Article Text

Abstract
Background The prognosis of patients with relapsed or progressive B cell (CD20+) non-Hodgkin’s lymphoma (B-NHL), including Burkitt lymphoma (BL), is dismal due to chemoradiotherapy resistance. Novel therapeutic strategies are urgently needed. N-820 is a fusion protein of N-803 (formerly known as ALT-803) to four single-chains of rituximab. This agent has tri-specific binding activity to CD20 and enhanced antibody-dependent cell-mediated cytotoxicity.
Methods We investigated the anti-tumor combinatorial effects of N-820 with ex vivo expanded peripheral blood natural killer (exPBNK) cells against rituximab-sensitive and rituximab-resistant CD20+ BL in vitro using cytoxicity assays and in vivo using human BL xenografted NOD/SCID/IL2rγnull (NSG) mice. We also investigated the cytokines/chemokines/growth factors released using ELISA and multiplex assay. Gene expression changes were examined using real-time PCR arrays.
Results N-820 significantly enhanced the expression of NK activating receptors (p<0.001) and the proliferation of exPBNK cells with enhanced Ki67 expression and Stat5 phosphorylation (p<0.001). N-820 significantly enhanced the secretion of cytokines, chemokines, and growth factors including GM-CSF, RANTES, MIP-1B (p<0.001) from exPBNK cells as compared with the combination of rituximab+N-803. Importantly, N-820 significantly enhanced in vitro cytotoxicity (p<0.001) of exPBNK with enhanced granzyme B and IFN-γ release (p<0.001) against BL. Gene expression profiles in exPBNK stimulated by N-820+Raji-2R showed enhanced transcription of CXCL9, CXCL1, CSF2, CSF3, GZMB, and IFNG. Moreover, N-820 combined with exPBNK significantly inhibited rituximab-resistant BL growth (p<0.05) and extended the survival (p<0.05) of BL xenografted NSG mice.
Conclusions Our results provide the rationale for the development of a clinical trial of N-820 alone or in combination with endogenous or ex vivo expanded NK cells in patients with CD20+ B-NHL failing prior rituximab containing chemoimmunotherapy regimens.
- translational medical research
- cytotoxicity
- immunologic
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Children, adolescents, and adults with de novo mature B cell (CD20+) non-Hodgkin’s lymphoma (B-NHL), including Burkitt lymphoma (BL) have excellent overall survival and event-free survival over the last two decades.1–3 Unfortunately, chemoradiotherapy resistance is developed and results in dismal prognosis in children and adolescents with de novo mature B-NHL who relapse or progress.1 4 5 Similarly, adults with recurrent/refractory BL have poor prognosis.6 CD20, a B cell marker, is highly expressed on the surface of mature B-NHL, which makes it an attractive therapeutic target.7 Rituximab, a monoclonal chimeric anti-CD20 antibody, has been widely used as a chemoimmunotherapeutic regimen in the frontline therapy for adults with CD20+ BL and diffuse large B cell lymphoma. The addition of rituximab to the cyclophosphamide, doxorubicin, vincristine, and prednisone backbone or to standard FAB/LMB therapy has greatly improved outcomes without significantly increasing toxicity in patients with B-NHL.2 3 8 However, patients who relapse have a poor clinical response after rituximab retreatment.9 10 Novel therapies are urgently needed to improve outcome and/or circumvent chemoimmunotherapy or rituximab resistance in these patients. Others and we have previously demonstrated that rituximab-resistant B-NHL cell lines provide an excellent model to evaluate novel targeted regimens.11 12
Natural killer (NK) cells are important component of innate immune system and show promise in patients with a variety of malignancies.13 NK cells express a vast repertoire of activating and inhibitory receptors on the surface, which regulate NK function and discriminate target cells from healthy ‘self’ cells in a major histocompatibility complex independent manner.14–16 CD16 (FcγRIII), one of NK activating receptors, is required for triggering NK-mediated antibody-dependent cellular cytotoxicity (ADCC).17 The ligation between CD16 and the antibody bounded tumor cells can activate and induce NK cells to secrete cytotoxic granules to kill and eliminate tumor cells.18
However, NK cells-based immunotherapy has been limited due to small numbers of active circulating NK cells in peripheral blood, short lifespan, lack of specific tumor targeting, exhaustion, inhibitory receptors induced inhibition, poor trafficking and poor tumor infiltration, and so on.13 19 It is both cumbersome and expensive to isolate enough primary NK cells from patients for adoptive cell therapy since NK cells make up only 10%–15% of the lymphocytes in peripheral blood.20 Our group and others have successfully expanded active NK cells in vitro by short-term culture with cytokines alone and co-culture with engineered feeder cells.21 22 A genetically engineered feeder cells expressing membrane bound IL-21 and 41BBL (K562-mbIL21-41BBL) has been developed by our group to successfully expand peripheral blood mononuclear cells (PBMCs) into NK cells.23 This approach results in over 35,000 fold expansion in NK cells and significant NK cell functional activation and increased telomere length.23 High doses of expanded NK cells using this feeder infused to high-risk acute myeloblastic leukemia after haploidentical transplantation showed promising outcome with a low relapse rate, a low incidence of viral reactivation and no excess graft-vs-host disease.24 We also successfully engineered expanded peripheral blood NK (exPBNK) cells with anti-CD20 chimeric antigen receptor (CAR) using CAR mRNA electroporation to enhance NK cells targeting to CD20+ B-NHL.12 25
N-803 (formerly known as ALT-803), a novel IL-15 superagonist, consists of an interleukin 15 superagonist mutein (IL-15N72D) and a dimeric IL-15 receptor alpha (IL-15Rα)/Fc fusion protein26 (online supplemental figure S1). N-820 is an antibody–cytokine fusion protein by fusing four single-chains of the tumor-targeting monoclonal antibody, rituximab to the N terminal of IL-15N72D and IL-15RαSuFc27 (online supplemental figure S1). N-820 exhibited the enhanced ADCC compared with rituximab against CD20+ B cell lymphoma.27 It showed higher complement dependent cytotoxicity and directly induced the apoptosis of tumor cells.27 A 64Cu labeled biodistribution study demonstrated that N-820 is cleared through both the hepatobiliary and renal pathways and has the ability to home to lymphoid tissues.27 28
Supplemental material
Supplemental material
The efficacy of ex vivo expanded NK cells in combination with N-820 against rituximab-resistant BL has not yet been investigated. Therefore, we performed in vitro and in vivo preclinical studies to investigate if N-820 significantly enhanced the function of exPBNK against rituximab-sensitive and rituximab-resistant BL.
Methods
Cell lines and reagents
Raji, Raji-2R, and Raji-4RH cells were generously provided by Matthew Barth, from Roswell Park Cancer Institute, Buffalo, New York, USA.11 K526-mbIL21-41BBL cells were generously provided by Dean A. Lee, from Nationwide Children’s Hospital, Columbus, Ohio, USA.23 Ramos, Daudi, and RS4;11 cells were purchased from ATCC. DOHH-2 cells were ordered from DSMZ. Raji, Raji-2R, Raji-4RH, and K526-mbIL21-41BBL cells were cultured in RPMI 1640 medium with 10% heat inactivated fetal bovine serum (FBS) at 37°C with 5% CO2. Obinutuzumab was generously provided by Christian Klein, from Roche, Zurich, Switzerland. N-820 and N-803 were generously provided by Hing Wong, Peter R. Rhode, John H. Lee, and Jeffrey T. Safrit, from ImmunityBio/Altor Bioscience, Culver City, California, USA. All fetal bovine serum used for cell culture and assays was heat-inactivated (heating to 56°C for 30 min) before use in order to inactivate complement. Leucocytes were obtained after informed consent from healthy donors at the New York Blood Center, New York, New York, USA. PBMCs were obtained by Ficoll gradient (Amersham Biosciences, Piscataway, New Jersey, USA) separation as we previously described.25
NK cell expansion
PBMCs were stimulated with irradiated genetically modified K562-mbIL21-41BBL cells as previously described.23 Expanded PBNK cells were isolated by negative selection using Miltenyi NK cell isolation kit (Miltenyi Biotec, Cambridge, Massachusetts, USA).25
In vitro cytotoxicity
Tumor targets were labeled with carboxyfluorescein succinimidyl ester (CFSE) (Thermofisher Scientific, USA, cat #C34570) or engineered to express luciferase. Briefly, exPBNK cells were incubated with target cells at different effector-to-target (E:T) ratios with IgG, N-803, N-820, rituximab, or rituximab+N-803 at 37°C for 2–3 days. For CFSE-labeled target cells, dead cells were stained with propidium iodide (Thermofisher Scientific, USA, cat #3566). Cytolytic activity was evaluated by flow cytometry analysis. Target cells were gated on CFSE+ and the live/dead cell percentage was visualized by propidium iodide exclusion. For target cells that express luciferase, living cells were monitored by measuring luciferase activity with a luminometer (Molecular Devices Multifilter F5 plate reader) as relative light units (RLU). Cells were treated with 1% Triton X-100 as a measure of maximal killing. Target cells incubated without effector cells were used to measure spontaneous death RLU. Percent lysis was calculated from the data with the following equation: % specific lysis = 100×(spontaneous death RLU–test RLU)/(spontaneous death RLU–maximal killing RLU). All tests were run in quadruplicate.
