Article Text

Abstract
Background Anti-programmed cell death ligand 1 (PD-L1)/programmed cell death 1 antibodies have shown clinical activity in platinum-treated metastatic urothelial carcinoma, resulting in regulatory approval of several agents, including avelumab (anti-PD-L1). We report ≥2-year follow-up data for avelumab treatment and exploratory subgroup analyses in patients with urothelial carcinoma.
Methods Patients with previously treated advanced/metastatic urothelial carcinoma, pooled from two cohorts of the phase Ib JAVELIN Solid Tumor trial, received avelumab 10 mg/kg every 2 weeks until disease progression, unacceptable toxicity or withdrawal. End points included best overall response and progression-free survival (PFS) per RECIST V.1.1, overall survival (OS) and safety. Post hoc analyses included objective response rates (ORRs) in subgroups defined by established high-risk/poor-prognosis characteristics and association between time to response and outcome.
Results 249 patients received avelumab; efficacy was assessed in 242 postplatinum patients. Median follow-up was 31.9 months (range 24–43), and median treatment duration was 2.8 months (range 0.5–42.8). The confirmed ORR was 16.5% (95% CI 12.1% to 21.8%; complete response in 4.1% and partial response in 12.4%). Median duration of response was 20.5 months (95% CI 9.7 months to not estimable). Median PFS was 1.6 months (95% CI 1.4 to 2.7 months) and the 12-month PFS rate was 16.8% (95% CI 11.9% to 22.4%). Median OS was 7.0 months (95% CI 5.9 to 8.5 months) and the 24-month OS rate was 20.1% (95% CI 15.2% to 25.4%). In post hoc exploratory analyses, avelumab showed antitumor activity in high-risk subgroups, including elderly patients and those with renal insufficiency or upper tract disease; ORRs were numerically lower in patients with liver metastases or low albumin levels. Objective response achieved by 3 months versus later was associated with longer OS (median not reached (95% CI 18.9 months to not estimable) vs 7.1 months (95% CI 5.2 to 9.0 months)). Safety findings were consistent with previously reported 6-month analyses.
Conclusions After ≥2 years of follow-up, avelumab showed prolonged efficacy and acceptable safety in patients with platinum-treated advanced/metastatic urothelial carcinoma, including high-risk subgroups. Survival appeared longer in patients who responded within 3 months. Long-term safety findings were consistent with earlier reports with avelumab treatment in this patient population.
- clinical trials as topic
- programmed cell death 1 receptor
- urinary bladder neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Patients with advanced urothelial cancer (UC) who have disease progression after platinum-based chemotherapy have a poor prognosis. Established factors that predict shorter overall survival (OS) include Eastern Cooperative Oncology Group performance status (ECOG PS) ≥1, hemoglobin level <100 g/L and presence of liver metastasis; these factors are used to determine the Bellmunt risk score.1 Other factors associated with high-risk disease or poor response to chemotherapy include upper tract disease,2 renal insufficiency,3 older age4 and albumin level.5 6
Antibodies that bind to programmed cell death protein 1 (PD-1) or its ligand (PD-L1) are clinically active treatments for patients with advanced UC.7–14 Avelumab, an anti-PD-L1 antibody, has shown antitumor activity and acceptable safety in a range of advanced solid tumors.9 10 15–18 In a subgroup of 161 patients with platinum-treated, advanced/metastatic UC enrolled in a large phase I trial (JAVELIN Solid Tumor), the objective response rate (ORR) after ≥6 months of follow-up was 17% and responses occurred across various subgroups.10 These data led to the accelerated approval of avelumab by the US Food and Drug Administration in this setting.19
In studies of checkpoint inhibitors, the identification of potential surrogate end points for OS has been a topic of high interest across multiple tumor types. Systematic reviews and meta-analyses have concluded that, although responders to checkpoint inhibitors tend to have longer OS than non-responders, including patients with UC, the correlation was weak.20–22 However, few studies have assessed whether early response is associated with longer OS in UC. A retrospective analysis of first-line chemotherapy for UC concluded that early response may not predict OS.23 However, in studies of second-line avelumab in patients with other cancers (metastatic Merkel cell carcinoma or unresectable mesothelioma), objective response achieved within 2–3 months was associated with longer OS compared with patients without an early response.24 25
Here, we report an updated analysis of avelumab in patients with previously treated UC with a minimum of 24 months of follow-up. We also report exploratory post hoc analyses evaluating high-risk subgroups and the association between early response and long-term outcomes.
