Article Text

Abstract
Background Immunotherapy of cancer is successful but tumor regression often is incomplete and followed by escape. Understanding the mechanisms underlying this acquired resistance will aid the development of more effective treatments.
Methods We exploited a mouse model where tumor-specific therapeutic vaccination results in tumor regression, followed by local recurrence and resistance. In depth studies on systemic, local and tumor intrinsic changes were performed with flow and mass cytometry, immunohistochemistry, transcriptomics and several perturbation studies with inhibitors or agonistic antibodies in mice. Main findings were recapitulated in vaccinated patients.
Results Full tumor regression and cure of tumor-bearing mice is dependent on the magnitude of the vaccine-induced T-cell response. Recurrence of tumors did not involve classical immune escape mechanisms, such as antigen-presentation alterations, immune checkpoint expression, resistance to killing or local immune suppression. However, the recurrent tumors displayed a changed transcriptome with alterations in p53, tumor necrosis factor-α and transforming growth factor-β signaling pathways and they became immunologically cold. Remarkably, ex vivo cell-sorted recurrent tumors, directly reinjected in naïve hosts retained their resistance to vaccination despite a strong infiltration with tumor-specific CD8+ T cells, similar to that of vaccine-responsive tumors. The influx of inflammatory mature myeloid effector cells in the resistant tumors, however, was impaired and this turned out to be the underlying mechanisms as restoration of inflammatory myeloid cell infiltration reinstated the sensitivity of these refractory tumors to vaccination. Notably, impaired myeloid cell infiltration after vaccination was also associated with vaccine resistance in patients.
Conclusion An immunotherapy-induced disability of tumor cells to attract innate myeloid effector cells formed a major mechanism underlying immune escape and acquired resistance. These data not only stresses the importance of myeloid effector cells during immunotherapy but also demands for new studies to harness their tumoricidal activities.
- immunotherapy
- active
- tumor escape
- tumor microenvironment
- vaccination
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Immunotherapy has become clinically effective in multiple cancers. Blockade of the immune inhibitory checkpoints cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) as well as adoptive transfer of (genetically modified) T cells are the successful examples. In addition, therapeutic vaccines targeting tumor-specific antigens start to show clinical benefit, especially when used in combination with other therapies. Despite the many patients with complete responses, the majority of clinically responding patients show partial tumor regressions after immunotherapy.1–7 Several primary and acquired resistance mechanisms, including low tumor-specific antigen load, antigen processing and presentation defects, T-cell exclusion, and local immune suppression by suppressive myeloid cell populations, regulatory T cells and coinhibitory molecule expression, allow tumors to become therapy resistant, limiting the clinical efficacy of the immunotherapeutic approaches used.8
The concept of immune editing dictates that a non-curative immune response eventually drives tumor immune escape.9 Unfortunately, most of the tumor models in which systemic immunotherapy is tested show tumor growth delay but not actual tumor regression and by this deviate from what is observed in the clinic. We have shown that under optimal conditions, therapeutic vaccination with the human papillomavirus (HPV16) E743-77 synthetic long peptide (SLP) and the toll-like receptor 9 agonist CpG results in full tumor regression and cure of all mice with an established TC-1 tumor. However, under suboptimal vaccine conditions TC-1 tumors do regress but after a period regrow,10 11 hereby mimicking non-curative partial clinical responses.
To understand the mechanisms driving immune escape after non-curative immunotherapy, we performed a detailed analysis of extrinsic and intrinsic mechanisms that may foster the development of therapy-refractory tumors. Our data indicate that resistance to T-cell-based immunotherapy not necessarily resides in pathways directly involving interaction between T cells and tumor cells, but can also be the result of an impaired co-infiltration with innate immune cells. This stresses the need for a reappraisal of tumor-infiltrating innate immune cells during immunotherapy focusing not only on their potential immune suppressive roles but also on their tumoricidal and phagocytic activities.
Methods
Mice
C57BL/6 mice were purchased from Charles River Laboratories (L'Arbresle, France). All mice were 6–8 weeks old at the beginning of the experiment. Mice were housed in individually-ventilated cages under specific pathogen-free conditions. All animal experiments were approved and executed in compliance with the guidelines of Dutch and European committees.