Bio-Plex Pro human cytokine screening
Cell culture supernatants were collected after 3 days culture and were stored at −80 °C. The concentrations of cytokines/chemokines/growth factors were measured by the Bio-Plex Pro human cytokines screening panel 48 cytokines Assay (Bio-Rad Laboratories, Hercules, California, USA) according to the manufacturer’s instructions. The beads were read on a Luminex System (Bioplex 200, Bio-Rad) and the data were analyzed using Bioplex Manager Software.
ELISA
Granzyme B (eBioscience, San Diego, California, USA), IFN-γ (eBioscience), GM-CSF (Raybiotech, Peachtree Corners, Georgia, USA), and chemokine ligand 22 (CCL22) (Raybiotech) concentrations were analyzed by ELISA according to the manufacturers’ instructions. The absorbance at 450 nm was measured on a Molecular Devices Multifilter F5 plate reader.
Flow cytometry-based phenotyping of NK activating and inhibitory receptors
The exPBNK cells under different conditions were analyzed for phenotypic expression of activating and inhibitory NK receptors by flow cytometry as we previous described.25
Flow cytometry analysis of intracellular proteins and phosphoproteins
Fixed and permeabilized cells were stained with mouse anti-human primary antibodies Ki67-PE (Thermofisher, cat #12-5698-80), phospho-STAT5-PE (Tyr694) (Thermofisher, cat #12-9010-41), phospho-Akt-APC (Ser473) (Thermofisher, cat #17-9715-42). Cells were analyzed using MACSQuant Analyzer (Miltenyi Biotec). No stain, or isotype controls were used for gating as we previously described.12
N-820 and rituximab binding assays
Raji, Raji-2R, or Raji-4RH were incubated with 10 nM IgG, 10 nM N-803, 10 nM N-820, 10 nM rituximab, or 10 nM N-803+10 nM rituximab for 30 min. The samples were washed three times in phosphate buffered saline (PBS). The binding of N-820 or rituximab to CD20 on the surface of BL cells was measured by staining the cells with phycoerythrin (PE) conjugated anti-CD20 antibody (Miltenyi Biotec, cat #130-113-374).
ExPBNK cells were incubated with 10 nM IgG, 10 nM N-803, 10 nM N-820, 10 nM rituximab, or 10 nM N-803+10 nM rituximabfor 3 days. The binding of N-820 or rituximab on the surface of exPBNK cells was measured by fluorescein isothiocyanate (FITC) conjugated goat anti-mouse IgG F(ab’)2 specific antibody (Jackson ImmunoResearch, cat #115-096-003).
Sorting exPBNK cells
ExPBNK cells were co-cultured with Raji-2R at E:T=1:1 plus 10 nM N-820 or Raji-2R at E:T=1:1 plus 10 nM rituximab+10 nM N-803 for 3 days. Harvested mixed cells were stained for CD56-PE-Vio770 (Miltenyi, cat# 130-113-313). CD56+ cells were gated and isolated using a MoFlo XDP high-speed cell sorter (Beckman Coulter Inc., Miami, Florida, USA). A minimum of 1–10×105 sorted cells with a purity >90% were collected into cold tissue culture media.
Real-time PCR-based array analysis
The Human Cancer Inflammation & Immunity Crosstalk RT2 Profiler PCR Arrays (PAHS-181ZA, Qiagen, Frederick, Maryland, USA) were used to profile the expression of 84 genes involved in mediating communication between tumor cells and exPBNK cells mediated by N-820 compared with rituximab+N-803. Total RNA was isolated from exPBNK cells using Qiagen RNeasy Mini Kit by following manufacturer’s protocol. The first-strand cDNA was mixed with 2×RT2 SYBR Green qPCR Master Mix and ddH2O. The qPCR was performed on an Applied Biosystems (ABI) QuantStudio 5 PCR system according to the RT2 Profiler PCR Array instructions under the following conditions: 95°C for 10 min, then 40 cycles at 95°C for 15 s and 60°C for 1 min. Data were normalized against the house keeping genes by calculating the ΔCt for each gene of interest in the plate. Fold changes of gene expression, scatterplot and heatmap were analyzed and generated using Qiagen Gene Globe data analysis web port.
Construction of protein–protein interaction (PPI) network
STRING database in Cytoscape software V.3.7.2 was used to assess PPI information and converted the results visually by using Cytoscape.29 A confidence score >0.7 was set as significant. CytoHubba, a plug-in Cytoscape software, was used to identify the top 10 hub genes in PPI networks through the degree method.30
Xenograft models of rituximab-resistant human BL
Six to eight week old NOD/SCID/γ-chain-/- (NSG) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA) and were bred, treated, and maintained in the animal facility of New York Medical College (NYMC) with NYMC Institutional Animal Care and USE Committee (IACUC)-approved protocols. The animal experiments were conducted in accordance with the recommendations of the Guide for Care and Use of Laboratory Animals.
Luciferase-expressing tumor cells (Raji-2R-Luc and Raji-4RH-Luc) were generated as we previously described.25 Tumor cells were retro-orbitally or intraperitoneally administered into the NSG mice. Tumor engraftment and progression were evaluated using the Xenogen IVIS-200 system (Caliper Life Sciences, Hopkinton, Massachusetts, USA) as we have previously described.25 Mice were followed until death.
Statistical analyses
Statistical analyses were performed using the INSTAT statistical program (GraphPad, San Diego, California, USA). Average values are reported as the mean±SEM. Results were compared using the one-tailed unpaired Student’s t-test with p<0.05 considered as significant. Probability of survival in animal studies was determined by the Kaplan-Meier method using the Prism program 8.0 (GraphPad Software).
Results
N-820 enhanced the expression of NK activating receptors and proliferation of exPBNK with enhanced Ki67, p-Stat5 and CD25 expression
To investigate if N-820 affects the expression of receptors on exPBNK cells and stimulates the proliferation of exPBNK cells, we cultured purified exPBNK cells in medium with 10 nM N-820 for 3 days. 10 nM IgG and 10 nM N-803 were used as controls. The activating and inhibitory receptors on exPBNK cells were examined by flow cytometry. N-820 significantly enhanced the expression of NK activating receptors such as NKp44, CD16, NKp30, and NKG2D (figure 1A) as compared with IgG. N-820 showed similar activity to N-803 in stimulating NK receptor expression (figure 1A) except for NKp46 and KIR2D2/3. The proliferation of exPBNK cells was examined using CellTiter 96 AQueous One solution cell proliferation assay after 3 days culture with PBS, 10 nM IgG, 10 nM N-820, or 10 nM rituximab+10 nM N-803. N-820 significantly stimulated exPBNK proliferation as compared with IgG (p<0.001), N-803 (p<0.001), and rituximab+N-803 (p<0.002) (figure 1B). Consistent with the enhanced exPBNK proliferation, N-820 significantly stimulated Ki67 expression in exPBNK cells as compared with IgG (p<0.001), N-803 (p<0.001), and rituximab+N-803 (p<0.01) (figure 1C). IL-15 is known to rapidly induce STAT5 phosphorylation, which plays important roles in regulating NK-cell survival, proliferation, cytotoxicity, and maturation.31 As expected, N-820 significantly enhanced STAT5 phosphorylation in exPBNK cells as compared with IgG (p<0.001), N-803 (p<0.02), and rituximab+N-803 (p<0.02) (figure 1D). In addition to the IL-15–STAT5 pathway, accumulating data demonstrate that the PI3K–AKT–mTOR pathway is essential for the development, differentiation, and activation of NK cells.32 We examined AKT phosphorylation in exPBNK cells stimulated by IgG, N-820, N-803, or rituximab+N-803. There was no significantly difference of AKT phosphorylation in exPBNK cells among N-820, N-803, or rituximab+N-803 but N-820 significantly stimulated AKT phosphorylation in exPBNK cells as compared with IgG (p<0.001) (figure 1E,F).

N-820 enhanced the expression of NK activating receptors and proliferation of exPBNK with enhanced Ki67, p-Stat5, and CD25 levels. The percentages of NK activating and inhibitory receptors on exPBNK surface were monitored by flow cytometry analysis at day 3 (A). The NK proliferation was monitored by CellTiter 96 AQueous One solution cell proliferation assay (Promega) according to the manufacturer’s instruction (B). Intracellular Ki67 (C), Phosphorylated STAT5 (p-STAT5) (D) and phosphorylated Akt (p-AKT) (E) were monitored by flow cytometry analysis at day 3. Ki67 (p<0.01) and p-STAT5 (p<0.02) were significantly increased following N-820 versus rituximab (Ritux)+N-803 . Average values are reported as the mean±SEM. Representative histograms illustrate the expression levels of Ki67, p-STAT5, and p-AKT (F). Purified exPBNK cells were co-cultured with Raji, Raji-2R, Raji-4RH cells or without tumor cells in medium with 10 nM N-820, 10 nM N-803, or 10 nM IgG for 3 days. The percentage of CD25 on exPBNK surface was monitored by flow cytometry analysis at day 3. CD25 expression was significantly enhanced on exPBNK cells by N-820 versus rituximab+N-803 (Ritux+N-803) against Raji-2R and Raji-4RH, p=0.0012 and p=0.0016, respectively (G). Average values are reported as the mean ± SEM on the left panel. The representative histograms illustrate the expression levels of CD25 on exPBNK cells that were co-cultured with Raji, Raji-2R, Raji-4RH cells or without tumor cells in medium with 10 nM N-820 or 10 nM IgG for 3 days on the right panel. n=4 (G). *p<0.05, **p<0.01, ***p<0.001, ritux=rituximab.