Methods
Study design and procedures
JAVELIN Solid Tumor (NCT01772004) was a phase I, open-label, multicohort trial assessing avelumab monotherapy for various solid tumors. The study design and full methodology in UC cohorts have been reported previously.10 Briefly, patients with locally advanced or metastatic urothelial carcinoma, disease progression following platinum-based chemotherapy or platinum ineligible, and no prior immunotherapy, received avelumab 10 mg/kg by 1 hour intravenous infusion every 2 weeks until confirmed disease progression, unacceptable toxicity or other protocol-specified criteria for withdrawal occurred. Premedication with an antihistamine (diphenhydramine or equivalent) and acetaminophen was administered 30–60 min prior to each avelumab infusion. Dose modifications of avelumab (reductions or increases) were not permitted. Tumor response and progression were assessed per Response Evaluation Criteria in Solid Tumors V.1.1 by a blinded independent review committee. In exploratory post hoc analyses, clinical activity was assessed in subgroups defined by standard baseline characteristics or characteristics associated with high-risk disease or poor response to chemotherapy. PD-L1 expression in tumor samples was assessed using a research-only assay (Dako PD-L1 IHC 73-10 pharmDx), and PD-L1+ status was defined prospectively as expression on ≥5% of tumor cells, consistent with previous analyses.9 10 Adverse events (AEs) were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events V.4.0. Immune-related AEs (irAEs) were identified using a prespecified list of Medical Dictionary for Regulatory Activities preferred terms, followed by comprehensive medical review using predefined criteria. Infusion-related reactions (IRRs) and related events were identified via an expanded definition that included prespecified Medical Dictionary for Regulatory Activities terms (IRR, drug hypersensitivity, anaphylactic reaction, hypersensitivity and type 1 hypersensitivity) occurring on the day of or the day after the infusion, in addition to signs and symptoms of IRR that occurred on the day of infusion and resolved within 2 days.
The primary end point for this study was the occurrence of dose-limiting toxicities during the first 3 weeks of avelumab treatment in the dose-escalation part of the study (reported previously),26 and best overall response per RECIST V.1.1, adjudicated by independent end point review committee, in efficacy expansion cohorts. Secondary end points included duration of response (DOR), progression-free survival (PFS), OS, tumor PD-L1 expression and safety.
Statistical analysis
Time-to-event end points (DOR, PFS and OS) were analyzed using the Kaplan-Meier method. For post hoc subgroup analyses, ORR (proportion of patients with a complete response (CR) or partial response (PR)) and disease control rate (DCR; proportion of patients with CR, PR, stable disease (SD) or non-CR/non-progressive disease (PD)) were analyzed using event proportions, and DCR and PFS were assessed as sensitivity analyses. Additionally, a sensitivity analysis was performed for post hoc subgroups based on continuous covariates, including creatinine clearance and albumin concentration; an automated cut-off search was applied using recursive partitioning to test the robustness of cut-offs selected.27 This data-mining approach conditionally partitions the covariate space that best predicts the end point of interest. DCR was selected to avoid redundancy and use classification tree models. The partition algorithm selected the input variable that had the strongest association with disease control and implemented a binary split in the selected input variable; this process was repeated recursively. Post hoc analysis of the association between time to response (TTR) and DOR or OS was performed using landmark Kaplan-Meier analysis. Associations were assessed visually using a scatter plot matrix for TTR, DOR, PFS and OS (data not shown). Subgroups were defined based on whether response occurred by week 7 (first tumor assessment ±5 days; DOR analysis) or by 3 months (12 weeks; second tumor assessment ±5 days; OS analysis), and analyses included only those patients who remained on study at the defined time point. Univariate and multivariate Cox regression model analyses were also performed (data not shown). Following methodology based on previously published studies, further OS analyses were carried out that defined separate categories for patients with early response versus SD/late response versus other patients, and for patients who continued treatment beyond PD versus patients who discontinued treatment when PD occurred.28 OS analyses were confirmed by supplementary analyses, including one using an alternative landmark definition (week 7, as above) and another that excluded patients who were not evaluable by blinded IRC.