Tumor challenge and treatments
The tumor cell line TC-1 was generated by retroviral transduction of C57BL/6 lung epithelial cells with the HPV16 E6/E7 and c-H-ras oncogenes.12 Mice were inoculated subcutaneously with 105 TC-1 tumor cells in 200 µL phosphate buffered saline (PBS) containing 0.2% bovine serum albumin (BSA) on day 0. Tumor size (horizontal dimension×vertical dimension) was measured twice a week using a caliper. On day 7 post-tumor injection, mice were divided into groups with comparable tumor sizes. On day 8 and 22 post-tumor challenge, mice were vaccinated with SLP vaccine subcutaneously in the contralateral flank (suboptimal setting: all figures) or tail base (optimal setting). SLP vaccine contains 100 µg HPV16 E743-63 (GQAEPDRAHYNIVTFCCKCDS) covering both Th epitope and the CTL epitope with 20 µg CpG (ODN1826, InvivoGen) dissolved in 200 µL (flank) or 50 µL (tail base) PBS and emulsified with incomplete Freunds adjuvants (flank). Cisplatin is administered at 10 mg/kg in 300 µL saline intraperitoneally (i.p.). Mice were routinely weighed 2–3 times/week. To deplete myeloid cells, CSF1R inhibitor PLX3397 or control was incorporated into chow at 290 mg/kg, resulting in a daily delivery of ~45 mg/kg and was obtained from Plexxikon. Mice were euthanized when tumor size reached>2000 mm2 or when mice lost over 20% of total body weight. For i.p. antibodies treatments, anti-PD-1 clone RMP1-14 (150–200 µg/mouse, BioXcell), anti-PD-L1 clone MIH-5 (150 µg/mouse, purified in-house), OX-40 clone OX86 (150 µg/mouse, Bioceros) and 4-1BB clone 3H3 (150 µg/mouse, purified in-house), CXCR3 clone CXCR3-173 (200 µg/mouse i.p every 3 days, BioXcell), transforming growth factor-β (TGFβ) neutralizing clone 1D11.16.8 and interleukin 6 (IL-6) blocking clone MP5-20F3 (200 µg/mouse, every 3–4 days i.p. and intravenously, respectively, BioXcell). Antibodies were administered as indicated in figure legends. RG7112 (Chemgood, USA) 100 mg/kg daily by oral suspension in 200 µL of 0.5 w/v% methyl cellulose 400 (Wako Pure Chemical Industries; Osaka, Japan).
Flow cytometric analysis of splenic and intratumoral immune cells
Tumors were collected after transcardial perfusion and incubated with Liberase (Roche) in IMDM for 15 min at 37°C. Spleen and lymph nodes were incubated with 0.02 mg/mL DNAse and 1 mg/mL collagenase for 10 min at room temperature. Single-cell suspensions were prepared by mincing spleen and tumor pieces through a 70 µm cell strainer (BD Biosciences). Cells were resuspended in staining buffer (PBS+2% fetal calf serum +0.05% sodium azide) and incubated with various fluorescently labeled antibodies against: CD11b (clone M1/70), CD11c (clone N418), CD45.2 (clone 104), F4/80 (clone BM8), Ly6C (clone HK1.4), Ly6G (clone 1A8), EGR2 (clone erongr2) and iNOS (clone CXNFT). Antibodies were obtained from eBioscience and Biolegend. APC labelled- H-2Db tetramers containing HPV16 E749-57 peptide (RAHYNIVTF) were used as E7 tetramer (E7 Tm). For dead cell exclusion, 7-aminoactinomycin D (7-AAD; Invitrogen), Zombie Aqua (Biolegend) and Zombie NIR (Biolegend) were used. Interferon-regulatory factors (IRF)-1 Rabbit mAb clone: D5E4 and Phospho-NF-κB p65 clone Ser536 (Cell Signaling Technology, USA). For intracellular cytokine staining, single cell suspensions of spleens or tumors were plated in 96-well cell culture flat-bottom plates in the presence of dendritic cells preloaded with SLP and brefeldin A (4 µg/mL). After 5 hours incubation, cells were stained for surface markers and were fixed in 0.5% paraformaldehyde overnight. To measure Tregs, eBioscience FOXP3/Transcription Factor Staining Buffer Set (Invitrogen, USA) was used. Thereafter, cells were washed, stained for cytokines and subsequently analyzed by flow cytometry. Samples were measured with a Becton Dickinson (BD) LSRII flow cytometer, and results were analyzed using FlowJo software (Tree Star).