Previous studies have shown that IL-15 stimulation results in transient (hours to days) CD25 expression, a key component required to form the high-affinity heterotrimeric IL-2Rαβγ on the majority of NK cells correlated with STAT5 phosphorylation.33 Therefore, we examined CD25 expression stimulated by N-820. We found that N-820 with or without BL tumor cells significantly stimulated CD25 expression on exPBNK cells (p<0.001) as compared with IgG controls. N-820 significantly stimulated CD25 expression on exPBNK cells as compared with rituximab+N-803 without co-culturing with BL tumor cells (p<0.001) or with Raji-2R (p<0.0012) or Raji-4RH (p<0.0016) cells (figure 1G).
Cytokines and growth factors screen of exPBNK cells stimulated by N-820 versus by rituximab+N-803
To investigate the cytokines and growth factors that are significantly secreted by exPBNK cells stimulated by N-820 and compare with the cells stimulated by rituximab+N-803, exPBNK cells were cultured with 10 nM IgG, 10 nM N-820, or 10 nM rituximab+10 nM N-803 for 3 days. The concentrations of cytokines/chemokines/growth factors in the supernatants were measured by the Bio-Plex Pro human cytokines screening panel 48 cytokines Assay. We found that the levels of eotaxin, FGF, GM-CSF, HGF, INF-A2, INF-G, IL-1RA, IL-2RA, IL-8, IL-12P40, IL-13, IL-17, CXCL9, SCF, TNF-A, TNF-B, LIF, PDGF-BB, CXCL1, RANTES, MIP-1B were significantly increased from exPBNK cells by N-820 as compared with rituximab+N-803 (figure 2A,B), while CXCL12 and SCGF-B were significantly reduced by N-820 as compared with rituximab+N-803 (figure 2A).
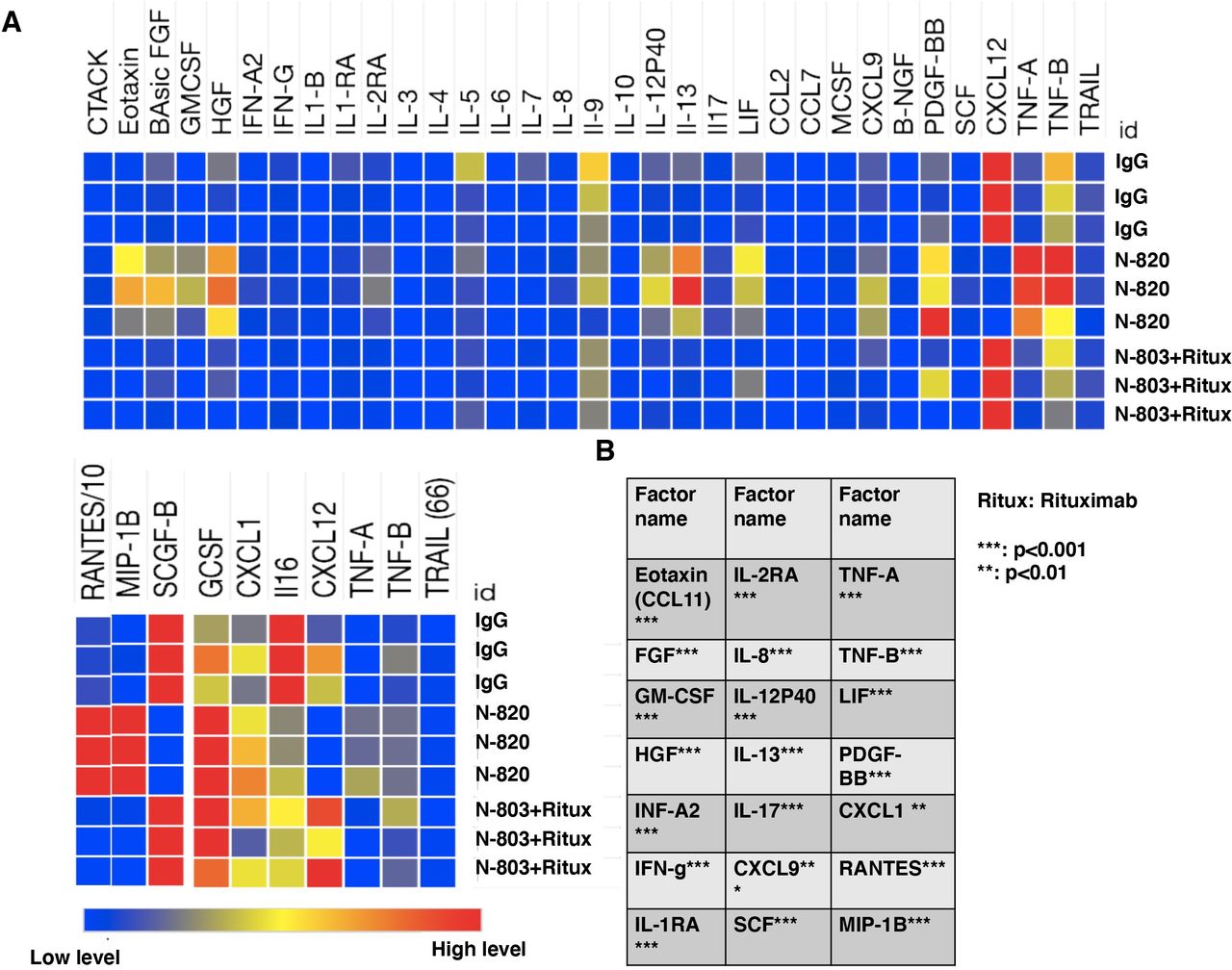
N-820 stimulates a variety of cytokines and growth factors secretion from exPBNK cells. Purified exPBNK cells were cultured in medium with 10 nM N-820, 10 nM N-803+10 nM rituximab, or 10 nM IgG for 3 days. The supernatants were collected after 3 days culture and used for Bio-Plex Pro human cytokines screening panel 48 cytokines assay. Heatmaps of cytokines/chemokines/growth factors abundance pattern in the supernants of exPBNK cells cultured in medium with IgG, N-820, N-803+rituximab(A). A list of secreted cytokines/chemokines/growth factors that were significantly enhanced in exPBNK cells by N-820 than N-803+rituximab (n=3) is shown (B). **p<0.01, ***p<0.001, Ritux=rituximab.
N-820 significantly enhanced in vitro cytotoxicity of exPBNK with enhanced granzyme B and IFN-γ release against rituximab-sensitive and rituximab-resistant BL
Since exPBNK cells express high level of CD16 (figure 1A) and N-820 stimulated the ADCC of primary NK cells,27 we investigated whether N-820 significantly stimulates the ADCC activity of exPBNK cells against rituximab-sensitive Raji and rituximab-resistant Raji-2R and Raji-4RH cell lines. Raji, Raji-2R, and Raji-4RH cells were treated with 10 nM IgG, 10 nM N-820, 10 nM N-820+exPBNK cells, or 10 nM IgG+exPBNK cells at E:T ratio=0.1:1, 1:1, and 3:1. We found that the combination of N-820 and exPBNK significantly killed Raji, Raji-2R, and Raji-4RH cells (p<0.001) as compared with N-820 alone or IgG alone (figure 3A) in an E:T ratio dependent manner. And N-820 significantly enhanced the ADCC activity of exPBNK cells (p<0.001) as compared with exPBNK+IgG against Raji, Raji-2R, and Raji-4RH (figure 3B). We further compared the activity of N-820 with the same doses of N-803, rituximab, or rituximab+N-803 on the cytotoxicity of exPBNK cells. We found that N-820 significantly enhanced the in vitro cytotoxicity of exPBNK as compared with N-803 (p<0.001), rituximab (p<0.001), and rituximab+N-803 (p<0.05 at low E:T ratios and p<0.001 at higher E:T ratios) in an E:T ratio dependent manner (figure 3B). Obinutuzumab is a humanized, type II anti-CD20 monoclonal antibody glycoengineered to enhance Fc receptor affinity.34 Considering the enhanced ADCC of obinutuzumab as compared with rituximab in the setting of rituximab-sensitive and rituximab-resistant BL,34–36 we compared the effect of N-820 on the ADCC activity of exPBNK with the same dose of obinutuzumab+N-803 against Raji, Raji-2R, and Raji-4RH cells. ExPBNK cells+10 nM N-820 showed similar killing activity as exPBNK cells+10 nM obinutuzumab+10 nM N-803 against Raji cells (p=0.26) (figure 3C). These studies demonstrated N-820 had enhanced cytolytic activity of exPBNK cells against Raji-2R cells (p=0.0611) and significantly enhanced cytolytic activity against Raji-4RH cells as compared with obinutuzumab+N-803 (p<0.01) (figure 3C). We also examined if N-820 enhances the anti-tumor effects of exPBNK against other CD20+B-NHL cells. We found that the combination of exPBNK cells with N-820 significantly killed CD20+ DOHH-2 (follicular lymphoma), Ramos (BL), or Daudi (BL) and significantly enhanced granzyme B release as compared with other control groups (online supplemental figure 2A). A CD20− acute leukemia cell line RS4;11 cells were used as a negative control. The combination of exPBNK cells with N-820 did not significantly killed RS4;11 and did not significantly enhanced granzyme B release as compared with the control groups (online supplemental figure 2B).