Results
Patient characteristics and disposition
Between September 3, 2014, and March 15, 2016, 329 patients with advanced/metastatic UC were screened for enrollment into two study cohorts; of those, 249 met eligibility criteria and received avelumab (table 1; online additional file 1). Detailed patient characteristics for the pooled population have been reported previously.10 Briefly, median age was 68 years (range 30–89 years), most patients were male (178 (71.5%)), and 124 (49.8%) had received ≥2 prior lines of therapy for advanced disease. At data cut-off on April 10, 2018, median follow-up was 31.9 months (range 24–43 months). Patients received a median of six doses of avelumab (range 1–93 doses) for a median of 2.8 months (range 0.5–42.8 months). Twelve patients (4.8%) remained on treatment at last follow-up. The most common reason for treatment discontinuation was disease progression (155 (62.2%)); other reasons were AEs (39 (15.7%)), death (13 (5.2%)), withdrawal of consent (13 (5.2%)), protocol non-compliance (2 (0.8%)) and other reasons (15 (6.0%)). Consistent with the approved label of avelumab and a previous report,10 seven platinum-naïve patients were excluded from efficacy analyses but were included in safety analyses.
Supplemental material
Patient demographics and baseline characteristics
Antitumor activity
Of 242 postplatinum patients, 40 had a confirmed response (CR in 10 (4.1%); PR in 30 (12.4%)), resulting in an ORR of 16.5% (95% CI 12.1% to 21.8%; online additional file 2). Data for best overall response were missing or not assessable in 43 patients (17.8%; no postbaseline tumor assessment in 35, postbaseline assessment but response not evaluable in 2, stable disease of insufficient duration in 5 and non-evaluable with PD >12 weeks after study assessment in 1). Median TTR was 2.7 months (range 1.3–11.0 months). At last follow-up, response was ongoing or considered SD in 20 of 40 responding patients (50.0%; online additional file 3A). Median DOR was 20.5 months (95% CI 9.7 months to not estimable), and the estimated proportion of responses lasting ≥12 months was 65.4% (95% CI 47.0% to 78.8%). Of 198 evaluable patients with a baseline and on-study tumor assessment, 54 (27.3%) had tumor shrinkage of ≥30% (online additional file 3B). In patients with ≤1 (n=118), 2 (n=73) or ≥3 (n=51) previous lines of therapy for advanced disease, ORRs (95% CIs) were 