To sort cells, single cells isolated from the digested tumors or sorted from the digested tumor for CD45+ (immune cells) and CD45- (tumor cells) were used. To sort tumor cells, live CD45-CD31-CD90.2-CD44+ cells were sorted with BD FACS Aria cell sorter.
Mass cytometry and data analysis
Mass cytometry (CyTOF) staining and acquisition was performed as generally described.13 In short, single-cell suspensions of TC-1 tumors were purified by Percoll gradient to remove aggregates and debris, stained with 37 mouse immune markers, and acquired on a Helios mass cytometer (Fluidigm Sciences). The metal-conjugated antibodies are listed in online supplementary file 1. After manual gating of alive singlet CD45+ cells using FlowJo, non-supervised clustering was performed by H-SNE analysis with default parameters using Cytosplore software.14 After clustering was performed based on all markers, cell types were manually assigned to clusters using the following criteria: CD8+ T cells (CD3+CD8a+), CD4+ T cells (CD3+CD8a-), inflammatory myeloid cells (CD11b+Ly6ChiF4/80low), tumor-associated macrophages (CD11b+F4/80hiLy6Clow), neutrophils (CD11b+Ly6G+), eosinophils (CD11b+Siglec-F+) and plasmacytoid dendritic cells (pDCs; Siglec-H+BST2+ B220+).
Supplemental material
RNAseq analysis
RNA was isolated with the NucleoSpin RNA XS kit (Macherey-Nagel, Düren, Germany). RNA-sequencing library preparation including ribosomal RNA depletion and sequencing were performed at GenomeScan B.V. (Leiden The Netherlands). At least 50 million, paired-end, 150 bp reads were generated per sample. These were mapped against the mouse reference genome (mm10 build) using GSnap.15 Read counts were generated per gene using htseq-count. Trimmed mean of M-values normalization was preformed using edgeR R package.16 Genes with overall low expression were filtered out, by only selecting genes with at last 0.7 counts per million in at least three samples. For comparisons between groups we selected genes included in the hallmarks of cancer pathways.17 Limma-Voom was used for statistical analysis.18 Genes were considered as significantly differentially expressed with a false discovery rate below 0.05. Pathway analysis of the hallmarks of cancer pathways was performed using the Ensemble of Gene Set Enrichment Analysis (EGSEA) R package that combines the results of several pathway analysis methods.19
Patient samples
Formalin-fixed paraffin-embedded biopsies of 24 HPV16+ vulvar high-grade squamous intraepithelial lesions (vHSIL) patients, treated with an experimental vaccine containing 13 SLPs covering the entire amino acid sequence of HPV16 oncoproteins E6 and E7, were cut in 4 um thick sections. These samples were stained with two multiplex immunofluorescence panels,20 one for T cells and one for myeloid cells. The slides were then scanned with the Vectra multispectral imaging system (PerkinElmer), and cells were, after manual training, automatically phenotyped and counted with inForm advanced image analysis software (PerkinElmer).
Immunohistochemistry
A 4 µm section of formalin-fixed, paraffin-embedded tumor tissue was mounted on adhesive slides. Following deparaffinization and rehydration, endogenous peroxidase activity was blocked with a 0,3% hydrogen peroxidase/methanol solution (Merck Millipore, Burlington, Massachusetts, USA) for 20 min. Microwave-mediated antigen retrieval was performed in 10 mM citrate (pH 6.0) buffer for 10 min. For TGFβ, slides were incubated with normal goat serum (1:10, DAKO Agilent Technologies, USA). For CD3, slides were incubated with SuperBlock (PBS) blocking buffer (Thermo Scientific). The slides were incubated for 30 min to reduce non-specific binding of the primary antibody. Murine anti-TGFβ clone TB21 (1:800, Anogen-Yes Biotech Laboratories, Canada), rabbit anti-β-catenin clone D10A8 (1:100, Cell Signaling Technology, USA) and rabbit antihuman CD3e clone D7A6E (1:200, Cell Signaling Technology, USA) were diluted in PBS/1% BSA and applied overnight For β-catenin, HRP-labeled, swine antirabbit immunoglobulins (DAKO Agilent technologies) were used to bind β-catenin antibody while poly-HRP labeled, goat antimouse/rabbit IgG was applied for TGFβ immunodetection. For CD3, following washing in PBS/0,05%Tween, slides were incubated for 60 min with biotinylated goat anti-rabbit, IgG (1:200, Aligent, USA). Next, VECTASTAIN Elite ABC-HRP Kit, Peroxidase (standard) (Vectorlabs, USA) was used to label the biotinylated goat antirabbit, IgG. Antibody binding was detected with DAB+chromogen (DAKO, Agilent Technologies, USA) after which nuclear counterstaining was done using haematoxylin (Thermo Fisher Scientific, USA).