Supplemental material
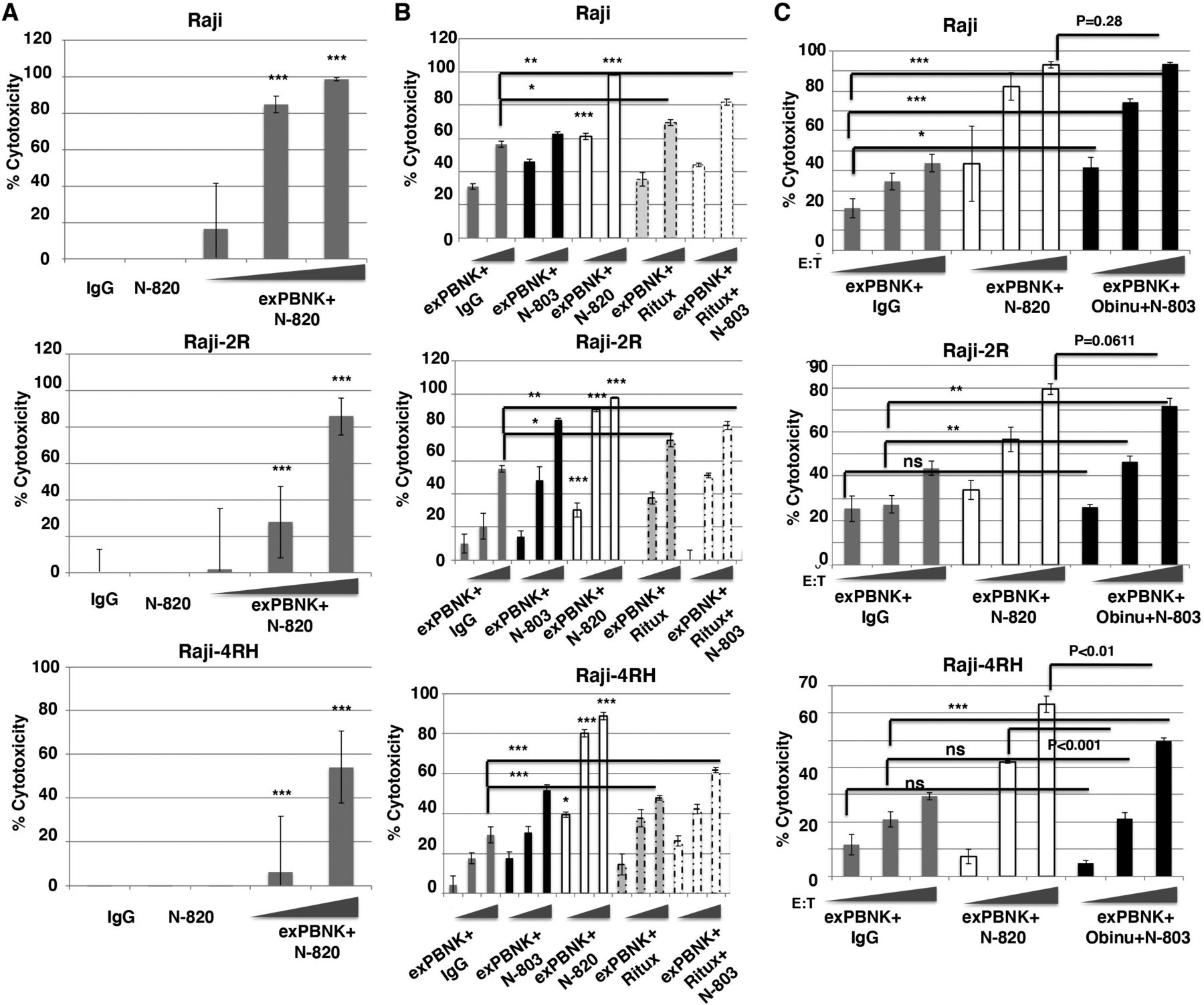
N-820 significantly enhanced in vitro cytotoxicity of exPBNK against rituximab-sensitive and rituximab-resistant BL cells. Expanded NK cells were isolated for in vitro cytotoxicity assays against rituximab-sensitive and rituximab-resistant BL cells. The combination of exPBNK cells with N-820 significantly killed rituximab-sensitive Raji and rituximab-resistant Raji-2R and Raji-4RH as compared with IgG or N-820 without exPBNK cells at E:T ratios=3:1 or 10:1 at day 3 (A). The combination of exPBNK cells with N-820 significantly killed Raji (E:T ratios=3:1 or 10:1), Raji-2R (E:T ratios=1:1, 3:1, or 10:1), and Raji-4RH (E:T ratios=1:1, 3:1, or 10:1) as compared with exPBNK+IgG, exPBNK+N-803, exPBNK+rituximab, or exPBNK+N-803+rituximab (B). ExPBNK cells+N-820 showed similar killing activity as exPBNK cells+obinutuzumab+N-803 against Raji cells (p=0.26) (C). ExPBNK cells+N-820 showed enhanced cytolytic activity against Raji-2R cells (p=0.0611) and significantly enhanced cytolytic activity against Raji-4RH cells (p<0.01) (n=4) (C). *p<0.05, **p<0.01, ***p<0.001, ns=not significant, Ritux=rituximab.
Previously, we reported the expression of CD20 was significantly reduced in Raji-2R and Raji-4RH as compared with Raji.25 The enhanced cytolytic activity of exPBNK cells+N-820 as compared with exPBNK cells+obinutuzumab+ N-803 against rituximab-resistant BL cells may be due to the higher affinity of N-820 to the CD20 target on the rituximab-resistant BL surface. We further confirmed that N-820 significantly enhanced exPBNK in vitro cytotoxicity as compared with controls in part by significantly enhancing the secretion of both granzyme B and IFN-γ. We found that 10 nM N-820 significantly enhanced granzyme B (figure 4A) and IFN-γ (figure 4B) release from exPBNK cells as compared with 10 nM IgG, 10 nM N-803, 10 nM rituximab, 10 nM rituximab+10 nM N-803, and 10 nM obinutuzumab against Raji, Raji-2R, and Raji-4RH (p<0.001) at E:T=1:1.