19.5% (12.8% to 27.8%), 16.4% (8.8%% to 27.0%) and 9.8% (3.3% to 21.4%), respectively. Median PFS was 1.6 months (95% CI 1.4 to 2.7 months), and PFS rates at 6 and 12 months were 26.5% (95% CI 20.9% to 32.5%) and 16.8% (95% CI 11.9% to 22.4%), respectively (figure 1A). Median OS was 7.0 months (95% CI 5.9 to 8.5 months), and OS rates at 12 and 24 months were 35.9% (95% CI 29.9% to 42.0%) and 20.1% (95% CI 15.2% to 25.4%), respectively (figure 1B).
Supplemental material
Supplemental material
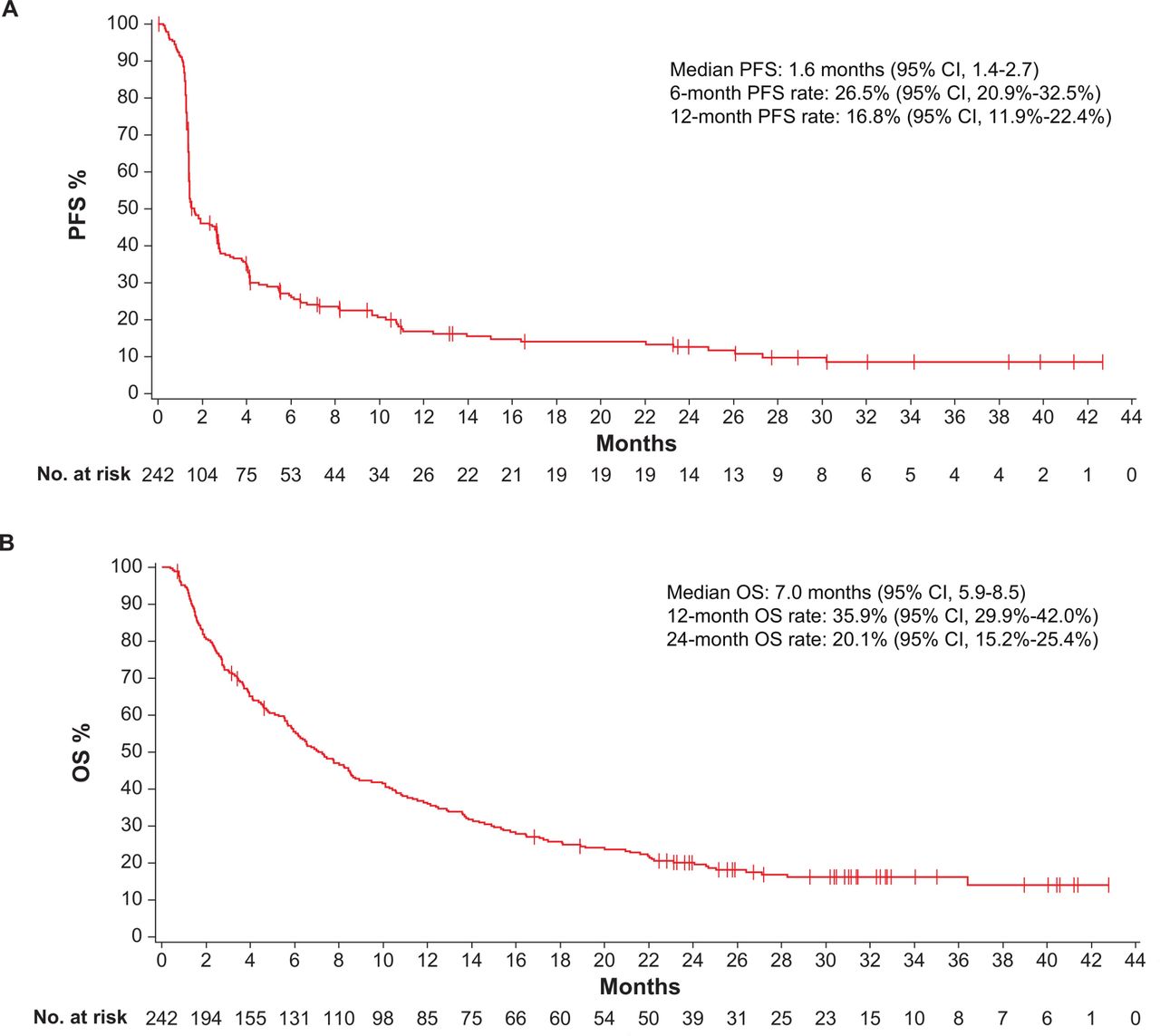
Survival in postplatinum patients after median duration of follow-up of 31.9 months (range 24–43 months). (A) Progression-free survival (PFS) and (B) overall survival (OS); n=242.
PD-L1 biomarker analysis
For PD-L1 expression, 214 patients (88.4%) were evaluable, of whom 84 (39.3%) had PD-L1+ tumors and 130 (60.7%) had PD-L1− tumors. Efficacy findings in PD-L1 subgroups are shown in online additional files 3B and 4.
Supplemental material
High-risk subgroup analyses
Responses to avelumab occurred in all subgroups defined by individual risk factors (figure 2). Factors associated with a higher ORR compared with respective comparator subgroups were absence of baseline visceral or liver metastases, older age (≥75 years) and high albumin (≥35 g/L). ORRs were similar within subgroups defined by renal function (creatinine clearance <60 or ≥60 mL/min) and tumor location (upper vs lower tract). In patients with a Bellmunt prognostic (or risk) score of 0, 1, 2 or 3,1 ORRs (95% CI) were 22.2% (12.0% to 35.6%), 21.4% (14.2%% to 30.2%), 6.9% (1.9% to 16.7%) and 0% (0% to 18.5%), respectively. DCR and PFS were analyzed in selected subgroups (online additional files 5 and 6). Similar to ORR analyses, no difference in DCR or PFS was found between subgroups defined by renal function or tumor location, and patients without baseline liver metastases had a higher DCR and longer PFS than those with liver metastases. In contrast to ORR analyses, subgroups defined by age or albumin level showed no differences in DCR or PFS. Data-driven cut-off searches yielded no new relevant subgroups, and cut-offs found using recursive partitioning were similar to predefined cut-offs (data not shown).
Supplemental material
Supplemental material

Objective response rates (ORRs) in selected high-risk and poor-prognosis subgroups. *Prespecified subgroups. †Lung, liver or bone metastases.
