Article Text

Abstract
Background Treatments for pancreatic ductal adenocarcinoma are poorly effective, at least partly due to the tumor’s immune-suppressive stromal compartment. New evidence of positive effects on immune responses in the tumor microenvironment (TME), compelled us to test the combination of gemcitabine (GEM), a standard chemotherapeutic for pancreatic cancer, with nicotinamide (NAM), the amide form of niacin (vitamin B3), in mice with pancreatic cancer.
Methods Various mouse tumor models of pancreatic cancer, that is, orthotopic Panc-02 and KPC (KrasG12D, p53R172H, Pdx1-Cre) grafts, were treated alternately with NAM and GEM for 2 weeks, and the effects on efficacy, survival, stromal architecture and tumor-infiltrating immune cells was examined by immunohistochemistry (IHC), flow cytometry, Enzyme-linked immunospot (ELISPOT), T cell depletions in vivo, Nanostring analysis and RNAscope.
Results A significant reduction in tumor weight and number of metastases was found, as well as a significant improved survival of the NAM+GEM group compared with all control groups. IHC and flow cytometry showed a significant decrease in tumor-associated macrophages and myeloid-derived suppressor cells in the tumors of NAM+GEM-treated mice. This correlated with a significant increase in the number of CD4 and CD8 T cells of NAM+GEM-treated tumors, and CD4 and CD8 T cell responses to tumor-associated antigen survivin, most likely through epitope spreading. In vivo depletions of T cells demonstrated the involvement of CD4 T cells in the eradication of the tumor by NAM+GEM treatment. In addition, remodeling of the tumor stroma was observed with decreased collagen I and lower expression of hyaluronic acid binding protein, reorganization of the immune cells into lymph node like structures and CD31 positive vessels. Expression profiling for a panel of immuno-oncology genes revealed significant changes in genes involved in migration and activation of T cells, attraction of dendritic cells and epitope spreading.
Conclusion This study highlights the potential of NAM+GEM as immunotherapy for advanced pancreatic cancer.
- immunomodulation
- lymphocytes
- tumor-Infiltrating
- T-Lymphocytes
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Patients with pancreatic ductal adenocarcinoma (PDAC) have a 5-year survival below 10%. Part of this poor outcome has been attributed to the fact that PDAC is characterized by a dense desmoplastic stroma that prevents drugs and immune cells from penetrating the pancreatic tumors.1 2 This stroma consists of different types of cancer activated fibroblasts (CAF)3 that not only produce extracellular matrix (ECM) components such as collagen but also crosstalk to immune cells.4 5 Among these inflammatory cells are tumor-associated macrophages (TAM) and myeloid-derived suppressor cells (MDSC) that promote immune suppression, tumor progression, angiogenesis, invasion and extravasation of tumor cells resulting in the development of metastases.6 Immunomodulatory therapies to counteract tumor progression are highly sought after but with limited success.
Gemcitabine (GEM) is an Food and Drug Administration (FDA)-approved drug for advanced pancreatic cancer (PDAC) that in combination with nab-paclitaxel,7 only modestly improves patient survival.8 9 Hence, more effective therapies are warranted. GEM is a deoxycytidine analog that inhibits DNA synthesis. New evidence indicates that antitumor activities of chemotherapy such as GEM also rely on several off-target effects, especially directed to the host’s immune system that contribute to tumor eradication. So far, studies that have been focusing on evaluating the effect of GEM on T cell responses to the tumors are limited. A few reports describe that GEM improves T cell activation in mice and humans with pancreatic cancer.10–14 GEM is known for its ability to reduce the MDSC population in tumor-bearing humans and mice.15 Finding drugs to combine with GEM that can strengthen the immune response would be a significant step forward.
Nicotinamide (NAM), the amide form of niacin (vitamin B3), is a nutrient provided by dietary source and supplement, and has little side effects.16 NAM is a precursor of the coenzyme NAM adenine dinucleotide and participates in the cellular energy metabolism in the mitochondrial electron transport chain.17 It has been shown that NAM kills pancreatic tumor cells through down regulation of SIRT-1, K-ras and Akt-1 expression,18 and that NAM sensitizes tumor cells to chemotherapy and radiotherapy through inhibition of poly (ADP-ribose) polymerase (PARP).19 New evidence has been reported that NAM affects immune responses positively. For instance, NAM increased the expansion of human T cells through mitochondrial activation20 and acted immunoprophylactic as well as immunotherapeutic in preclinical mouse models with hormone receptor-positive breast cancer.21 Also, peritumoral and infiltrating CD4 and CD8 T cells were significantly increased in melanomas on NAM treatment compared with a placebo.22 Together this suggests that NAM may qualify as an effective therapeutic add on in PDAC. Moreover, it is safe to use and showed cancer preventative actions in a phase three clinical trial of non-melanoma skin cancer.23
Based on this information we evaluated the effect of combining GEM with NAM on pancreatic cancer and focused on the immune system. We found a significant decrease in the growth of primary tumors and metastases, as well as an increase in the survival time of NAM+GEM treated mice, in correlation with a significant influx of immune cells into the pancreatic tumors, and with a significant increase in CD31-positive blood vessels. The immune infiltrate was characterized by T cells that formed peritumoral lymph node-like structures (LNS) predominantly in the NAM+GEM group, and intratumoral LNS sometimes in the NAM or GEM groups. CD4 and CD8 T cells were activated by NAM+GEM to the tumor-associated antigen (TAA) survivin, which is highly expressed by tumors and metastases in the Panc-02 model,24 one of the experimental models used here. In addition, the number of TAM and MDSC in the tumor decreased. Potential immunoregulatory mechanisms of NAM+GEM based on Nanostring analysis will be discussed.
Methods
Animals
C57BL/6 female mice aged 3 months were obtained from Charles River. These mice were used to generate the Panc-02 model. Male and female KPC-wild type mice were maintained at Einstein, which have the same genetic background as KPC mice (LSLKrasG12D+/- LSL-p53R17+/- Pdx1 Cre)25 but are negative for KrasG12D and Tp53R172H mutations and for Cre recombinase. These mice were used to generate the orthotopic KPC mice. In addition, a nude mouse model was used, that is, Fox n1nu/J (cat#000819), Jackson Laboratories, lacking T cells.
Cell lines
The Panc-02 cell line was derived from a methylcholanthrene-induced ductal adenocarcinoma growing in a C57BL/6 female mouse.26 Panc-02 cells were cultured in McCoy’s5A medium supplemented with 10% fetal bovine serum, 2 mM glutamine, non-essential amino acids, 1 mM sodium pyruvate and 100 U/mL Pen/Strep. The KPC tumor cell line was derived from a pancreatic KPC tumor (LSLKrasG12D+/- LSL-p53R172-/-; Pdx1-Cre) in our lab, which was kindly provided by Jacco van Rheenen, Cancer Genomics, Utrecht, The Netherlands.27
Mouse models
Orthotopic Panc-02 model
Orthotopic Panc-02 tumors were generated in immune competent C57BL/6 mice as we described previously.14 Briefly, mice were anesthetized with ketamine (Mylan Institutional LLC)/xylazine (Akorn Animal Health) (respectively, 100 mg and 10 mg/kg, ip), the hair was removed at the location of the spleen, and the skin was sterilized with betadine, followed by 70% alcohol. The animal was covered with gauze sponge surrounding the incision site. A 1 cm incision was made in the abdominal skin and muscle just lateral to the midline and directly above the spleen/pancreas to allow visualization. The spleen/pancreas was gently retracted and positioned to allow injection of Panc-02 tumor cells (106/50 µL phosphate-buffered saline (PBS)) directly into the pancreas, from the tail all the way to the head of the pancreas. To prevent leakage of injected cell suspension, the injection site was tied off after tumor cell injections with dissolvable suture. The spleen/pancreas were then replaced within the abdominal cavity, and both muscle and skin layers closed with sutures. Following recovery from surgery, mice were monitored and weighed daily. A palpable tumor appeared within 10 days in the pancreas. In this model, tumor cells migrate via the blood stream to the liver and grow into small metastases visible by the naked eye. Sometimes, some tumor cells leaked into the peritoneal cavity. Matrigel was not used here since it may prevent dissemination of tumor cells and the development of metastases. The tumor weight was measured, and the number of metastases was counted after euthanizing the mice.