TGFβ expression was mostly present in the tumor stroma at the rim of the tumors. Differential TGFβ intensities in the rim of the tumors were analyzed by the Image Management Systems 3.3.1 (PHILIPS) by measuring the percentage of the tumor rim expressing TGFβ compared the to the total perimeter of the tumor, The expression was normalized to the negative control (no primary antibody) for each tumor. β-catenin expression was detected in both tumor and stromal cells and, furthermore, could display cytoplasmic or nuclear localization. Therefore, tumor and stromal compartments were scored separately and intracellular localization was also noted. Staining intensity was differentiated into 0 (negative), 1 (weak), 2 (positive) and 3 (strong).
qPCR analysis
RNA was isolated from the cells with an RNeasy kit (Qiagen) according to manufacturer’s protocol. Isolated RNA was quantified with Nanodrop (Thermo Scientific). Transformation of RNA into cDNA was performed with a high capacity RNA-to-cDNA kit (Applied Biosystems). cDNA (5–10 ng per well) was analyzed by SYBR green real-time PCR with 10 nM primers using a CFX96 or CFX384 Real-Time System C1000 Thermal Cycler (Bio-Rad). The primers used for qPCR are shown in the table below. To measure the chemokine and chemokine receptors, mouse chemokines and receptors RT2 Profiler PCR Array Mouse Chemokines and receptors was purchased from Qiagen (online supplementary file 2).
CTL assay
Naïve C57BL/6 mice were vaccinated with SLP vaccines as described above on day 0 and 14. Splenocytes were isolated 8 days after the last vaccination and restimulated in vitro with dendritic cells (DCs) loaded with SLP or irradiated TC-1 tumor cells. After restimulation, splenocytes were harvested by using ethylenediaminetetraacetic acid (EDTA) and CD8 T cells were isolated by using the mouse CD8+ T lymphocyte enrichment set (BD Biosciences) and used as effector cells. Tumor cells isolated from the untreated or relapsed tumor-bearing mice were exposed to IFNγ (10 IU, Prospec) 1 day before CTL assay and were use as target cells. Target cells were labeled with 100 µL 51Cr for 1 hour, washed and plated into a 96-well round-bottom plate at a density of 2.000 tumor cells/well with different ratios of effector cells. After 4 hours incubation, supernatants of the cells were harvested and the percentage of 51Cr release was measured by a gamma counter.
TGFβ and IL-6 measurements
TGFβ and IL-6 were measured in the supernatants of tumor cells. In brief, digested tumors were seeded in 96-flat bottom plate. After overnight, the supernatants were collected and stored at −80°C until measurement. Total TGFβ levels were determined using commercially available DuoSet ELISAs (R&D systems, USA) after acidification of the samples to activate latent TGFβ as described before.21 22 IL-6 was measured using a mouse Bio-Plex Pro Mouse Cytokine 23-plex immunoassay (Bio-Rad, Herculus, California, USA) according to manufacturer’s protocol.
Statistical analysis
Statistical analyzes were performed using Prism (GraphPad). Survival data were analyzed by Kaplan-Meier and log-rank (Mantel-Cox) tests. Statistical significance was determined using the Mann-Whitney test or unpaired t-test as described in the figure legends, depending on the parametricity of the data. Two-sided p values≤0.05 were considered statistically significant.