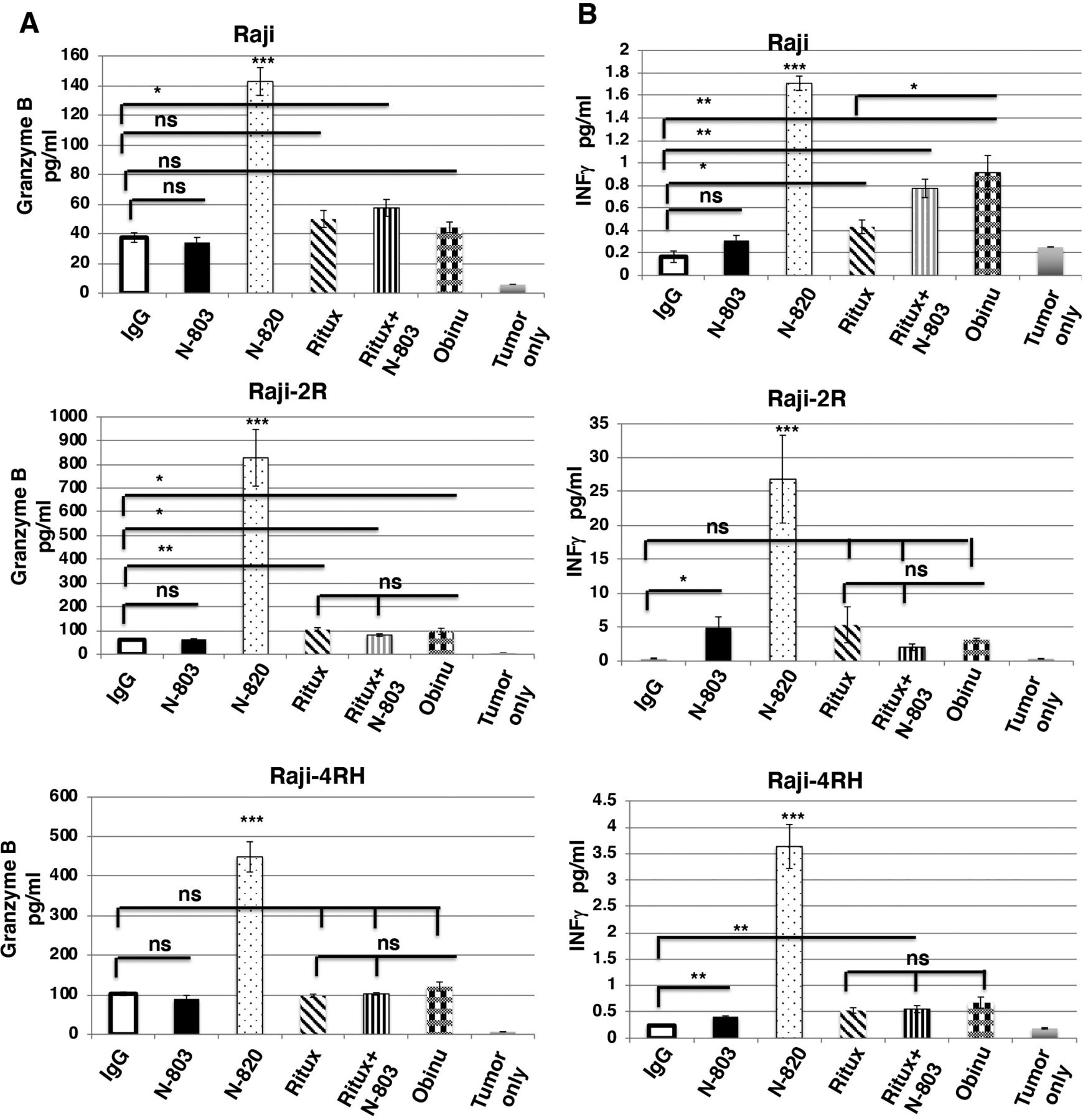
N-820 significantly enhanced granzyme B and IFN-γ release from exPBNK cells against rituximab-sensitive and rituximab-resistant BL cells. The purified exPBNK cells were co-cultured with Raji, Raji-2R, or Raji-4RH in combination with 10 nM IgG, 10 nM N-803, 10 nM N-820, 10 nM rituximab, 10 nM N-803+10 nM rituximab, or 10 nM obinutuzumab for 3 days. The supernatants from the culture were used for ELISA. Supernatants from Raji, Raji-2R, or Raji-4RH without exPBNK cells were used as controls. N-820 significantly enhanced granzyme B release from exPBNK cells as compared with other agents (A). N-820 significantly enhanced IFN-γ release from exPBNK cells as compared with other agents (n=4) (B). *p<0.05, **p<0.01, ***p<0.001, ns=not significant, Ritux=Rituximab, Obinu=Obinutuzumab.
N-820 has significantly higher binding ability to Raji, Raji-2R, and Raji-4RH and exPBNK cells
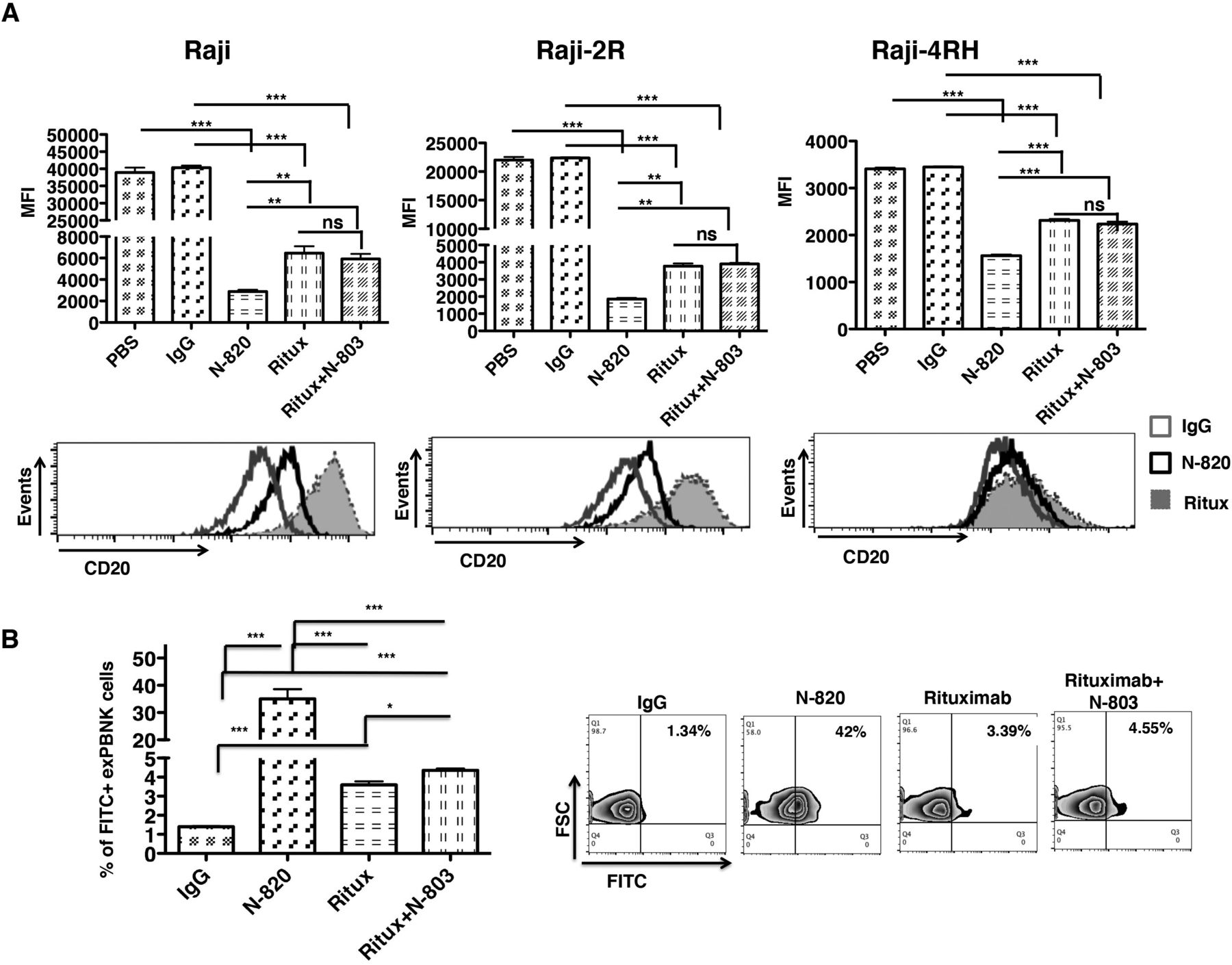
We investigated the binding ability of N-820 to CD20 on Raji, Raji-2R, or Raji-4RH cells. Raji, Raji-2R, or Raji-4RH cells were incubated with 10 nM IgG, 10 nM N-803, 10 nM N-820, 10 nM rituximab, or 10 nM N-803+10 nM rituximab for 30 min. After PBS wash, the cells were stained with PE conjugated anti-CD20 antibody (clone LT20, which recognizes the similar epitope of CD20 as rituximab).37 N-820 significantly blocked the accessibility of CD20 to another anti-CD20 antibody LT20 on the surface of Raji as compared with the IgG (p<0.001) and rituximab (p<0.01) (figure 5A, left panels). N-820 significantly blocked the accessibility of CD20 to LT20 on the surface of Raji-2R (figure 5A, middle panels) and Raji-4RH (figure 5A, right panels) as compared with the IgG (p<0.001) and rituximab (p<0.001), indicating N-820 may have a significant higher binding ability to CD20 in rituximab-sensitive and rituximab-resistant BL cells.

The binding activity of N-820 to BL cells and to exPBNK cells. Raji, Raji-2R, or Raji-4RH were incubated with 10 nM IgG, 10 nM N-803, 10 nM N-820, 10 nM rituximab, or 10 nM N-803+10 nM rituximab for 30 min. The samples were washed three times in PBS. The cells were stained with phycoerythrin (PE) conjugated anti-CD20 antibody (clone LT20). The top panels show the median fluorescence intensity (MFI) of PE conjugated anti-CD20 antibody on Raji, Raji-2R, and Raji-4RH after incubating with N-820 and the indicated controls by flow cytometry analysis. The lower panels show the representative histograms of PE conjugated anti-CD20 antibody on Raji, Raji-2R, and Raji-4RH after incubating with N-820 and the indicated controls by flow cytometry analysis (A). The purified exPBNK cells were incubated with 10 nM IgG, 10 nM N-803, 10 nM N-820, 10 nM rituximab, 10 nM N-803+10 nM rituximab for 3 days. After PBS wash, the cells were stained with fluorescein isothiocyanate (FITC) conjugated goat anti-mouse anti-F(ab’) antibody (B). The left chart shows the percentage of FITC+ exPBNK cells. The right panel shows the representative contour plots of each group. Ritux=rituximab, **p<0.01, ***p<0.001.