Orthotopic KPC model
Orthotopic KPC tumors were generated similarly, but now the KPC tumor cells (105/50 µL PBS) were injected into the pancreas of immune competent ‘KPC-wild type’ mice. KPC-wild type mice have the same genetic background as the KPC transgenic mice (C57BL/6xFVBxJ129) but lack the expression of Kras and p53 mutations and the Pdx-Cre, and allow the generation of pancreatic tumors and metastases in the liver. In this orthotopic model, a palpable tumor appeared within 10 days, and the metastases are blood born, like in the orthotopic Panc-02 model.
Orthotopic Panc-02 model in nude mice
Nude mice (Fox n1nu/J (000819) Jackson Laboratories) with a C57BL/6 background were used. These mice are immune incompetent because they lack T cells. Tumors were generated orthotopically in the pancreas by injection with Panc-02 cells as described above. A palpable tumor appeared within 10 days, and the metastases are blood born, like in the orthotopic Panc-02 model.
Protocol for treatment of mice with NAM+GEM
The optimal dose of NAM (Green Labs Nutrition, Poland) was determined by testing different concentrations of NAM (100, 50 and 25, 12.5 and 6.25 mg/200 µL/dose). The highest dose without any physiological side effects appeared to be 25 mg/200 µL. Based on these results, we decided to use 20 mg/200 µL as the optimal dose used in all experiments. First, tumors and metastases were generated as described above. NAM (20 mg/200 µL/dose) was administered orally and GEM (1.2 mg/200 µL/dose) was administered ip, alternately, starting 10 days after tumor cell injection and continued for 2 weeks as outlined for all three pancreatic cancer models in online supplemental figure S1A–C. Preparation of NAM: 1 gram of NAM was dissolved in 10 mls of apple juice. Black pepper of 16 mg was added to the solution to prevent glucuronidation. One dose consisted of 20 mg of NAM in 200 µL apple juice. The NAM solution was aliquoted and stored at −20°C until use. Preparation of GEM (Fresensius Kabi; Gemita): 1 g of GEM was dissolved in 100 mls of endotoxin-free saline, diluted until 6 mg/mL, and then aliquoted and stored at −20°C until use. One dose consisted of 1.2 mg in 200 µL saline.
Supplemental material
Supplemental material
Survival study
Orthotopic Panc-02 mice were treated with NAM+GEM as outlined in online supplemental figure S1A. At the end of treatments, mice were monitored without any further treatment until they succumbed spontaneously, or were terminated on appearance of severe premorbid symptoms requiring euthanasia as specified by our approved animal use protocol.
Flow cytometry
Immune cells from spleens of mice were isolated as described previously.28 Immune cells were also isolated from pancreatic tumors using Dispase (Roche cat#049422078001) and Collagenase (Sigma-Aldrich cat #C0130) as we described previously.29 Anti-CD3 (BD Bioscience, cat # 560527) and anti-CD8 antibodies (BD Bioscience, cat # 552877) were used to identify CD8 T cells, anti-CD3 and anti-CD4 (BD Bioscience, cat # 552051) to identify CD4 T cells, anti-CD11b (BD Bioscience, cat # 553312) and anti-Gr1 (BD Bioscience, cat # 553127) to identify MDSC, and anti-CD11b and anti-F4/80 (eBioscience, cat # 17-4801-82) to identify TAM. Appropriate isotype controls were included for each sample. A total of 50 000–1 00 000 cells were acquired by scanning using a LSR-II Fluorescence Activated Cell Sorter system special order (Beckton and Dickinson), and analyzed using FlowJo V.7.6 software. Cell debris and dead cells were excluded from the analysis based on scatter signals and use of Fixable Blue or Green Live/Dead Cell Stain Kit (BD Bioscience, cat # 564406).
Elispot
Immune cells from spleens or tumors were isolated from treated and control Panc-02 mice for ELISPOT (BD Biosciences, cat# 551083) analysis, as described previously.29 30 To detect T cell responses to survivin, 105 spleen cells were transfected with pcDNA-3.1-survivin or pCDNA3.1 alone (negative control) or nothing (negative control). The frequency of IFNγ-producing spleen cells was measured 72 hours later using an ELISPOT reader (CTL Immunospot S4 analyzer). To determine the frequency of IFNγ-producing CD4 and CD8 T cells, spleen cells were depleted for CD4 and CD8 T cells using magnetic bead depletion techniques according to the manufacturer’s instructions (Miltenyi Biotec).
Immunohistochemistry
Tumors were dissected from pancreas and immediately fixed with buffered formalin, and the tissue was embedded in paraffin. Sections (5 µm) were sliced and placed on slides, then deparaffinized at 60°C for 1 hour, followed by xylene, an ethanol gradient (100%–70%), water, and PBS. Slides were then incubated for 30 min in 3% hydrogen peroxide, followed by boiling in citrate buffer for 20 min. Once the slides were cooled, washed, and blocked with 5% goat serum, the sections were incubated with primary antibodies such as anti-CD4 (Cell Signaling Technology, cat # 25229) (1:100 dilution), anti-CD8α (1:400 dilution)(Cell Signaling Technology, cat # 98941), anti-Perforin (Cell Signaling Technology, cat # 31647)(1:300 dilution), anti-Granzyme (Cell Signaling Technology, cat # 44153) (1:200 dilution), anti-CD31 (Cell Signaling Technology, cat # 77699) (1:100 dilution), or anti-α-Smooth muscle actin (αSMA) antibodies (Cell Signaling Technology, cat # 19245)(1:400), followed by incubation with secondary antibody (mouse anti-goat IgG-HRP) (Cell Signaling Technology, cat# 8114S), and SignalStain Boost immunohistochemistry (IHC) Detection Reagent (Cell Signaling Technology, cat# 8114S). Subsequently, the slides were incubated with 3,3'-diaminobenzidine (DAB) (Vector Laboratories, cat# SK-4100), counterstained with hematoxylin, dehydrated through an ethanol gradient (70%–100%) and xylene, and mounted with Permount. The slides were scanned with a 3D Histech P250 High Capacity Slide Scanner to acquire images and quantification data. Secondary antibodies without primary antibodies were used as negative control.
CD4 and CD8 T cell depletions in vivo in orthotopic Panc-02 model
CD4 and CD8 T cells were depleted in C57BL/6 mice with orthotopic Panc-02 tumors during GEM+NAM treatment. Briefly, 10 days after tumor cell injection (when tumors were approximately 5 mm2), mice were treated with NAM+GEM and with 300 µg of anti-CD4 (Clone GK1.5; Cat#BE0003-1: BioXCell) or anti-CD8 (Clone YTS169.4; Cat$BE0117: BioXCell) antibodies (five injections every third day), as outlined in online supplemental figure S1D. All mice were euthanized 2 days after the last anti-CD4 or anti-CD8 treatment and analyzed for tumor weight and number of metastases. As control, isotype-matched rat antibodies against HRPN were used (Clone LTF-2; Cat#BE0090: BioXCell).
Nanostring technology
Pieces of 3 mm3 tumors were submerged in 5 vol of RNAlater (Invitrogen) (n=5 samples/group). Total RNA was isolated from these tissues using TRIzol (Life Technologies) as we described previously.30 The RNA concentrations in the samples were measured using a NanoDrop 2000 instrument (Thermo Fisher Scientific) and checked for quality on agarose gels. All RNA samples used in this study exhibited optical density (OD) 260/280 ratios greater than 1.9 and RNA integrity numbers greater than 8.5.