Association between early response and long-term outcomes
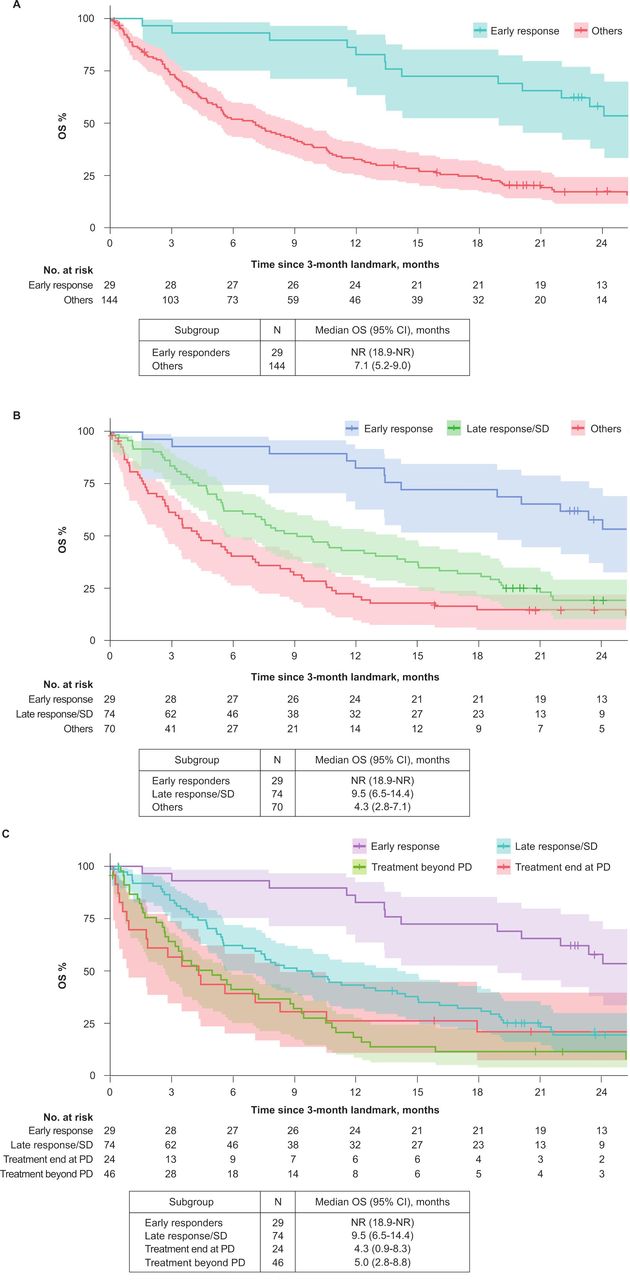
In a post hoc exploratory analysis comparing DOR in patients who had achieved an objective response (CR or PR) either by week 7 (first tumor assessment; n=15) or after that time point (n=25), the median DOR was 25.9 months (95% CI 9.0 months to not estimable) vs 20.5 months (95% CI 8.1 months to not estimable), respectively. CIs were large due to the small sample size. Post hoc landmark analyses were also performed to assess the association between response by 3 months of treatment (second tumor assessment) and OS. In patients who had an objective response by this time point, the median duration of OS beyond 3 months was not reached (95% CI 18.9 months to not estimable) vs 7.1 months (95% CI 5.2 to 9.0 months) in patients who did not respond by 3 months (figure 3). To investigate this observation in more detail, patients without response by 3 months were further categorized according to whether they had SD by 3 months and/or achieved a later response, or whether their best response was PD or not evaluable; median duration of OS beyond 3 months in these subgroups was 9.5 months (95% CI 6.5 to 14.4 months) and 4.3 months (95% CI 2.8 to 7.1 months), respectively (figure 3). Further analyses found that OS was similar between patients with PD who continued treatment beyond investigator-confirmed PD and those who did not (figure 3), although this analysis might be biased because the decision to continue treatment would have also been based on patient characteristics at the time of PD. OS findings were confirmed by a sensitivity analysis that used an earlier landmark time point (7 weeks).