N-820, rituximab, and N-803 all contain IgG Fc region (online supplemental figure 1), which can bind to CD16. We further examined the binding ability of N-820 to exPBNK cells. ExPBNK cells were incubated with 10 nM IgG, 10 nM N-803, 10 nM N-820, 10 nM rituximab, 10 nM N-803+10 nM rituximab, or 10 nM obinutuzumab. After incubation for 3 days, N-820 can still be detected and significantly binds to exPBNK cells compared with controls (p<0.001) (figure 5B).
Gene expression profiles of signal pathways in exPBNK cells stimulated by N-820+Raji-2R compared to rituximab+N-803+Raji-2R
Expanded NK cells from three different donors were co-cultured with Raji-2R cells at E:T ratio=1:1 with IgG, N-820, or rituximab+N-803. After 3 days, NK cells were sorted with 93%–95% purity (figure 6A). The important genes involved in the effect of N-820+BL on exPBNK cells were identified using the Human Cancer Inflammation & Immunity Crosstalk RT2 Profiler PCR Array. The present study found that 60 genes had at least a twofold increase in exPBNK cells stimulated by N-820+Raji-2R as compared with the gene expression levels in exPBNK cells stimulated by rituximab+N-803+Raji-2R and three genes (CCL22, CXCR4, and CXCR5) were downregulated (figure 6B). Consistent with the results of cytokines and growth factors screened in figures 2 and 4, the transcription levels of IL-17, CXCL9, CXCL1, CSF2, CSF3, GZMB, and IFNG were enhanced in exPBNK cells stimulated by N-820+Raji-2R as compared with those stimulated by rituximab+N-803+Raji-2R. A heat map provides a graphical representation of fold expression in exPBNK cells stimulated by N-820+Raji-2R against exPBNK cells stimulated by rituximab+N-803+Raji-2R, red and green color represent increasing or decreasing genes, respectively (figure 6C). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis shows that the differentially expressed genes (DEGs) are related to apoptosis, NK-mediated cytoxicity, HIF-1, NK-kappa B, Jak-STAT, TNF, PI3K-Akt, Toll-like receptor, chemokine signal pathways, and cytokine–cytokine receptor interaction (figure 6D). PPI network was constructed with fourfolds upregulated DEGs using the STRING database imported in Cytoscape software (V.3.7.2) and revealed 25 nodes (figure 6E). Using the plug-in CytoHubba app in Cytoscape software, we evaluated the degree and betweenness in the PPI network and screened the hub genes. The top 10 hub genes showing significant interaction were cytokine and chemokine genes: IL-6, IL-10, IL-4, IL-17a, CXCL10, CXCL11, CXCL9, CXCL1, CXCL2, and CXCL5 (figure 6E), which are consistent with the KEGG pathway analysis results (figure 6D) that chemokine signal pathways and cytokine–cytokine receptor interaction are the two top enhanced events.
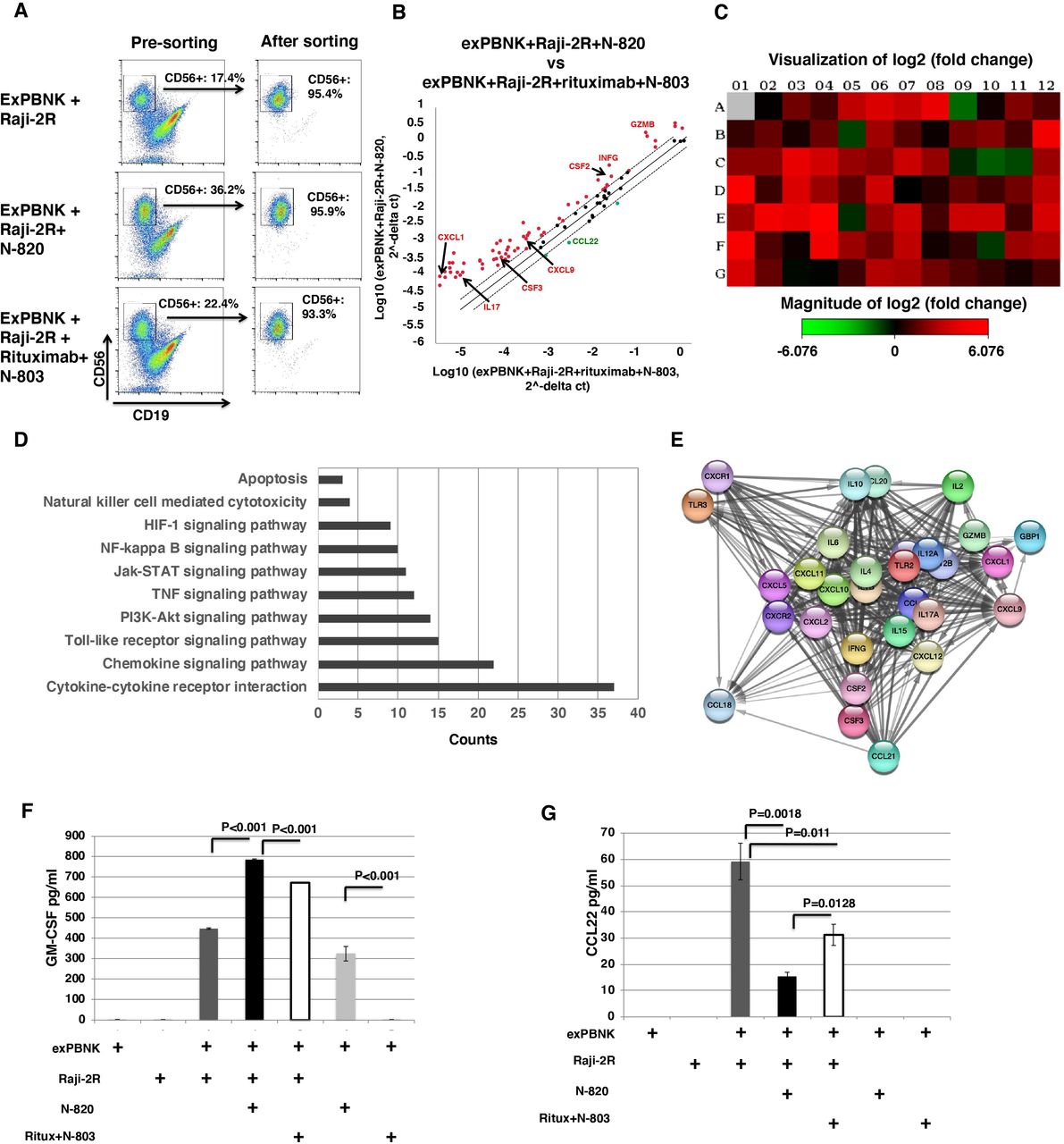
Effects of N-820+Raji-2R on gene expression profiles of signal transduction in exPBNK cells compared with N-803+Rituximab+Raji-2R. The expression changes of the 84 genes in exPBNK cells co-cultured with N-820+Raji-2R or N-803+Rituximab+Raji-2R for 3 days were determined by real-time PCR-based array analysis (n=3). The exPBNK only, exPBNK+Raji-2R and exPBNK+N-803+Rituximab+Raji-2R groups were used as controls. The representative dot plots of flow cytometry show the purity of exPBNK cells before sorting and after sorting after 3 days co-culture with Raji-2R, N-820+Raji-2R, or N-803+rituximab+Raji-2R (A). In the scatter plot, the central line indicates unchanged gene expression. The gene expression changes are showed in red dot for upregulated genes, or green dot for downregulated genes in exPBNK cells stimulated by N-820+Raji-2R as compared with exPBNK cells stimulated by rituximab+N-803+Raji-2R (B). In the heat map, upregulated (red) and downregulated (green) genes are shown in exPBNK cells co-cultured with N-820+Raji-2R as compared with N-803+rituximab+Raji-2R control group (C). The KEGG pathway enrichment analysis of DEGs is shown (D). The horizontal axis represents the count of enriched DEGs. The vertical axis represents the different KEGG pathways. KEGG, Kyoto encyclopedia of genes and genomes; DEG, differentially expressed gene (D). The Protein–protein interaction (PPI) network was constructed with fourfold upregulated DEGs using the STRING database imported in Cytoscape software (V.3.7.2) (E). ELISA quantitation shows that GM-CSF release was significantly enhanced when exPBNK cells were co-cultured with N-820+Raji-2R as compared with other groups including the group with rituximab+N-803+Raji-2R (n=4) (p<0.001) (F). ELISA quantitation shows that CCL22 release was significantly enhanced when exPBNK cells were co-cultured with Raji-2R and the addition of 10 nM N-820 to the co-culture significantly reduced the released CCL22 level as compared with the group with rituximab+N-803 (n=4) (p=0.0128) (G). Ritux=rituximab.