A total of 770 immune-related mouse genes were analyzed using the nCounter Mouse PanCancer Immune Profiling Panel (NanoString), which are categorized in various pathways (online supplemental table S1).
Supplemental material
RNA hybridization
RNA in situ hybridization (RNAscope, ACD Bio-Techne, Minneapolis, Minnesota, USA) was performed on formalin-fixed paraffin-embedded sections using the RNAScope 2.5 HD Detection Kit RED according to manufacturer’s instructions. Probes used were directed against mouse Ccl21a (cat no 489921) and mouse Fcer1a (cat no 886381). Scans of whole tissues were acquired with Aperio CS2 (Leica Biosystems). Images analyzes and quantifications were performed on whole slides with HALO Image Analysis (Indica Labs). Images were acquired using EVOS M7000 Imaging System (Thermo Fisher Scientific).
Bioinformatics analysis
Differential expression analyzes of mRNA expression data in 20 samples were performed by using the DESeq2 R package V.1.20.0. A total of 770 immunology-related mouse genes, created from the nCounter Mouse PanCancer Immune Profiling Panel (NanoString), were then implemented as candidate genes in this study. GSVA was employed to detect the variation values of the GO term pathways in each group using the R package GSVA.
Statistical analysis Nanostring
All experiments were repeated in triplicate unless otherwise stated. Statistical analyzes were performed using GraphPad Prism software V.7 (GraphPad Software), and statistical significance was defined as p<0.05. Two-way analysis of variance was performed on the experimental data for tumor volumes and mouse weights. The results are presented as the mean±SE of the mean.
Differentially expressed genes were compared with Student’s t-test, and adjusted p values of less than 0.01 and fold changes of greater than 2 were considered to indicate significant dysregulation. p values were adjusted by the Benjamini-Hochberg (BH) method. Adjusted p values are also called q values. Data were analyzed using R (V.3.5.1).
Statistical analysis tumors, metastases and immunological responses
To statistically compare the effects NAM+GEM on the growth of metastases and tumors or on immune responses in the mouse models, the Mann-Whitney U test was applied using Prism. *p<0.05, **<0.01, ***<0.001, ****<0.0001. Values of p<0.05 were considered statistically significant. Survival studies were analyzed using Mantel-Cox test. p<0.05 is significant.
Results
NAM+GEM significantly improves clinical parameters, including survival, in mouse PDAC
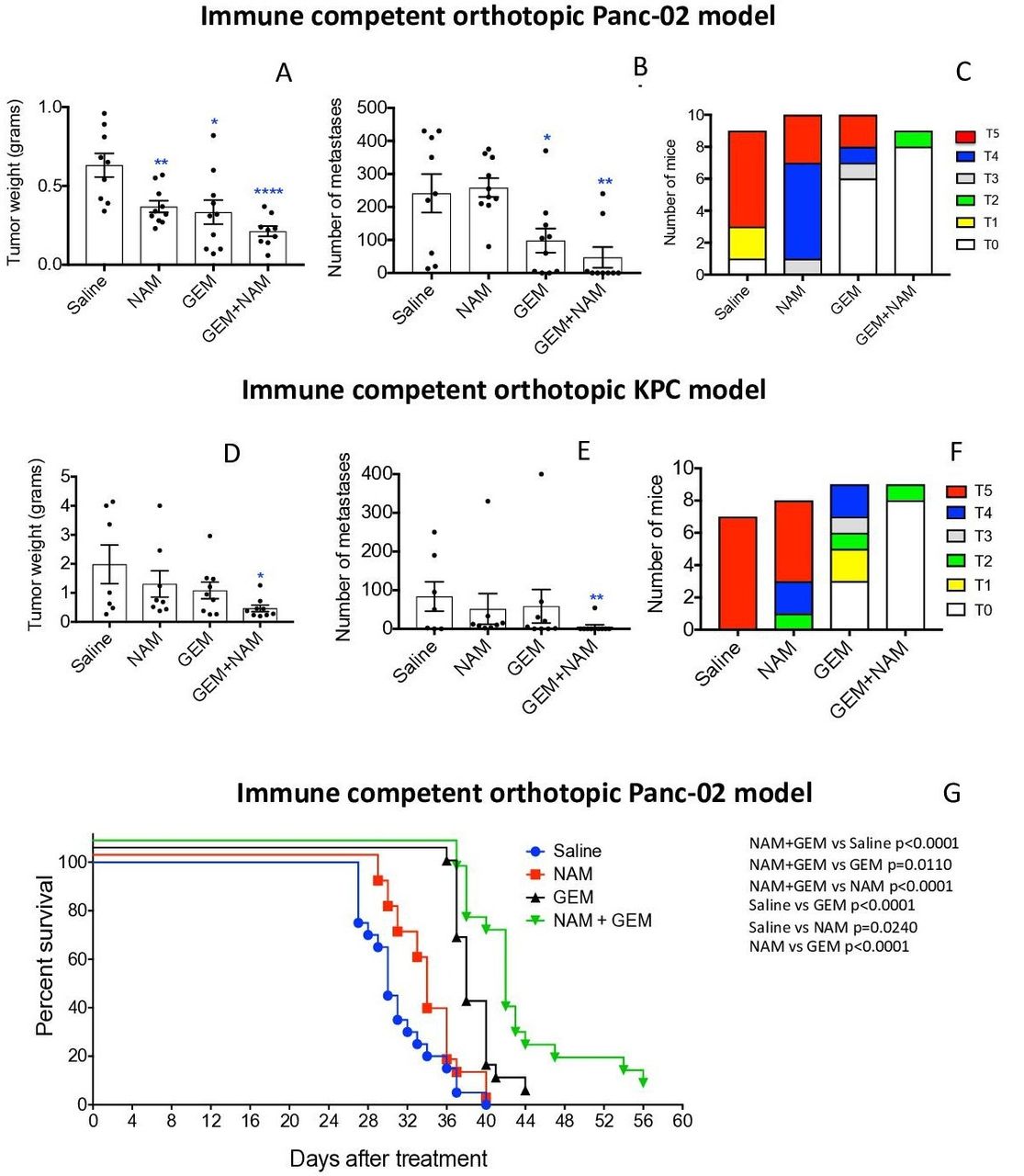
The effect of low doses of NAM (20 mg/dose) and GEM (1.2 mg/dose) on pancreatic cancer was analyzed in two different tumor models, that is, an immune competent orthotopic Panc-02 model, and an immune competent orthotopic KPC model. First, we tested NAM+GEM in the immune competent orthotopic Panc-02 model. Briefly, 106 tumor cells were orthotopically injected into the pancreas and 10 days later the treatment with NAM+GEM started as outlined in online supplemental figure S1A (at this time point tumors are palpable in the pancreas) and the effect of treatment on tumors and metastases was analyzed. As shown in figure 1AB, a significant reduction in tumor weight (p<0.0001) and metastases (p<0.01) was observed in the NAM+GEM group compared with the saline group, while ascites production was relatively high in all control groups (grade 0–5) but relatively minor in the NAM+GEM group (grade 0–2) (figure 1C). The tumor weight and number of metastases in mice treated with the vehicle (negative control: Apple Juice+Pepper), were not significantly different from the saline-treated mice (online supplemental figure S2). Tumors and metastases are depicted in online supplemental figure S3AB.