Results
Non-curative immunotherapy drives the development of immune-resistant tumors
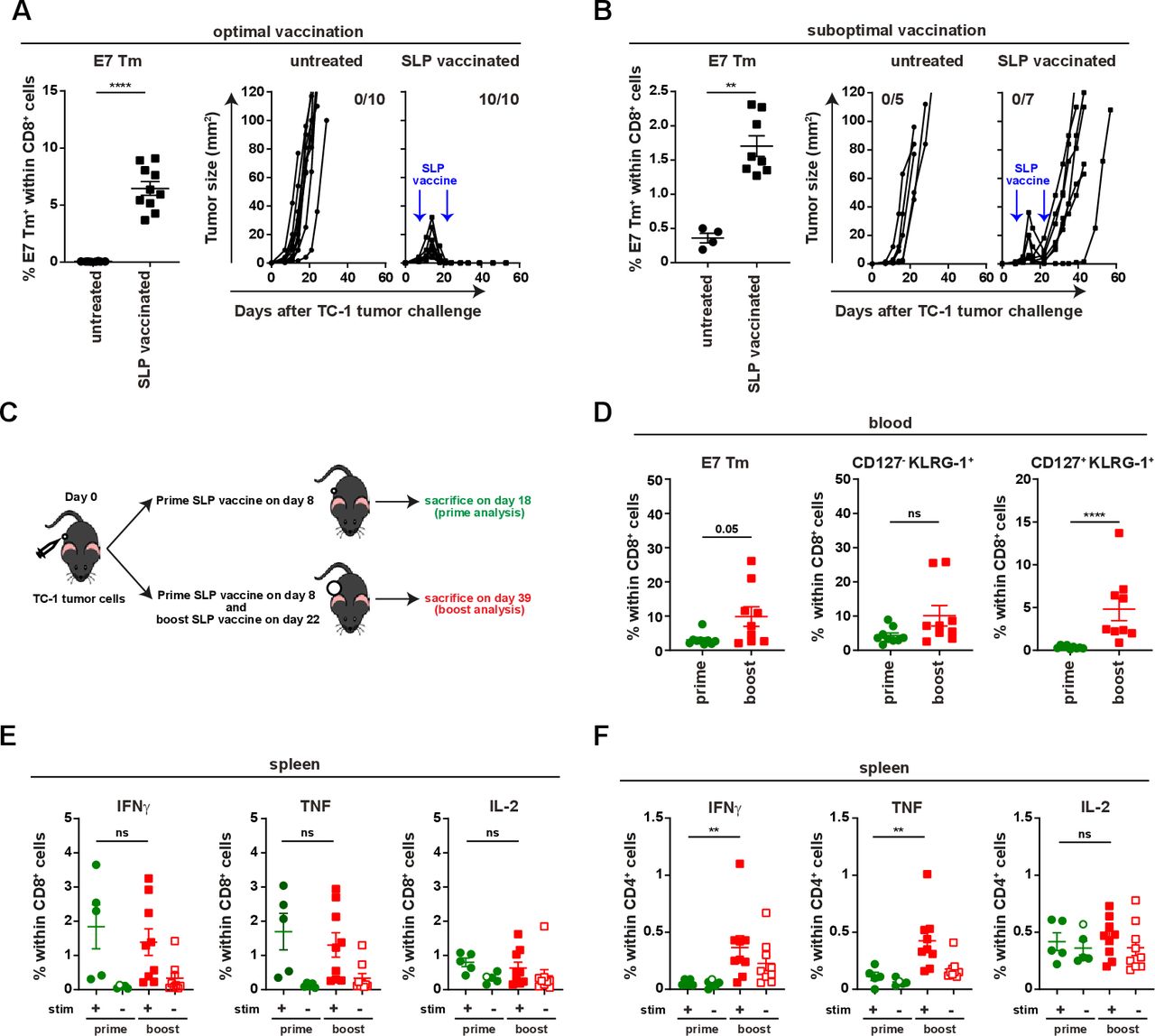
Vaccination with the HPV16 E743-77 SLP, containing both a CTL epitope and a helper T (Th) epitope, and CpG resulted in a strong T-cell response and the full eradication of almost all established TC-1 tumors in mice receiving tail-base vaccination, hereby reaching draining lymph nodes at both flanks (optimal vaccine condition) (figure 1A and online supplementary file 3A,B). In contrast, vaccination in the contralateral flank (suboptimal vaccine condition)—resulting in a lower E7-specific T-cell response—still induced tumor regression in all mice between days 16 and 22 but from day 28 onwards the tumors recurred (figure 1B and online supplementary file 3A–C). Amplification of the tumor-specific T-cell response by booster vaccination at day 22 could not prevent tumor recurrence (figure 1B–D). This was not due to the incapacity of tumor-bearing animals to respond to this booster vaccination as the magnitude of circulating tumor-specific (E7)-specific CD8+ and CD8+CD127+KLRG-1+ effector T cells increased in blood (figure 1D and online supplementary file 3D–E). In addition, splenic and lymph node-derived T cells were fully capable to produce IFNγ, tumor necrosis factor (TNF) and IL-2 after E7 stimulation (figure 1E and online supplementary file 4A,B). Moreover, the numbers of splenic CD4+ IFNγ, TNF and IL-2 producing T cells were increased after booster vaccination (figure 1F and online supplementary file 4C,D). Thus, a suboptimal vaccine-induced tumor-specific T-cell response fosters therapy resistance. Since the suboptimal vaccine condition clearly mimics the clinical situation, it provides an excellent model to study acquired resistance to immunotherapy.