CSF2 encodes an immune regulator GM-CSF.38 Using ELISA, we further confirmed that GM-CSF was significantly released from exPBNK cells stimulated by N-820 as compared with rituximab+ALT803 (p<0.001) (figure 6F), which is consistent with the results from the cytokine screening (figure 2). GM-CSF was also significantly released from exPBNK cells stimulated by Raji-2R cells as compared with exPBNK cells alone. More importantly, GM-CSF was significantly released from exPBNK cells stimulated by N-820+Raji-2R as compared with rituximab+N-803+Raji-2R (p<0.001) (figure 6F). We further confirmed these findings using Raji-4RH cells as tumor targets (data not shown).
CCL22 induces regulatory T cell migration to malignant cells through the CCR4 receptor, leading to inhibition of antitumor immunity and tumor progression.39 We found that the CCL22 secretion level was significantly enhanced when co-culturing exPBNK cells with Raji-2R cells as compared with exPBNK or Raji-2R alone (figure 6G) in part suggesting that the crosstalk between exPBNK cells and Raji-2R cells potentially inhibits antitumor immunity in the tumor microenvironment. The addition of N-820 (p=0.0018) or rituximab+N-803 (p=0.011) to the exPBNK+Raji-2R co-culture significantly reduced the secreted CCL22 levels (figure 6G). Consistent with reduced CCL2 transcription, N-820 significantly reduced CCL22 protein expression compared with rituximab+N-803 when added to the exPBNK+Raji-2R co-culture (p=0.0128) (figure 6G).
N-820 combined with exPBNK cells significantly inhibited rituximab-resistant BL cells growth and extended the survival of rituximab-resistant BL xenografted NSG mice
We further confirmed the anti-tumor effects of N-820 combined with exPBNK cells in human rituximab-resistant BL xenografted NSG mice. 1×106of Raji-2R-Luc cells were injected in NSG mice on day 0. After confirming tumor engraftment at day 7, 1×107 exPBNK cells+10 mg/kg IgG (n=12), 1×107 exPBNK cells+5 mg/kg N-820 (n=8), 1×107 exPBNK cells+10 mg/kg N-820 (n=12), 10 mg/kg N-820 (n=8), or PBS (n=12) was intraperitoneally injected to each mouse. NK cells were administered once a week for 3 weeks. IgG and N-820 were injected two times a week for 3 weeks. We found that the combination of exPBNK+10 mg/kg N-820 significantly reduced the tumor burden measured by bioluminescence signal intensity as compared with the groups treated with PBS (p=0.0141), exPBNK (p=0.0005), 10 mg/kg N-820 (p=0.0212) in Raji-2R xenografted mice (figure 7A–C). More importantly, the combination of exPBNK+10 mg/kg N-820 significantly extended the survival of mice as compared with the group treated with PBS (p=0.0029), exPBNK (p=0.0406), exPBNK +5 mg/kg N-820 (p=0.0421), 10 mg/kg N-820 (p=0.0088) (figure 7D,E). We further used NSG mice xenografted with another rituximab-resistant cell line, Raji-4RH, to confirm these results. We found that 1×107 exPBNK cells+10 mg/kg N-820 (n=8) decreased tumor growth as compared with the tumor burden in the control groups (figure 7F) and significantly enhanced the survival of Raji-4RH xenografted NSG mice as compared with the group treated with exPBNK cells+10 mg/kg rituximab+0.2 mg/kg N-803 (n=7, p=0.0159), the group treated with exPBNK cells+10 mg/kg IgG (n=5, p<0.001), and the group treated with PBS (n=5, p<0.001) (figure 7G).
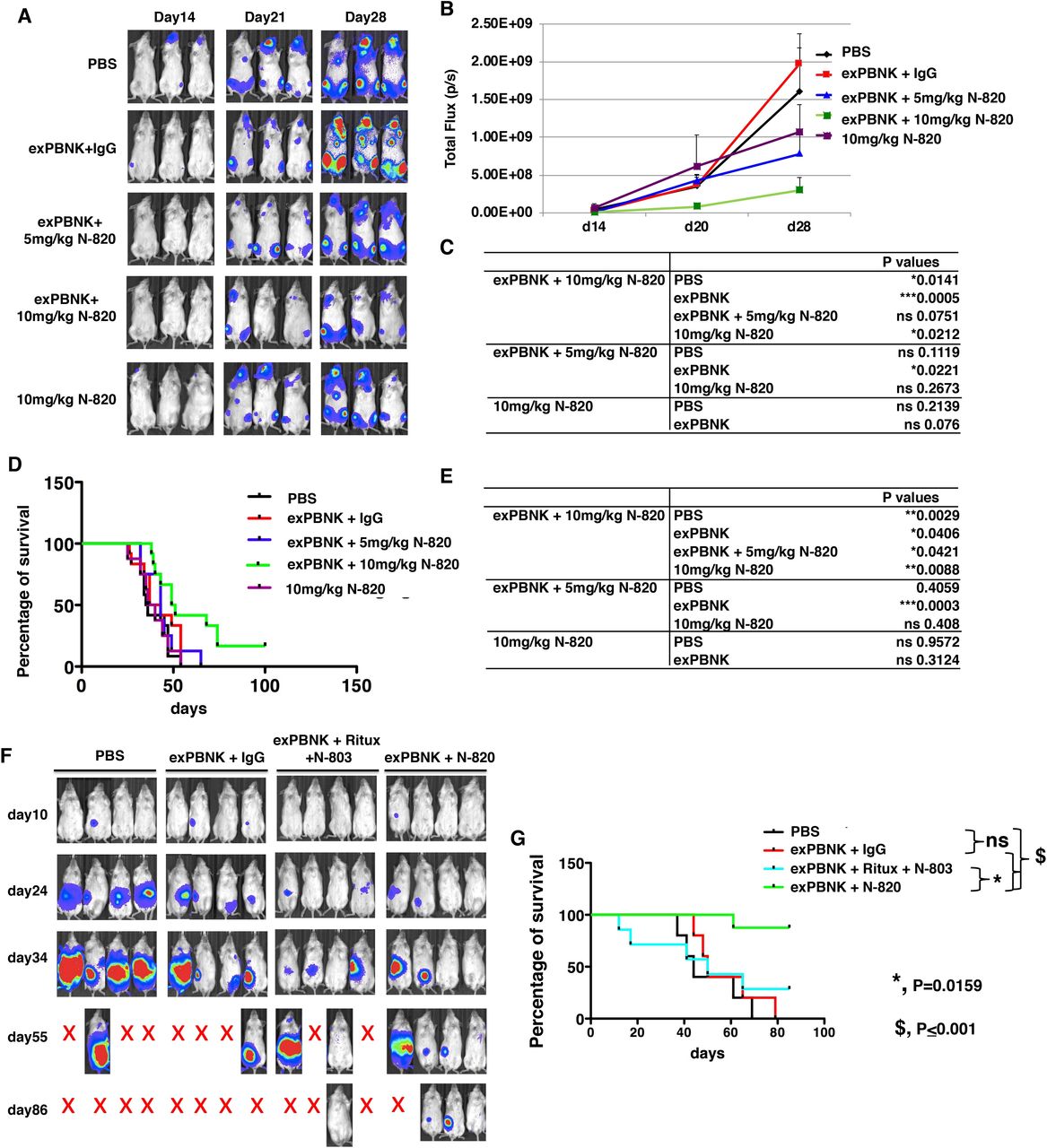
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
N-820 combined with exPBNK cells significantly inhibited rituximab-resistant BL cells growth and extended the survival of rituximab-resistant BL xenografted NSG mice. Raji-2R-Luc xenografted NSG mice were treated with PBS (n=12), exPBNK cells+10 mg/kg IgG (n=12), exPBNK cells+5 mg/kg N-820 (n=8), exPBNK cells+10 mg/kg N-820 (n=12), or 10 mg/kg N-820 (n=8). Photons emitted from luciferase-expression cells were measured in regions of interest that encompassed the entire body and quantified using the living image software. Three representative bioluminescence images of mice of each group are shown (A). Signal intensities (total flux) at the time points from two independent experiments were plotted as mean±SEM (B) and the p values of the comparisons are shown (C). ExPBNK cells+10 mg/kg N-820 treated mice had significant lower bioluminescent signal intensities as compared with the groups treated with PBS, exPBNK cells+IgG, or 10 mg/kg N-820. Raji-2R-Luc xenografted mice were followed until death. The Kaplan–Meier survival curves for all groups pooled from two independent experiments were generated following therapy initiation using animal sacrifice as the terminal event (D). The p values of comparison of survival between groups are shown on the right table (E). The combination of exPBNK+10 mg/kg N-820 significantly extended the survival of Raji-2R-Luc mice compared with other groups. *p<0.05, **p<0.01, ***p<0.001. Raji-4RH-Luc xenografted NSG mice were treated with PBS (n=5), exPBNK cells+10 mg/kg IgG (n=5), exPBNK cells+10 mg/kg rituximab+0.2 mg/kg N-803 (n=7), or exPBNK cells+10 mg/kg N-820 (n=8). Whole mouse luciferase activity was measured at various time points. Photons emitted from luciferase-expression cells were measured in regions of interest that encompassed the entire body and quantified using the Living Image software. Four representative bioluminescence images of mice of each group are shown in the top panel (F). Raji-4RH-Luc xenografted mice were followed until death. The Kaplan–Meier survival curves for all groups were generated following therapy initiation using animal sacrifice as the terminal event. Comparison of survival between groups is shown (G). The combination of exPBNK+N-820 significantly extended the survival of Raji-4RH-Luc mice compared with the group treated with the combination of exPBNK+rituximab+ N-803, exPBNK+IgG, or PBS. Ritux=rituximab, *p<0.05, $p<0.001.