Supplemental material
Supplemental material

NAM+GEM significantly improved clinical parameters, including survival, in mouse PDAC. Orthotopic Panc-02 model. Tumors and metastases were generated and treatments with NAM+GEM and controls were performed as outlined in online supplemental figure S1A. At the conclusion of the experiment tumor weight (A) and number of metastases (B) was measured, as well as the grade of ascites (T0–5) (C). Average of two experiments with n=10 mice per group. Orthotopic KPC model. Tumors and metastases were generated and treatments with NAM+GEM and controls were performed as outlined in online supplemental figure S1B. At the conclusion of the experiment tumor weight (D) and number of metastases (E) was measured, as well as the grade of ascites (T0–5) (F). Average of two experiments with n=10 mice per group. Significant differences were determined by Mann-Whitney U test. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Error bars represent SE of the mean (SEM). Survival study orthotopic Panc-02 model. Tumors and metastases were generated and treatments with NAM+GEM and controls were performed as outlined in online supplemental figure S1A. At the end of treatments mice were monitored until death, and survival was assessed (G) when defined clinical endpoints were reached. The results of 2 experiments were averaged with n=10 mice per group. Significant differences were determined by the Mantel-Cox test. GEM, gemcitabine; NAM, nicotinamide; PDAC, pancreatic ductal adenocarcinoma.
Second, we tested the NAM+GEM treatment in the immune competent orthotopic KPC model. Briefly, 105 KPC tumor cells were injected into the pancreas (the tumors in this model are more aggressive than in the orthotopic Panc-02 model because of the homozygous p53R172-/- mutation), and 10 days later the treatment with NAM+GEM was started (at this time point tumors are palpable in the pancreas) and continued for 14 days as outlined in online supplemental figure S1B. As shown in figure 1DE, the tumor weight in the NAM+GEM group was significantly reduced (p<0.05) as well as the number of metastases (p<0.01), while the ascites production was high in all control groups (grade 0–5), and minor in the NAM+GEM group (grade 0–2) (figure 1F). Tumors and metastases are depicted in online supplemental figure S4AB. Several mice died in the saline, NAM, or GEM group because of the aggressive cancer, but none in the NAM+GEM group.
Supplemental material
To analyze the clinical relevance of the combination therapy, we tested the effect of NAM+GEM on the survival rate of orthotopic Panc-02 mice. Briefly, 106 tumor cells were injected into the pancreas and 10 days later the NAM+GEM treatment was started and continued for 14 days as described above. Mice were monitored for the following weeks without any further treatment. Here, we show that NAM+GEM significantly improved the survival time of the orthotopic Panc-02 mice not only compared with the saline group but also compared with all other control groups ( figure 1G ).
NAM+GEM increases the influx of immune cells and activates T cells while reducing TAM and MDSC
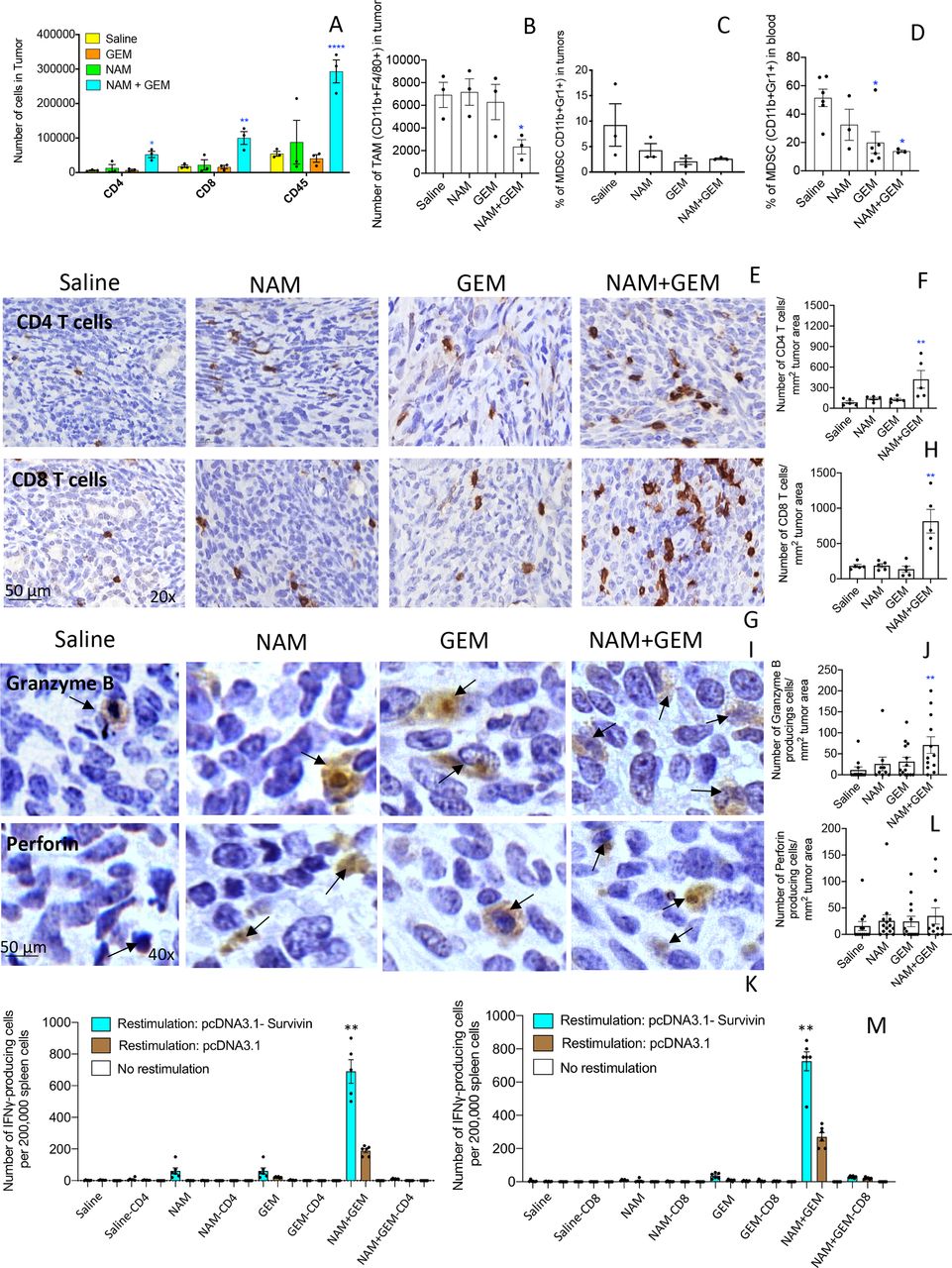
In skin cancer prevention, it has been shown that NAM significantly increased peritumoral and infiltrating CD4 and CD8 T cells compared with a placebo.22 Here, we tested the effect of the combination of GEM+NAM on immune cells in the orthotopic Panc-02 tumors. At the end of treatment, whole tumors were digested with collagenase and dispase to generate single cell suspensions as we described previously.14 Subsequently, the cell population was analyzed by flow cytometry. We found that the CD45+ cells (leucocytes) (p<0.0001), CD4+ (p<0.05) and CD8+ T cells (p<0.01) significantly increased in the NAM+GEM group compared with the saline group (figure 2A). We also analyzed the number of TAMs in the Panc-02 tumors and found a significant reduction in the NAM+GEM group, but not in the GEM only group, compared with saline (p<0.05) (figure 2B). The MDSC population was also reduced in tumors of the NAM+GEM group, although this was not statistically significant because of high variability in the saline treated mice (figure 2C). However, the MDSC population in blood was reduced by GEM alone (p<0.05) (figure 2D), supporting our results in a previous study.14

NAM+GEM increased the influx of immune cells and activates T cells while reducing TAM and MDSC. Immune cells (CD45+) were isolated from whole tumors and analyzed by flow cytometry for the presence of CD4 and CD8 T cells (A), TAM (B) and MDSC (C). In addition, blood was also analyzed for MDSC (D). Subsequently, tumor tissues were analyzed by IHC for the presence of CD4 and CD8 T cells (E, G), and quantified (F, H). 5 fields in each group were analyzed and the number of CD4 and CD8 T cells was calculated per mm2. n=5 mice per group. The results were averaged and analyzed by Mann-Whitney U test. *p<0.05, **p<0.001 is significant. Granzyme B (I) and perforin (K) were analyzed by IHC, and quantified (J, L). Ten fields in each treatment group were analyzed and the number of perforin and granzyme B-producing cells was calculated per mm2. n=5 mice per group. The results were averaged and analyzed by Mann-Whitney U test. *p<0.05, **p<0.001 is significant. finally, spleen cells from NAM+GEM-treated and control mice were restimulated with survivin in an ELISPOT assay (M). CD4 and CD8 T cells were depleted by magnetic beads technology. The spleens of five mice were pooled in each treatment group. The spots of six wells were averaged and analyzed by Mann-Whitney U test. **P<0.01 is significant. GEM, gemcitabine; IHC, immunohistochemistry; MDSC, myeloid-derived suppressor cells; NAM, nicotinamide; TAM, tumor-associated macrophage.