Booster vaccination amplifies the E7-specific response but does not prevent relapse. Mice were injected with TC-1 tumor cells on day 0 and vaccinated with prime and boost SLP vaccine on day 8 and 22 subcutaneously in tail base (optimal vaccination) (A) or contralateral flank (suboptimal vaccination) (B). Percentage of E7 Tm+ cells within CD8+ cells and tumor outgrowth of untreated mice and SLP vaccinated mice that received the vaccination in optimal (A) or suboptimal (B) vaccination. (C) The scheme of the experiment shown in D–F. (D) The percentage of E7 Tm+, CD127-KLRG-1+, CD127+KLRG-1+ CD8+ T cells in blood at day 16 (prime analysis) and day 36 (boost analysis). (E, F) The percentage of IFNγ+, TNF+ and IL-2+ cells within CD8+ T cells (E) or CD4+ T cells (F) in spleen. Data presented in A, B, D–F are mean±SEM, and statistical analysis was performed using Mann-Whitney U test. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. IFNγ, interferon-γ; IL-2, interleukin 2; ns, not significant; TNF, tumor necrosis factor.
Classical immune escape mechanisms do not play a role in tumor recurrence
Loss of tumor antigen or major histocompatibility complex (MHC) I expression can mediate immune resistance.23 24 However, all the relapsed tumors retained the expression of E7 and H2-Db, which presents the E749-57 T-cell epitope, at comparable levels to the TC-1 tumor cell line and tumor cells isolated from both untreated and relapsed tumors were efficiently killed by CD8+ T cells isolated from vaccinated mice but not by T cells isolated from non-vaccinated mice (online supplementary file 5A–C).
Another obvious immune escape mechanism is formed by the PD-L1/PD-1 inhibitory pathway.25 26 Vaccination of TC-1 tumor-bearing mice resulted in a strong increase in the number of intratumoral leukocytes including CD8+ T cells and myeloid cells.27 Among these leucocytes, the number of highly activated, type 1 cytokine-producing CD8+ T cells expressing several coinhibitory molecules increased (online supplementary file 5D–F). This coincided with a lower percentage of TC-1 tumor cells expressing the proliferation marker Ki-67 (online supplementary file 5G) suggesting that the T cells still exert their function. Comparison of PD-1, TIM-3 and LAG-3 expression on CD8+ T cells including tumor-specific T cells revealed that almost all intratumoral CD8+ T cells (~90%–100%) expressed PD-1 both at the start of the regression and at the time of recurrence (online supplementary file 6A,B). We previously demonstrated that blockade of NKG2A did not prevent TC-1 recurrences.28 PD-L1 and PD-L2 levels were not increased on recurrent tumor cells but intratumoral myeloid cells showed high PD-L1 expression during tumor regression, which may facilitate the subsequent relapse (online supplementary file 6C,D). Nevertheless, recurrence could not be prevented by using PD-1 or PD-L1 blocking antibodies either before or at the time of regression (online supplementary file 6E,F).