{kind=link}
{kind=link}
{kind=link}
Landmark analysis of overall survival beyond 3 months in postplatinum patients according to objective response (complete or partial response (CR/PR)) at 3 months. (A) Comparison of patients with (i) CR/PR by 3 months or (ii) no CR/PR by 3 months. (B) Comparison of patients with (i) CR/PR by 3 months, (ii) CR/PR after 3 months or stable disease (SD) by 3 months or (iii) best response of progressive disease (PD; before/after 3 months) or not evaluable. (C) Comparison of patients with (i) CR/PR by 3 months, (ii) CR/PR after 3 months or SD by 3 months, (iii) best response of PD with end of treatment at PD or (iv) best response of PD with treatment beyond progression. NR, not reached.
Safety
Safety findings were consistent with the 6-month analysis of this population (N=249).10 Treatment-related AEs (TRAEs) of any grade occurred in 177 patients (71.1%), which included 11 additional patients compared with the 6-month analysis (online additional file 7). Grade ≥3 TRAEs occurred in 29 patients (11.6%; 8 additional patients compared with the 6-month analysis). The most common grade ≥3 TRAE was fatigue (four (1.6%)); all other grade ≥3 TRAEs occurred in one to two patients only. irAEs occurred in 51 patients (20.5%; 17 additional patients compared with the 6-month analysis), including 12 (4.8%) who had grade ≥3 events (online additional file 8). The most commonly occurring categories of irAEs (any grade) were immune-related rash (28 (11.2%), including various rash/pruritus AEs) and immune-related thyroid disorders (13 (5.2%), including hypothyroidism, hyperthyroidism and blood thyroid-stimulating hormone increased). IRRs and related events (based on an expanded definition) occurred in 78 patients (31.3%), including grade ≥3 events in three (1.2%). Treatment-related death occurred in one patient (0.4%), due to pneumonitis in a patient with ongoing, treatment-unrelated Clostridium difficile colitis and diverticulitis, as reported previously.
Supplemental material
Supplemental material
In an exploratory post hoc analysis, frequencies of the TRAEs examined were generally similar in high-risk patient subgroups compared with the overall population (table 2). In particular, patients with renal insufficiency or upper tract disease did not experience significant increases in serum creatinine, patients with liver metastases did not have an increased risk of hepatic events, and incidences of pneumonitis, gastrointestinal events (diarrhea and colitis), endocrine events (hypothyroidism and adrenal insufficiency) and IRRs were not elevated in any subgroup examined.
Treatment-related adverse events and infusion-related reactions in high-risk subgroups
Discussion
In this updated analysis with ≥2 years of follow-up, avelumab showed durable antitumor activity in patients with previously treated advanced UC. The confirmed ORR in postplatinum patients was 16.5%, including CR in 4.1%. The median DOR was 20.5 months, and 65.4% of responses were maintained for ≥12 months. Since this trial was initiated, the therapeutic landscape in advanced UC has changed following the regulatory approval of avelumab and four other anti-PD-L1/PD-1 agents in the USA and other countries. The efficacy of avelumab cannot be compared directly with other agents because of differences in patient populations, time frames of enrollment and study designs; however, ORRs appear to be consistent across approved anti-PD-L1/PD-1 agents in the postplatinum setting.7 8 11 13 Although the ORR with avelumab was numerically higher in patients with PD-L1+ versus PD-L1− tumors (23.8% vs 12.3%, respectively), ORRs in both subgroups exceeded the historical, ~10% ORR benchmark for postplatinum chemotherapy11 29 and median OS was similar between subgroups. In phase III trials of other anti-PD-1/PD-L1 agents in the postplatinum setting, PD-L1 expression was not found to be a predictive biomarker.11 13 In first-line treatment of patients with cisplatin-ineligible UC, however, agents approved as monotherapy without prior chemotherapy (atezolizumab and pembrolizumab) are indicated only for patients with PD-L1+tumors.
In the exploratory post hoc analyses, avelumab showed antitumor activity in subgroups defined by characteristics associated with a poor prognosis or high-risk disease, suggesting that various known prognostic factors are not associated with response to avelumab. Compared with the overall population, ORRs with avelumab were numerically lower in patients with liver or visceral metastases or low albumin levels, but not in patients with low creatinine clearance levels or upper tract disease. Historically, patients with these characteristics have had poor outcomes with chemotherapy1 6 30; thus, avelumab and other anti-PD-1/PD-L1 agents should be considered as treatment options for these patients. Unlike ORR findings, DCRs and PFS were similar in subgroups defined by albumin level. From a statistical perspective, trying to identify subgroups of patients with enhanced treatment effects introduces a high probability of finding false positives, particularly when there are no adjustments for multiple testing. To reduce the risk of false positives, subgroups should be defined prior to analysis, or a specific data-driven selection procedure should be predefined.27 For these analyses, a tree-based cut-off search with recursive partitioning was used. Despite using these established approaches, findings from the post hoc analyses should be interpreted with caution.