To confirm the flow cytometry data, we analyzed which T cells infiltrated the pancreatic tumors by IHC, an approach we described previously.14 As shown in figure 2E–H and online supplemental figure S5AB, the number of CD4 and CD8 T cells was significantly higher in the tumors of orthotopic Panc-02 mice treated with NAM+GEM (p<0.01 compared with the saline group).
Supplemental material
We also analyzed the production of perforin and granzyme B in the pancreatic tumors through IHC. We found a significant increase in the production of granzyme B (p<0.01) (figure 2IJ and online supplemental figure S5C) but less robust in perforin in the pancreatic tumors of mice treated with NAM+GEM compared with saline (figure 2KL and online supplemental figure S5D).
Since a strong decrease was observed in the size of the pancreatic tumors accompanied with a strong influx of CD4 and CD8 T cells, we analyzed whether these T cells exhibited antitumor reactivity. For this purpose, spleen cells of NAM+GEM-treated and control mice were restimulated with survivin, a TAA expressed by the Panc-02 tumors,24 and analyzed by ELISPOT and magnetic bead technology. It appeared that CD4 and CD8 T cells were highly activated in the NAM+GEM group but not in the other groups (figure 2M).
CD4 T cells eradicate tumors and metastases in orthotopic Panc-02 mice
Since the number and the activity of both CD4 and CD8 T cells was significantly increased in the orthotopic Panc-02 tumors of NAM+GEM-treated mice in correlation with a significant reduction of tumors and metastases, we analyzed in further detail whether this NAM+GEM effect could be reduced by CD4 or CD8 T cells depletions in vivo, as outlined in online supplemental figure S1D. As shown in figure 3A, the average tumor weight in mice treated with NAM+GEM plus CD4 antibodies was significantly increased by 58% compared with the NAM+GEM alone group (p=0.0397, Mann-Whitney U test), while the tumors in mice depleted for CD8 T cells were increased by 22% compared with the NAM+GEM alone group but this was not significant (p=0.3016, Mann-Whitney U test). As expected, the tumors in mice treated with NAM+GEM plus the isotype control (negative control) were not significantly different from the tumor in mice treated with NAM+GEM only (p=0.6111, Mann-Whitney U test). Similarly, the number of metastases in mice treated with NAM+GEM plus CD4 antibodies significantly increased by 97% compared with mice treated with NAM+GEM alone (p=0.0238, Mann-Whitney U test), while the number of metastases in mice treated with NAM+GEM plus antibodies to CD8 T cells was increased by 66% but this was statistically not significant (p=0.4048, Mann-Whitney U test) (figure 3B). Also, here the number of metastases in mice treated with NAM+GEM plus the isotype control was not significantly different compared with mice treated with NAM+GEM only (p=0.6825, Mann-Whitney U test). Flow cytometry analysis of the spleen confirmed that CD4 and CD8 T cells were efficiently depleted in vivo (figure 3C). Pictures of tumors of all mice are shown in online supplemental figure S6.
Supplemental material

CD4 T cells significantly reduced pancreatic cancer by NAM+GEM in immune competent mice, while little effect of NAM+GEM was observed in nude mice. Orthotopic Panc-02 tumors were generated in immune competent C57BL/6 mice and depleted for CD4 and CD8 T cells during NAM+GEM treatment as outlined in online supplemental figure S1D. Antibodies to CD4 T cells (300 µg/200 µL) or to CD8 T cells (300 µg/200 µL) or isotype control were administered IP every 3rd day for 2 weeks. At the conclusion of the experiment the average tumor weight (A) and number of metastases (B) was determined in the NAM+GEM-treated/T cell-depleted compared with NAM+GEM-alone treated mice, with n=5 mice per group. Mann-Whitney U test. *P<0.05, **p<0.01 is significant. depletion of CD4 and CD8 T cells was confirmed by flow cytometry in the spleen (C). Orthotopic Panc-02 tumors were generated in nude mice (Fox n1nu/J) and treated with NAM+GEM as outlined in online supplemental figure S1C. At the conclusion of the experiment the average tumor weight (D) and number of metastases (E) was determined in the NAM+GEM-treated compared with the saline mice with n=5 mice per group. Mann-Whitney U test. GEM, gemcitabine; NAM, nicotinamide; ns, non-significant.
No significant effect of NAM+GEM was observed on orthotopic Panc-02 tumors in nude mice
After demonstrating that in NAM+GEM treatment CD4 T cells significantly contributed to eradication of the pancreatic cancer, we also determined the contribution of NAM+GEM to eradication of tumors and metastases in nude mice (lacking T cells). For this purpose, we generated in addition to the mouse models in figure 1, a third mouse model, that is, orthotopic Panc-02 tumors in nude mice (Fox n1nu/J (000819)) in a C57BL/6 background, followed by NAM+GEM treatment and compared with Saline treatment. As shown in figure 3D,E, the average tumor weight decreased by 40% and number of metastases by 45%. However, this was statistically not significant. Pictures of tumors of all mice are shown in (online supplemental figure S7).