Discussion
In this study, we carried out the first investigations of the combinatorial anti-tumor effects of N-820, a novel targeted cytokine–antibody fusion protein, with ex vivo expanded NK cells against rituximab-sensitive and rituximab-resistant CD20+ BL in vitro and in human BL xenografts. We demonstrated that N-820 significantly enhanced the expression of NK activating receptors and proliferation of exPBNK and it also significantly enhanced the secretion of cytokines, chemokines, and growth factors including FGF, GM-CSF, IL-13, IL-17, CXCL9, SCF, TNF-A, RANTES, MIP-1B from exPBNK cells as compared with rituximab+N-803. More importantly, N-820 significantly enhanced the in vitro cytotoxicity of exPBNK with enhanced granzyme B and IFN-γ release against rituximab-sensitive and rituximab-resistant BL. To study how the expanded NK cells response differently to the stimulation from BL+N-820 from the stimulation from BL+rituximab+N-803, we investigated the transcriptional profiles of expanded NK cells to identify signature difference between N-820+Raji-2R and rituximab+N-803+Raji-2R. Moreover, N-820 combined with exPBNK cells significantly inhibited rituximab-resistant BL cells growth and extended the survival of rituximab-resistant BL xenografted NSG mice.
IL-15 is a 15 kDa cytokine that possesses structural and functional similarities to IL-2, and is active in both cis and trans conformations.40 However, a rapid renal clearance and a short plasma half-life may diminish the effect of recombinant IL-15. N-803, the IL-15 superagonist, has been reported to have prolonged in vivo pharmacokinetics and increased in vivo biological activity compare to IL-15.26 The combination of N-803 with rituximab resulted in significantly reduced tumor cell burden and increased survival of mice xenografted with CD20+ B cell lymphomas.41 Based on the preclinical results, the first in human phase I combination of N-803 (ALT-803) with rituximab for patients with relapsed or refractory indolent non-Hodgkin’s lymphoma was recently conducted (NCT02384954).42 In this trial, the investigators observed 1 complete response, 2 stable disease (45% and 36% tumor volume decrease), and 2 progressive disease of the 5 rituximab-refractory patients.42 New strategies or novel agents are needed for these patients who did not respond to retreatment with rituximab. Our data demonstrate the in vitro and in vivo effectiveness of the combination of N-820 with expanded NK cells in inhibiting the growth of the rituximab-sensitive and rituximab-resistant BL cells (figures 3 and 7), suggesting that the combination of N-820 with ex vivo expanded NK cells can overcome the rituximab-resistance.
We and others previously reported that the rituximab-resistant cells have reduced CD20 expression.11 25 Our data indicate that N-820 may have significant higher binding ability to both rituximab-sensitive and rituximab-resistant BL cells and expanded NK cells (figure 5), which may in part explain the higher ADCC activity of N-820 than rituximab to both rituximab-sensitive and rituximab-resistant BL tumor cells. As a fusion protein using IL-15 superagonists as a scaffold (online supplemental figure 1), N-820 significantly enhanced the expression of NK activating receptors such as NKp44, CD16, NKp30, and NKG2D (figure 1A) as compared with IgG. The enhanced expression of NK activating receptors also in part contribute to the enhanced natural cytotoxicity of exPBNK by N-820.
The role of cytokines in controlling primary NK cell responses has been extensively studied.19 Our data show that both N-820 and N-803 significantly enhance NK activating receptors expression (figure 1A), stimulate NK proliferation (figure 1B,C) and promote in vitro cytotoxicity (figure 3B). However, little is known about the differences in biological and functional consequences of ex vivo expanded NK cells primed by N-820 and N-803. Considering there are ScFv fragments of rituximab in N-820, we investigated the consequences of N-820 or N-803+rituximab priming on the cytokines/chemokines/growth factors secretion profile of expanded NK cells using a human cytokine screening panel consisting of 48 cytokines. Consistent with previous reports that activated NK cells produce a variety of immunoregulatory cytokines/chemokines/growth factors such as IFN-γ, TNF-α, MIP-1α/β, RANTES, and GM-CSF,43 44 N-820 primed ex vivo expanded NK cells produced significantly more of these cytokines, chemokines, and growth factors as compared with N-803+rituximab primed expanded NK cells, suggesting in part that N-820 may facilitate NK cells to recruit other immune cells, such as T cells, to tumor sites and induce activation and proliferation of these cells.
Understanding the pathways activated in expanded NK cells in response to tumor stimulation is important to maximize the therapeutic potential of NK cells in cancer patient’s treatment. The IL-15- Jak/STAT pathway plays an indispensable role in stimulating NK cells development, maturation, survival, proliferation, and/or function.31 NF-κB activation was identified to be important for perforin expression in activated NK cells.45 PI3K–AKT–mTOR pathway was identified as an essential pathway for modulating the development, differentiation, and activation of NK cells.32 Consistently with the roles of these pathways in NK cells, our KEGG pathway analysis showed Jak/STAT, NF-κB, and PI3K–AKT–mTOR pathways were significantly activated in ex vivo expanded NK cells by N-820+Raji-2R as compared with N-803+rituximab+Raji-2R (figure 6D), suggesting in part an enhanced priming effect of N-820+tumor cells as compared with rituximab+N-803+tumor cells. We also found that the HIF-1 signaling pathway was significantly enhanced in expanded NK cells by N-820+Raji-2R as compared with N-803+rituximab+Raji-2R (figure 6D). Recently, it was reported that HIF-1α was required for cytokine production and cytotoxicity on NK cell activation and HIF-1α depletion in NK cells delayed tumor growth due to non-productive angiogenesis.46 ,47 The anti-tumor responses of ex vivo expanded NK cell should be further investigated under hypoxia in combination with N-820 in the future.
In conclusion, these results demonstrate the significant effects of the combination of N-820 with ex vivo expanded NK cells against rituximab-sensitive and rituximab-resistant BL in vitro and in vivo. Future studies will aim to further contribute to our understanding of the mechanisms of lymphoma escape/progression from the combinatorial effect of N-820 and ex vivo expanded NK-mediated killing of CD20+ NHL. Overall, the current results provide a rationale for the development of a clinical trial of N-820 alone or in combination with endogenous or ex vivo expanded NK cells in patients with CD20+ B-NHL previously treated with rituximab containing chemoimmunotherapy regimens.
Acknowledgments
The authors would like to thank Erin Morris, BSN and Virginia Davenport, RN for her excellent assistance with the preparation of this manuscript and Janet Ayello, MS for her assistance with purchasing research reagents. The authors would like to thank Dean A. Lee for providing the K562-mbIL21-41BBL cells, Matthew Barth for providing Raji-2R and Raji-4RH cell lines, Christian Klein from Roche for providing Obinutuzumab and Patrick Soon-Shiong, John H. Lee, and Jeffrey T. Safrit from ImmunityBio/Altor Bioscience for providing N-820 and N-803.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @DrPatSoonShiong
Presented at Presented in part at American Society of Hematology (2018), BMT Tandem meeting (2018) and at Pediatric Blood & Marrow Transplant Consortium (2019).
Contributors YC and MSC conceived and designed the study; YC, GN, and NKS developed the methodology, performed the analysis and interpreted the data. YC, JMR, and MSC wrote, reviewed and revised the manuscript. PS-S, JL, JTS, MB, and DL provided administrative, technical, and material support. All authors approved the final manuscript for submission.
Funding The research for this study was supported by the grants from Pediatric Cancer Research Foundation (to Cairo), and New York Medical College School of Medicine Translational Science Institute Children Health Translational Research Award (to Chu/Rosenblum).
Competing interests DL reports personal fees and other from Kiadis Pharma, Courier Therapeutics, and Caribou Biosciences outside the submitted work; In addition, DL has a patent broadly related to NK cell therapy of cancer with royalties paid to Kiadis Pharma. PS-S is a majority shareholder of ImmunityBio, Inc. and Altor BioScience, LLC, related to N-820. JTS is an employee of NantKwest, an affiliated company to ImmunityBio, Inc and Altor BioScience, LLC, related to N-820. Othe co-authors declare they have no conflict of interest.
Patient consent for publication Not required.
Ethics approval (NSG) mice were bred, treated, and maintained in the animal facility of New York Medical College (NYMC) with NYMC IACUC-approved protocols. The animal experiments were conducted in accordance with the recommendations of the Guide for Care and Use of Laboratory Animals.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.