFα. Subsequent administration of CID mitigated the late T cell expansion and avoided discernible toxicity while maintaining tumor control (figure 6A–D). On day 32 after CAR T cell infusion, the CLL-1 CAR +iC9-IL15 T cells remaining after CID treatment were 92% CD4 and 8% CD8 (figure 6E) and produced no late/long-term toxicity. Overall, this dual treatment of anti-TNFα and CID enabled CLL-1 CAR +iC9-IL15 T cells to safely produce long-term anti tumor activity, significantly prolonging survival (p<0.0001 compared with untreated CLL-1 CAR-IL15 controls) (figure 6F). To determine whether this combination therapy would be effective in a second model of AML, we repeated the experiments using an HL60 AML xenograft. As shown in online supplemental figure 5, toxicity could be successfully controlled and anti-tumor activity maintained.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sequential anti-TNF alpha and CID administration in vivo. (A) Total T cell counts in each experimental group (anti-TNF alpha (1.5 mg/kg) administration on days 1, 4, 7, 9, 11, 13 and 15 marked with red arrows followed by CID (100 ug/mouse) on day 15 marked with blue arrow). (B) IL15 producing T cell counts in each experimental group (anti-TNF alpha (1.5 mg/kg) administration on days 1, 4, 7, 9, 11, 13 and 15 marked with red arrows followed by CID (100 ug/mouse) on day 15 marked with blue arrow). (C) tumor cell counts in each experimental group. (D.) Serum human TNFα levels in peripheral blood (anti-TNFα (1.5 mg/kg) administration on days 1, 4, 7, 9, 11, 13 and 15 marked with red arrows followed by CID (100 ug/mouse) on day 15 marked with blue arrow). (E) C4/CD8 T cell distribution in mouse from CLL-1 CAR+IL15+Anti-TNF+CID group on day 32. (F) Kaplan-Meier curve showing the survival of mice in each experimental group. P values were determined by log-rank Mantel-Cox test (n=7). CAR, chimeric antigen receptor; CLL, C-type lectin-like molecule 1; CID, chemical inducer of dimerization; IL-15, interleukin 15; TNF, tumor necrosis factor.
Discussion
We have shown that introduction of the transgenic cytokine IL15 into CLL-1 directed CAR T cells greatly increased their expansion, persistence and anti tumor activity in xenograft and PDX models of AML. However, coexpression of transgenic IL15 in the CAR induced CRS with lethal levels of TNFα. Fortunately, discernible CRS could be averted and anti tumor activity maintained by early prophylactic administration of a clinically available TNFα antibody and later depletion of the IL15 secreting subpopulation of CAR-T cells by CID-mediated activation of an embedded iC9 safety switch. Overall, these studies provide a path toward the safe clinical evaluation of CLL-1 CAR-IL15 T cells to treat CLL-1-positive AML.
In animal models, CLL-1 CAR T cells with a 41BB costimulatory endodomain effectively controlled CLL-1-positive AML but had limited activity against established disease.19 Initially, to improve their kinetics in vivo, we examined whether the speedier expansion of CLL-1-CD28 CART cells versus CLL-1-41BB T cells that we observed in vitro was matched in our mouse models. Disappointingly, although CLL-1 CD28 CART cells did indeed expand more rapidly and improve survival compared with CLL-1-41BB T cells in an aggressive tumor model, the benefits were marginal since many animals relapsed due to insufficient CAR T cell persistence.
It is now clear that long-term CAR T cell persistence is key to achieving durable responses.26 ,27 28 Thus, to sustain killing and maintain a less terminally differentiated population of T cells, we transduced CD28z-CD8 CAR-T cells with a second vector encoding soluble IL15, a prosurvival cytokine.11 12 29 30 T cells expressing transgenic IL15 have increased viability and proliferative capacity in vitro due to reduced cell death and higher expression of the anti-apoptotic gene Bcl-2 compared with control CARs18 31 with safety and superior activity in several animal models.9 32 Transgenic IL15 can also increase the persistence and anti tumor activity of human CAR-NK and and CAR-iNKT cells and clinical studies using these cells have shown safety and preliminary evidence of activity against lymphoid and solid malignancies.33 34 In our study, the expansion of CLL-1 CAR +IL15 CAR T cells was superior to CLL-1 CAR T cells and also enabled these more extensively expanding transgenic cells to maintain a less differentiated phenotype. Consistent with their slower progression to a functionally exhausted phenotype, the IL15 CART had greater anti tumor activity in long-term (recursive) cytotoxicity assays in vitro.