Supplemental material
NAM+GEM affects the stromal architecture with reduced ECM proteins and increased endothelial cells
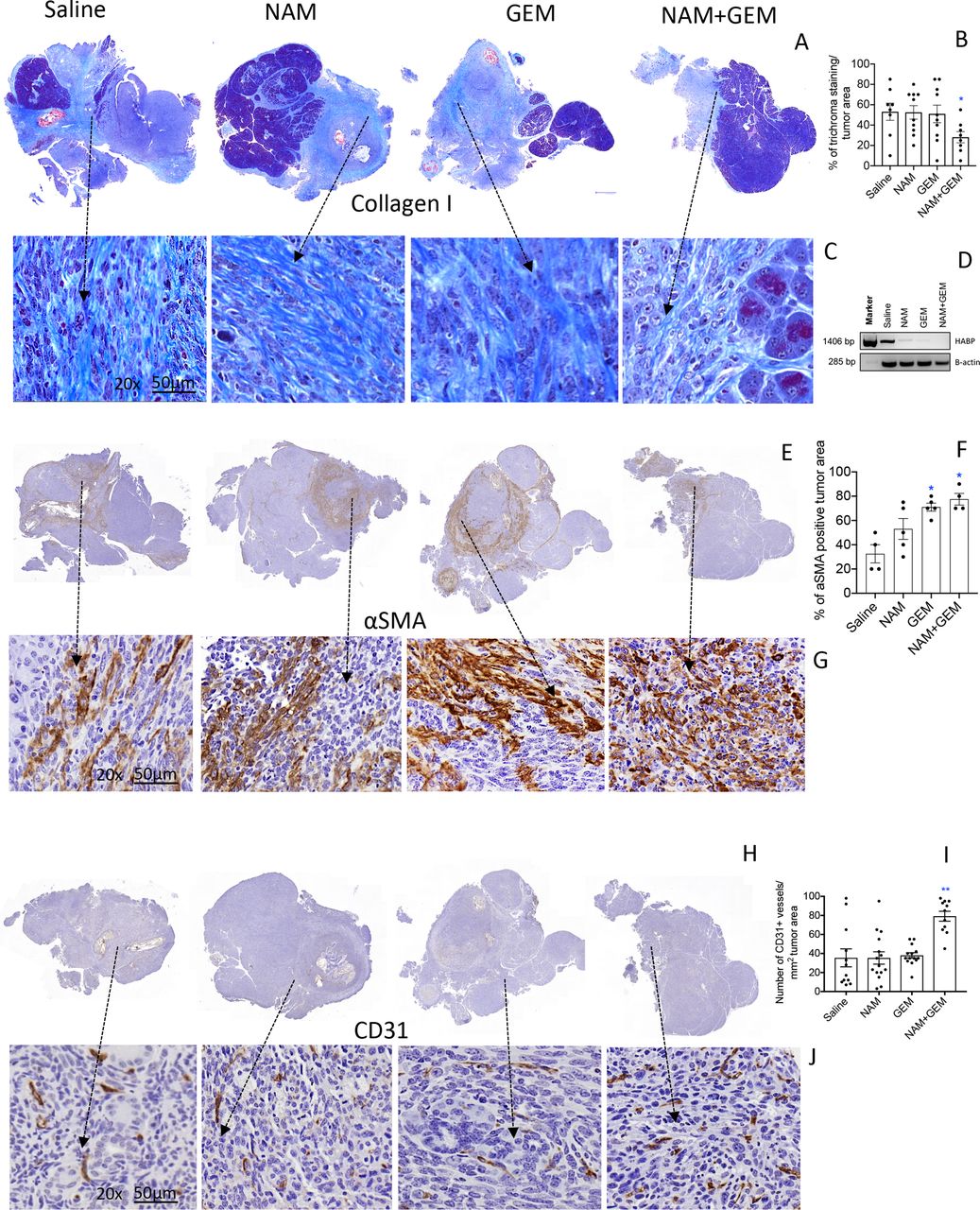
The immunomodulatory effects observed in the above experiments demonstrates that T cell infiltration is improved in the NAM+GEM group. This compelled us to look at altered architecture of the tumor surrounding stroma such as the ECM. For instance, collagen I, produced by CAFs, is known to prevent penetration of drugs and immune cells into the pancreatic tumors, particularly when cross-linked by hyaluronic acid fibrils. Here we showed a significant decrease (p<0.05) in collagen I by Trichrome staining in the tumor area of NAM+GEM-treated mice compared with the saline group (figure 4A–C and online supplemental figure S8A). Moreover, we found a reduction in the expression of the hyaluronic acid binding protein (HABP) through RT-PCR in the NAM+GEM group compared with the saline group (figure 4D). Also, NAM or GEM separately reduced the expression of HABP.
Supplemental material

NAM+GEM decreased the production of collagen I and HABP, and increased αSMA and CD31, in pancreatic tumors of orthopic Panc-02 model. Tumors were analyzed for collagen I by trichrome staining (A) and quantified (B) and shown in more detail (C). n=5 mice per group and the results of 5 fields were averaged. HABP fibrils was analyzed by RT-PCR (D). αSMA protein was by analyzed by IHC (E), and quantified (F), and shown in more detail (G). For both trichrome and αSMA the percentage of positive areas were determined and the results of n=5 mice per group was averaged. The presence of blood vessels in the pancreatic tumors was analyzed by IHC using anti-CD31 antibodies (H) and quantified (I) and shown in more detail (J). n=5 mice per group and the results of 10 fields were averaged. Mann-Whitney U test. *P<0.05, **p<0.01 is significant. Error bars represent SEM. αSMA, α-smooth muscle actin; GEM, gemcitabine; HABP, hyaluronic acid binding protein; IHC, immunohistochemistry; SEM, SE of the mean.
αSMA, a protein expressed by a myofibroblastic CAF subpopulation, was significantly increased (p<0.05) in the tumor area of NAM+GEM-treated mice compared with the saline group (figure 4E–G and online supplemental figure S8B). Also, GEM-treated mice showed a significant increase in the αSMA compared with the saline group. Various reports have been published about the function of αSMA, which will be discussed later.
To allow the influx of immune cells into the tumors, blood vessels will be required. Therefore, we analyzed the tumors for CD31 expression. We found that CD31 was most abundantly expressed in the tumor areas of NAM+GEM treated mice (figure 4H–J and online supplemental figure S8CD).
LNS are often observed in patients with PDAC. Some reported a correlation with improved outcome and others a correlation with a worse outcome.31 32 Here, we found peritumoral LNS in the NAM+GEM group (4/4) with CD4 and CD8 T cells (figure 5A), and less frequently intratumoral LNS in NAM (3/5) and GEM (2/5) groups but not in the saline group (online supplemental figure S9). IHC showed CD31-positive vessels in both peritumoral and intratumoral LNS (figure 5B). More detail about peritumoral and intratumoral LNS in the different treatment groups is shown in (online supplemental figure S9).
Supplemental material