In other exploratory landmark analyses, there was no difference in response durability with avelumab in patients with response by week 7 compared with those who had a later response. However, treatment response (PR or CR) by 3 months appeared to be associated with longer OS than SD or later response (after 3 months), and OS in both subgroups was longer than in those who did not respond or achieve SD. Nevertheless, we also observed that some patients had long durations of stable disease with avelumab and some benefitted from continued treatment beyond initial RECIST-defined progression. Given the hypothesis-generating nature and limitations of the exploratory analyses, patients should remain on treatment until disease progression or unacceptable toxicity, per US FDA or local labeling.
With extended follow-up, avelumab continued to show an acceptable safety profile, including a low rate of grade ≥3 TRAEs, and there were no new safety signals compared with an earlier analysis in this population or studies of avelumab in other tumor types. Additionally, occurrence of TRAEs in high-risk subgroups was consistent with the overall patient population.
Overall, these findings confirm the efficacy and safety seen in prior analyses of avelumab in patients with advanced UC and disease progression following platinum-based therapy. Initial results from this study led to a randomized phase III trial (JAVELIN Bladder 100; NCT02603432) to assess avelumab plus best supportive care as maintenance therapy versus best supportive care alone in patients with advanced UC without disease progression after 4–6 cycles of first-line platinum-based chemotherapy. It has been reported that the trial met its primary end point by showing significantly longer OS in the avelumab arm versus control arm (median 21.4 vs 14.3 months, respectively; HR 0.69 (95% CI 0.56 to 0.86); p=0.0005).31 Together, these findings provide a further illustration of the clinical activity of avelumab in the treatment of advanced UC. Following the regulatory approval of avelumab as first-line maintenance and its inclusion in treatment guidelines,19 32 33 it is likely that an increasing proportion of patients with advanced UC will receive avelumab in the first-line maintenance setting.
Acknowledgments
The authors would like to thank the patients and their families, investigators, co-investigators and study teams at each of the participating centers and at Merck KGaA, Darmstadt, Germany, and EMD Serono, Inc.; a business of Merck KGaA, Darmstadt, Germany. Medical writing support was provided by ClinicalThinking and funded by Merck KGaA, Darmstadt, Germany, and Pfizer.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @apolo_andrea, @gulleyj1
Correction notice This article has been corrected since it was published online. The affiliations were re-ordered.
Contributors Conception and design: ABA, JM, GP, MR, JLG. Provision of study materials or patients: ABA, JAE, JRI, MA, MSG, RA, TG, LD, K-WL, MT, PS, DW, AR, JLG, MRP. Collection and assembly of data: all authors. Data analysis and interpretation: all authors. Manuscript writing: all authors. Final approval of manuscript: all authors.
Funding This trial was sponsored by Merck KGaA, Darmstadt, Germany, as part of an alliance between Merck KGaA and Pfizer Inc. ABA and JLG received research funding from the National Cancer Institute’s Center for Cancer Research.