Unexpectedly, however, when we tested the approach in vivo in xenograft models of AML, the superior initial tumor control in mice receiving CLL-1 CAR +IL15 CAR T cells was, followed by rapid onset of a progressive and lethal CRS, with hypothermia and tachycardia associated with multiorgan damage. Levels of TNFα were profoundly elevated in these animals (around 3000 pg/mL during toxicity), while other proinflammatory cytokines were no different from animals receiving CLL-1 CAR T without the IL15 transgene.
CRS is a systemic inflammatory response (SIRS) that correlates with high levels of inflammatory markers and cytokines and is a common feature of many CAR T cell therapies.35 Increased production and release of IL6 is the main driver of CRS related to CD19 CAR T cell activation, the setting in which this phenomenon has been most extensively studied. After CD19 CAR T cell administration, levels of IL6 and IFN-gamma increase in association with peak CD19 CAR numbers and activation.36 37 In severe CRS, levels of IL8, sIL1R, sIL2R, soluble glycoprotein 130 (sgp130), sIL6R, macrophage inflammatory protein, monocyte chemoattract protein 1, TNFα, and granulocyte-macrophage colony stimulating factor may also be elevated.37
CRS and associated cytokine release are difficult to model precisely in murine systems, due in part to differences in the sequence of cytokine production and the patterns of cross-talk between cytokines and innate and adaptive immune target cells. Nonetheless, our AML xenografts showed undoubted differences from studies of CRS triggered by CD19-CARs in both murine and human disease. We detected a rapid rise in TNFα and elevation of IL2 rather than of IL1 and IL6, a pattern that more closely mimics macrophage activation syndrome (MAS) than T cell-related CRS,38 although TNFα can also be a component of the pro-inflammatory cascade in severe CRS.
MAS is an immune dysregulation producing abnormal activation of macrophages and T cells.39 We observed no such MAS in mice treated with CLL-1 CAR in the absence of transgenic IL15, so its presence must be a critical initiating component. The contribution of IL15 to this phenomenon parallels the observation of early toxicity in mice accompanied by hypothermia and liver injury when administered an IL15 superagonist (IL15/IL15Ralpha complex). However, in those studies NK cells, which produce proinflammatory cytokines such as IFN-gamma rather than TNFα, appeared to mediate the immune toxicity. Thus, although one of the etiologic agents and the clinical manifestations may be shared between these two mouse models, the mechanisms are likely different.40–42
The TNFα pathway is a double-edged sword in immune regulation. Optimal levels of TNFα have a pivotal role in inducing and sustaining innate immunity against bacterial infection and promoting subsequent tissue repair. However, excessive TNFα production has been associated with a number of pathologies. Antibody blockade of TNFα was initially used to control the onset of acute respiratory distress syndrome (ARDS) after septicemia43 since dysregulated secretion promotes cytokine storms, triggers cell death and activates tissue resident neutrophils and macrophages.44 In patients with SIRS and sepsis-induced ARDS, TNFα modifies epithelial and endothelial cells, fibroblasts to increase recruitment and adhesion of neutrophils, platelets and lymphocytes, further amplifying cytokine release, thrombosis and tissue damage.45 46 Although the clinical benefits of TNF-blockade were of promise in these intital studies, TNFα antibodies were ultimately approved for use in the setting of chronic inflammation including rheumatoid arthritis and inflammatory bowel disease. Nonetheless the recent CoVID-19 pandemic and associated ARDS associated with induction of cytokines such as IL1 and TNFα is a reminder that the cytokine we observed as a fundamental mediator of lethality in this mouse model of CART treatment of AML has a well-documented and substantive role in acute and lethal human disease.47