Peritumoral and intratumoral LNS in pancreatic tumors of orthotopic Panc-02 model. Detail of a peritumoral LNS (A). Peritumoral LNS, characterized by well-developed follicular structures with occasional germinal centers. CD4+ and CD8+ T cells are concentrated in the deep cortical zone/paracortex, where they were arranged in dense sheets, and are scattered within follicles and surrounding tissue. The peritumoral LNS is surrounded by a fibroblast layer (red arrows). For more detail about peritumoral and intratumoral LNS (see online supplemental figure S9). Immunostaining for CD31 highlights the presence of vessels within peritumoral and intratumoral LNS in pancreatic tumors (black arrows) (B), which may allow the T cells to migrate to the tumor areas. LNS, lymph node-like structures.
Immuno-oncology gene expression profiling shows enhanced immune reaction and reduced pro-tumorigenic markers
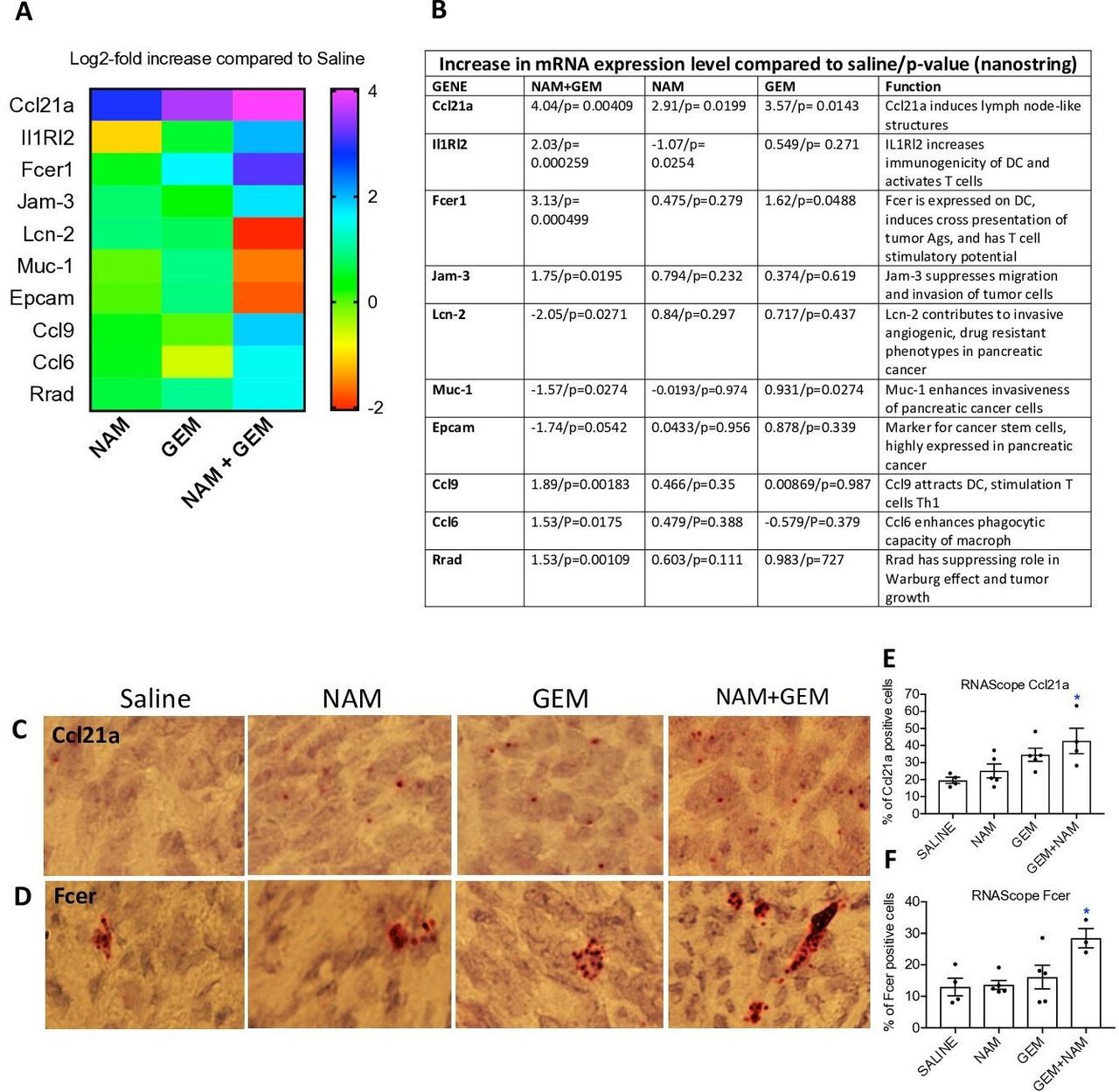
To obtain more insight in the pathways potentially involved in the above-described changes, we analyzed tumors of all treatment groups of the orthotopic Panc-02 model by Nanostring Technology and assessed specifically an immune-oncology gene panel (online supplemental table S1). We found significant changes in expression of genes relevant for immune responses and the progression/regression of the pancreatic cancer in the NAM+GEM group compared the saline group. This includes, a 16-fold increase in the expression of Ccl21a, which is involved in T cell migration and recruitment of LNS, a fourfold increase in Interleukin-1 Receptor-Like 2 (IL1RL2), which is involved in activating T cells and increasing DC immunogenicity33, a 10-fold increase in Fcer-1 which is involved in epitope spreading34, a fourfold increase in Ccl9 which is involved in attracting DC and activating T cells35. Chemokine (C-C) ligand 21a (Ccl21a) was also increased in the NAM or GEM only groups but less robust than in NAM+GEM combination. In another study, we also found that GEM improved the migration of T cells to pancreatic tumors.14 Finally, we found a fourfold reduction in Lipocalin-2 (Lcn-2)36 and a less robust but still significant reduction in Mucin-1 (Muc-1)37 and Epithelial cell adhesion molecule (Epcam)38. These genes are involved in the promotion of invasive angiogenic drug resistant tumor cells, invasiveness and tumor progression. A heatmap of the most relevant genes is shown in figure 6A and an explanation of the function of genes in figure 6B and refs 33–42.
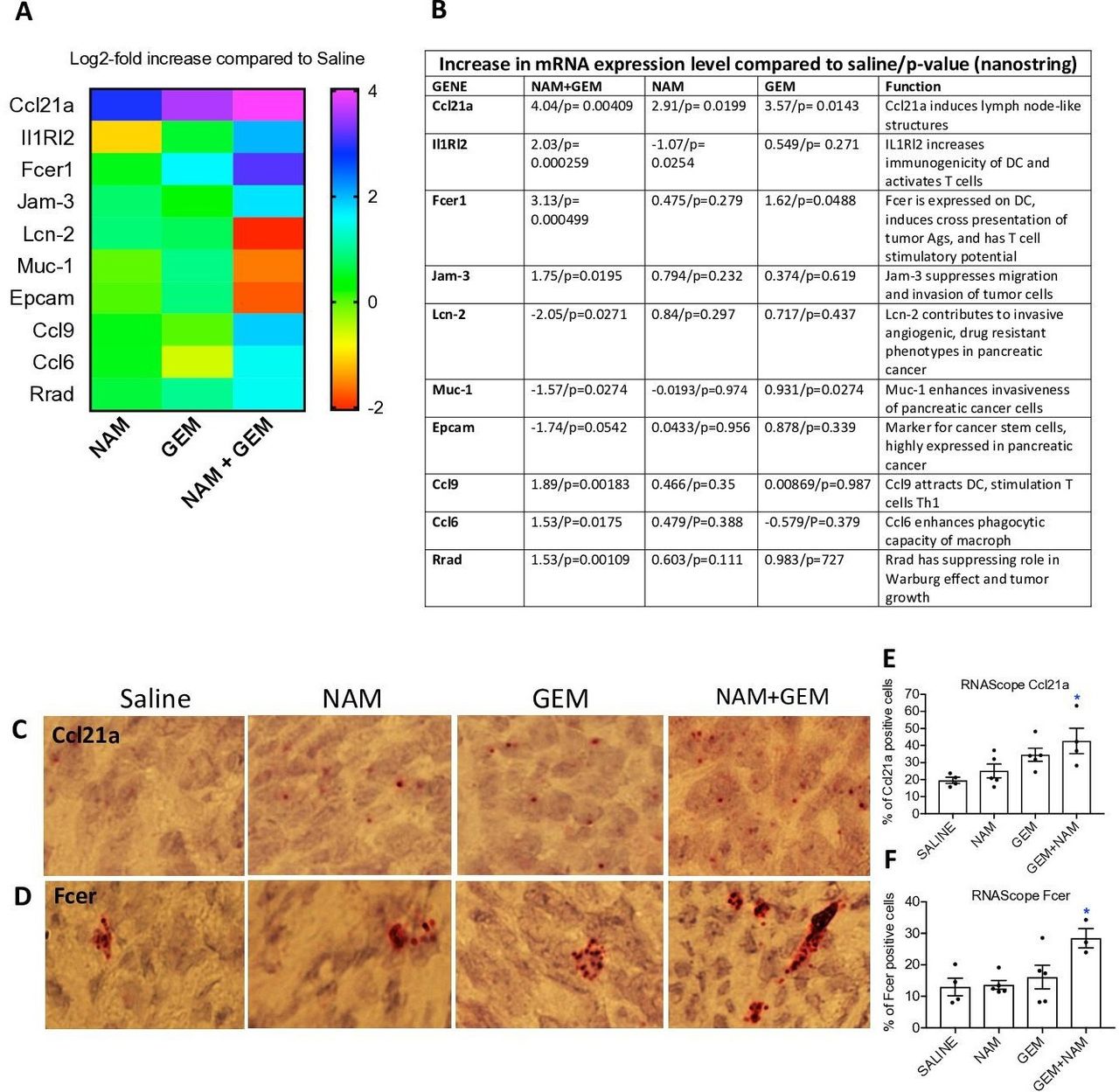
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
NAM+GEM increased the expression of genes involved in T cell migration and activation, and reduced the expression of genes involved in invasion of tumor cells. Orthotopic Panc-02 tumors of the different treatment groups were analyzed by Nanostring through gene expression profiles involved in T cell migration, activation, epitope spreading and invasion of tumor cells. A heatmap of relevant genes is shown (A), and the function of each gene and p values (B). Gene expression levels in the NAM+GEM group were compared with the saline group and analyzed by ANOVA p<0.05 is significant. To confirm the Nanostring data, sections of tumors in the different treatment groups were analyzed for the expression of Ccl21a and Fcer by RNAscope (C D), and quantified (E F). Whole tissues were quantified using HALO Image Analysis; the number of cells with red dot(s) was determined. Images were acquired using ×40 magnification. Data are presented as SE of the mean (SEM); statistical analysis was performed using a one-way ANOVA. *p<0.05 is significant. ANOVA, analysis of variance; GEM, gemcitabine; NAM, nicotinamide.