Competing interests ABA has no relationships to disclose. JAE has no relationships to disclose. JRI has received research funding from Aileron Therapeutics, ARMO BioSciences, AstraZeneca, BioMed Valley Discoveries, Bristol Myers Squibb, Calithera Biosciences, Celldex, eFFECTOR Therapeutics, Genentech/Roche, GlaxoSmithKline, Immunocore, Janssen Oncology, MedImmune, Merck and Co., Novartis, Pfizer, Phosplatin Therapeutics, Roche, and TESARO. MA has no relationships to disclose. MSG reports having stocks and other ownership interests in Caremission and WCCT Global; has received research funding from AbbVie, Acetylon, Aduro Biotech, Advaxis, Amgen, Array BioPharma, BeiGene, BioLineRx, Calithera Biosciences, CanBas, Celgene, Celldex, Corcept Therapeutics, CtyomX Therapeutics, Deciphera, Driver Group, Endocyte, ESSA, Five Prime Therapeutics, FujiFilm, Genentech/Roche, Gilead Sciences, GlaxoSmithKline, Halozyme, Hengrui Therapeutics, Inanovate, Incyte, Lilly, Lilly/ImClone, MabVax, Macrogenics, MedImmune, Merck KGaA (Darmstadt, Germany), Merrimack, Millennum, Minneamrita Therapeutics, Nektar, Novartis, Novita Pharmaceuticals, OncoMed, Pfizer, Phoenix Biotech, Plexxikon, Proderm IQ, Samumed, Seattle Genetics, Sirtex Medical, Strategia Therapeutics, Syndax, SynDevRx, TESARO, Tokai Pharmaceuticals, Tolero Pharmaceuticals, Toray Industries, TRACON Pharma, Trovagene, and Verastem; and reports patents, royalties or other intellectual property for patient-on-patient selection for clinical trials. RA has no relationships to disclose. TG has received research funding from Ferring Pharmaceuticals. LD has no relationships to disclose. K-WL has received research funding from Array BioPharma, ASLAN Pharmaceuticals, AstraZeneca/MedImmune, Five Prime Therapeutics, Green Cross Corp., LSK BioPharma, MacroGenics, Merck KGaA (Darmstadt, Germany), MSD, Ono Pharmaceutical, Pfizer, and Pharmacyclics. MHT reports serving as a consultant or advisor for ArQule, Array BioPharma, Bayer, Blueprint Medicines, Bristol Myers Squibb, Eisai, Loxo, and Novartis; and is a member of a speaker’s bureau for Bristol Myers Squibb, and Eisai. PS reports serving as a consultant or advisor for Blueprint Medicines, Eisai, Ellipses Pharma, Epizyme, Ipsen, Lilly, Loxo, PIQUR Therapeutics, and Plexxikon; and has received research funding from Blueprint Medicines, Boehringer Ingelheim, CoBioRes, Eisai, Exelixis, G1 Therapeutics, Lilly, Novartis, PharmaMar, and Plexxikon. DW reports serving as a consultant or advisor for Merck and Co.; and has received reimbursement for travel, accommodation and expenses from Merck and Co. AR has received honoraria from Bristol Myers Squibb, Ipsen, Merck KGaA (Darmstadt, Germany), MSD, Novartis, and Pfizer; reports serving as a consultant or advisor for Bristol Myers Squibb, Ipsen, Novartis, Pfizer, and Roche; has received research funding from Novartis and Pfizer; and has received reimbursement for travel, accommodation and expenses from Bristol Myers Squibb, MSD, Novartis, and Pfizer. JM reports employment at EMD Serono Research & Development Institute, Inc.; a business of Merck KGaA, Darmstadt, Germany. GP reports employment at EMD Serono, Inc.; a business of Merck KGaA, Darmstadt, Germany. MR reports employment at EMD Serono Research & Development Institute, Inc.; a business of Merck KGaA, Darmstadt, Germany. JLG has received research funding from Astellas/Medivation, Bavarian Nordic, Bristol Myers Squibb, EMD Serono (a business of Merck KGaA, Darmstadt, Germany), Incyte, Janssen, Merck & Co, Immunity Bio, and Pfizer. MRP is a member of a speaker’s bureau for Celgene, Exelixis, Genentech/Roche, and Taiho Pharmaceutical.
Patient consent for publication Not required.
Ethics approval The trial was conducted in accordance with the ethics principles of the Declaration of Helsinki and the International Council for Harmonization Guidelines on Good Clinical Practice. The protocol was approved by the institutional review board or independent ethics committee of each center. All patients provided written informed consent before enrollment.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. For all new products or new indications approved in both the European Union and the USA after January 1, 2014, Merck KGaA, Darmstadt, Germany, will share patient-level and study-level data after deidentification, as well as redacted study protocols and clinical study reports from clinical trials in patients. These data will be shared with qualified scientific and medical researchers, on researchers’ request, as necessary for conducting legitimate research. Such requests must be submitted in writing to the company’s data sharing portal. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-anddevelopment/healthcare/clinical-trials/commitment-responsible-data-sharing.html. Where Merck KGaA has a coresearch, codevelopment or comarketing/copromotion agreement or where the product has been out-licensed, it is recognized that the responsibility for disclosure may be dependent on the agreement between parties. Under these circumstances, Merck KGaA will endeavor to gain agreement to share data in response to requests.