We do not yet know the exact mechanisms by which the interaction of CLL-1/IL15 CAR T cells with AML blasts produces this excessive inflammatory response. Neither CLL1-IL15 CAR T cells nor any of the AML cell lines tested produced TNFα alone in culture. Moreover, transgenic IL15 has not produced equivalent effects in vitro or in vivo from T cells expressing CARs with the same CD28 costimulatory domain but targeting different antigens (eg, CD199 or GD210 33). Indeed, CLL1-IL15 CAR T cells only minimally elevated TNFα during equivalent short and long term in vitro culture with AML cell lines. Higher levels of TNFα were secreted from CLL-1 IL15 CAR T cells were co cultured with primary blasts in vitro. However, the lethal effects of TNFα produced in vivo may result from interactions between the species-crossreactive cytokines released by the human CAR-T cells with murine components including vascular endothelium.48
Only clinical studies will reveal whether the interaction between IL15-secreting CD33-directed CAR T cells and CD33+ blast cells will lead to a similar release of TNF in humans, and if it does, whether or not it is unique to this pairing of target antigen and AML. Nonetheless, our data flag TNFα as an additional potential driver of CRS following CAR-T cell administration. We describe two approaches that will interrupt the resultant toxicity. In the first, we incorporated a clinically validated suicide gene, iC9, that induces apoptosis on exposure to a CID, an analog of which has been extensively studied in stem cell transplantation for AML in humans.49–51 Early activation of this suicide gene rapidly induced apoptosis of IL15-producing T cells in vitro6 and in vivo, but also led to loss of leukemic control and resurgent disease.
Since excessive TNFα secretion was the dominant perturbation in cytokine levels in affected mice, we next used a TNFα antibody to avert or delay toxicity while allowing sustained antileukemic activity. Monoclonal antibodies are widely and successfully used to prevent or treat CRS after CAR-T cell treatment. For instance, a blocking antibody to the IL6 receptor (tocilizumab) was approved to treat CRS after administration of CD19-CAR T cells.52 53 In our AML models, the toxicities associated with high levels of TNFα can be controlled by administration of the cognate blocking antibody. TNFα, however, is not the primary driver of the T cell proliferative response, since expansion of T cells continued unabated even after administration of high and frequent doses of blocking antibody. The mice were, therefore, not protected long term from CLL-1 IL15 CART cell toxicities, and subsequently died from a shock-like syndrome.
Ultimately, combining early administration of TNFα antibody, short-term administration of which should have limited general immunosuppressive activity followed by later depletion of proliferating CAR-T cells by activation of the safety switch, should permit both sufficient CART expansion, persistence and potency to avoid disease relapse and sufficient control of unwanted T cell activity to prevent both early and late toxicity.
Since our in vivo studies were conducted in immune-deficient NSG-SGM3 mice, we do not know whether the in vitro safety or the in vivo toxicity is more likely to predict treatment outcomes in humans.35 If clinical studies demonstrate that the benefits we observed treating human AML with CLL-1-CAR-IL15 T cells in mice are similar in patients, then TNFα antibody may need to be added to the repertoire of cytokine-neutralizing agents54–56 required to ensure both safe and effective CAR T cell treatments. Combination of such antibodies with regulation of T cell expansion and survival with a clinically validated safety system such as described here using iC956 may be optimal.
Acknowledgments
We thank Catherine Gillespie for critical review of the manuscript.
References
Footnotes
Contributors PAA, EA, MM and MKB designed the study; PAA, EA, HEH, MM and MKB decided on methodology; PAA, MKM, EA, MS, HT, FM, NW, AMS and MSR provided materials and performed experiments; PAA, MKM, EA, MM and BWS acquired and analyzed results; PAA, EA, MM and MKB wrote and reviewed the manuscript and all authors critically reviewed the manuscript.
Funding This work was supported by Leukemia and Lymphoma Society SCOR (#7001-19) (HEH and MKB). PAA and EA were supported by Scientific and Technological Research Council of Turkey 2219 grant.
Competing interests HEH is a cofounder of Allovir and Marker Therapeutics, has served on advisory boards for Tessa Therapeutics, Gilead Biosciences, Novartis, Kiadis and PACT Pharma and received research support from Kuur Therapuetics and Tessa Therapuetics. MKB is a cofounder of Allovir and Marker Therapeutics, reports personal fees from Tessa Therapeutics and Allogene.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as online supplementary information.