To validate the nanostring data, we tested the expression of Ccl21 (involved in the formation of LNS and T cell migration) by RNA hybridization (RNAscope) in the tumors of the orthothopic Panc-02 model. We found a significantly higher number of cells expressing Ccl21 in the NAM+GEM group compared with the saline group, but also NAM or GEM showed increased numbers of Ccl21-positive cells compared with the saline group, although this was not significant (figure 6C,E). A similar pattern was observed for Fcer. The highest number of cells expressing Fcer was found in the NAM+GEM group compared with the saline group, and less in the NAM or GEM groups (figure 6D,F).
Discussion
The success of cancer immunotherapies such as checkpoint inhibitors and CAR T cells in PDAC has been underwhelming, due to low immunogenicity of the tumor, strong immune suppression at multiple levels, and inefficient activation of T cells in the TME.8 9 11–14 Here, we demonstrate that a novel combination of NAM+GEM in mice with pancreatic cancer not only reduced the pancreatic cancer (tumors and metastases) but also significantly improved the survival time compared with all control groups. We found suggestive evidence that tumor cells were killed by T cells through epitope spreading. This is based on the strong CD4 and CD8 T cell responses to survivin, which is expressed by the Panc-02 tumor cells,24 and the 10-fold increase in Fcer mRNA in the tumors of NAM+GEM treated mice, which is responsible for epitope spreading, and specifically the significant eradication of pancreatic cancer by CD4 T cells through NAM+GEM treatment in the immune competent orthotopic Panc-02 model. CD4 T cells are known to kill tumor cells through different mechanisms, that is, through perforin and Granzyme B,43 or indirectly through IFNγ-activated M1 macrophages,44 or by helping B cell activation producing antigen-specific antibodies resulting in antibody-dependent cytotoxicity.45 Such mechanisms need to be further analyzed in detail. CD8 T cells also eradicated the tumors and metastases but considerably less robust than CD4 T cells. Also, a reduction in the pancreatic tumors was observed in nude immune incompetent mice lacking T cells, but this was statistically not significant. Most likely tumor cell kill here was caused directly by NAM or GEM through down regulation of SIRT-1, K-ras and Akt-1 expression,18 inhibition of PARP,19 or through inhibition of DNA synthesis, respectively. A schematic view of potential immune mechanisms of NAM+GEM in pancreatic cancer is shown in online supplemental figure S10.
Supplemental material
Immune suppression is one of the hallmarks of PDAC. Unfortunately, checkpoint inhibitors do not improve the outcome of therapy in PDAC patients, which we also observed when adding anti-PD-L1 antibodies to the NAM+GEM treatment (data not shown). However, the TAM and MDSC populations were significantly reduced in the pancreatic tumors and blood, respectively, of the NAM+GEM-treated mice compared with the saline group. This most likely has contributed to the improved T cell responses to survivin in the NAM+GEM group as well.
An interesting observation was the increased number of perforin and particularly granzyme B-producing cells in the tumors of the NAM+GEM group in IHC analysis. Granzyme B is not only known to be produced by T cells and NK cells, but also by B cells and macrophages.46 47 More detailed studies are required to identify the cell type(s) that produce granzyme B here. Other studies have shown that the production of granzyme B plays an important role in ECM remodeling.48 Granzyme B cleaves fibronectin, which is involved in the formation of collagen I fibrils.48 Collagen I is produced by CAFs.49 These fibrils form cross-links between the collagen fibers, which in turn may prevent drugs and immune cells from penetrating into the pancreatic tumor.1 50 Cleaving fibronectin leads to degradation of Collagen I density and may lead to a better infiltration of immune cells. We observed a significant decrease in Collagen I density and in HABP in the NAM+GEM-treated group, in correlation with an influx of immune cells into the pancreatic tumors, and increase in CD31 positive blood vessels in the tumor areas.
LNSs are often found in cancer although their function is not well understood. Tertiary lymphoid structures in close proximity to pancreatic tumors have been correlated with increased patient survival in PDAC.31 Conversely, tumor draining lymph nodes (intratumoral LNS) were found to correlate with immune suppression, a more metastatic character, and poor outcome in PDAC patients.32 Our study showed both peritumoral and intratumoral LNS in the TME of the orthotopic Panc-02 model. The peritumoral LNS contain well-developed follicular structures, germinal centers and T cells, all surrounded by a fibroblast layer, at the periphery of the tumor, and the intratumoral LNS are located inside the tumor with an accumulation of T cells in the tumor areas. While in NAM+GEM-treated mice a positive correlation was found between the peritumoral LNS and improved T cells responses to TAA, efficacy, and survival, no such correlation was observed for intratumoral LNS. None of these effects were found in the NAM or GEM group only. We found peritumoral LNS in all tumors of mice treated with NAM+GEM, while in the NAM or GEM groups intratumoral LNS only were observed. The T cell responses in the tumors of NAM or GEM treated mice were not significantly different from the saline group. It has been suggested that peritumoral LNS protects T cells from immune suppression.51 Based on these and our results we speculate that the peritumoral LNS are a place where T cells can be activated without inhibition by immune suppression, while T cells in the intratumoral LNS are exposed to the same immune suppression as T cells in the TME. However, this hypothesis needs to be tested in studies specifically designed for it.
High αSMA expression is characteristic of myofibroblastic CAFs.49 However, different correlations of αSMA expression with survival have been reported. One study in PDAC patients reported that high expression levels of αSMA in tumor stroma promoted invasion and cellular migration or was associated with a worse outcome,52 while others found that high expression of αSMA is associated with improved outcome.53 Also, deletion of αSMA+ fibroblasts in transgenic mice led to invasive undifferentiated tumors and reduced survival.54 In our study, we found that αSMA increased in the NAM+GEM group compared with the saline group and this correlated with significant improved survival of the orthotopic Panc-02 mice. This heterogeneity and plasticity of αSMA in the reported studies may have been the result of different types of therapy. Therefore, more detailed studies are required to obtain better insight in the role of αSMA in PDAC in relation to different therapies. It is yet unclear how these data, which are snapshots in time, need to be interpreted in a context of CAF heterogeneity and plasticity.5
Conclusions
In conclusion, we have demonstrated that NAM+GEM significantly reduced pancreatic cancer in different mouse tumor models, and significantly improved the survival time compared with all control groups. This correlated with an altered tumor architecture and immune responses in the TME. Collagen I and HABP was significantly reduced by NAM+GEM, while more T cells infiltrated the pancreatic tumors, and CD4 T cells significantly eradicated the pancreatic tumors and metastases in vivo. Also improved T cell responses to survivin were observed, in correlation with reduction in TAM and MDSC in the tumor microenvironment. The results of this study, and its promising effect in a phase 3 clinical trial to prevent skin cancer,23 as well as the immunoprophylactic and immunotherapeutic success of NAM in the preclinical hormone receptor-positive breast cancer models21 suggest that NAM as addition to GEM, which is the backbone of standard care for PDAC patients, could be a promising new lead for the treatment of pancreatic cancer.
Acknowledgments
We greatly thank Ms Hong Zhang, Department of Pathology, and Dr Vera DeMarais, Director of Light Microscopy and Image Analysis, Department of Structural and Cell Biology, Einstein for providing outstanding training and support regarding the IHC and image analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors BCS performed and designed most of the experiments and analyzed the data. KM, OL and AB presented in figures 1-6 and contributed experimentally to the data and online supplemental figures. AB contributed to the IHC analysis. BCS, KM, OL, LM and IR contributed with critical discussions and design of experiments. CG conceived, designed and supervised the study and wrote the manuscript. All authors reviewed the manuscript.
Funding This work was supported by the Anticancer Fund, a private donation of Janet and Marty Spatz, NCI cancer center support P30CA013330 (Flow Cytometry Core, Pathology Core), and IR was supported by the FWO Odysseus Program (Research Foundation Flanders), and OL is recipient of a Fellowship (FWOTM904, Research Foundation Flanders).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data sharing not applicable as no datasets generated and/or analyzed for this study. We do not have datasets generated in this manuscript.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.