Vaccine-resistant recurrent tumors display impaired CD8+ T cell infiltration
Most likely, tumor cells deployed other means to escape the immune response. Therefore, we quantified the different subsets of immune cells in the tumor microenvironment at the time of tumor regression (day 18) or relapse (day 39), when there was no overt difference in the sizes of regressing and relapsed tumors (online supplementary file 7A–C). Overall leucocyte infiltration was strongly reduced and altered in composition in the relapsed tumor leading to a near absence of CD8+ T cells, including E7-specific CD8+ T cells (online supplementary file 8A). The percentage of CD4+ T cells and Tregs, both present in low quantities among the tumor infiltrating immune cells, was not altered (online supplementary file 8C). Interestingly, the cytokine production of the intratumoral CD8+ T cells in the recurrent tumors, although present at much lower numbers, was similar to that of the CD8+ T cells in regressing tumors while the percentage of type 1 cytokine-producing CD4+ T cells was increased (online supplementary file 8D). The significantly lower presence of CD8+ T cells in recurrent tumors could reflect a lower capacity of tumors to attract these T cells and/or a failure of the T cells to proliferate and expand locally. To explore potential defects in the attraction of T cells to the tumor, the RNA expression of 84 chemokines and chemokine receptors from all cells present in the tumors from non-treated, regressed and recurrent tumors was measured (online supplementary file 9). This showed that T cell-attracting chemokines, including CXCL9, were among the abundantly expressed cytokines in regressing tumors but absent in recurrent tumors. In addition, the expression of its receptor CXCR3 appeared lower on the relapsed tumor-infiltrating CD8+ T cells, although not significant (online supplementary file 10A). However, antibody-mediated blocking of CXCR3 delayed the onset of tumor regression but did not negatively influence overall survival of the mice (online supplementary file 10B, C), indicating that CXCL9 was not absolutely required. As we previously showed that tumor cells can produce several chemokines after activation by IFNγ-producing tumor-infiltrating T cells,27 29 the lower expression of chemokines in relapsed tumors most likely is a reflection of reduced numbers of tumor-infiltrating immune cells and not the cause for immune escape. Indeed, TC-1 tumor cells from relapsed tumors were still responsive to stimulation in vitro with IFNγ and TNF and produced several cytokines (online supplementary file 10D) indicating that these signaling pathways were still intact. In addition, the capacity of the intratumoral T cells to locally expand was assessed by their expression of Ki-67 showing that the percentage of CD8+ T cells expressing Ki-67 is lower in recurrent than in regressing tumors (online supplementary file 8B). To overcome this problem, we aimed to amplify the T-cell costimulatory signals OX40 or 4-1BB30–32 by administration of agonistic antibodies at the time of regression. This did not prevent tumor recurrence or improve the survival of vaccinated mice, and further addition of PD-1 blockade had no effect (online supplementary file 11A–C).
Escaped tumors display a changed transcriptome
Next, we investigated the alterations in cancer cells using RNA sequencing on flow cytometry sorted fractions of cancer cells derived from untreated and relapsed tumors. Gene Set Enrichment Analysis of the Hallmark gene set collection17 showed differential expression of 160 of the 2086 genes analyzed between untreated and relapsed samples. Top 100 differentially expressed genes are shown in online supplementary file 12. In the relapsed tumors, strong upregulation of epithelial-mesenchymal transition (EMT) genes and downregulation in the TNF and p53 pathways was observed (figure 2A,B; online supplementary file 13). It has been extensively shown that several signaling pathways, including TGF-β and Wnt/β-catenin, play a role in EMT,33 34 hence we confirmed the transcriptome data at the protein level. While the expression of cytoplasmic and nuclear β-catenin was about twofold higher in the stroma of recurrent tumors (online supplementary file 14A), the expression of TGF-β was strongly increased in a layer of cells—presumably fibroblasts—surrounding the tumor (figure 2C) and increased quantities of TGF-β were measured in the supernatant of ex vivo recurrent tumors (figure 2D). In accordance with the down regulation of the p53 pathway, the expression levels of mdm2, ccng1, ei24 and pidd were lower in recurrent tumors (figure 2E). Recently, TGF-β production and signaling in fibroblasts was shown to restrict T-cell infiltration of tumors,35 36 however, administration of TGF-β-blocking antibodies from the regression phase onwards did not prevent tumor recurrence or altered survival (figure 2F and online supplementary file 14B). Similarly, activation of p53 signaling by using the MDM2 small-molecule antagonist RG7112 alone or in combination with TGF-β-blocking antibody had no effect (figure 2G and online supplementary file 14C). Altogether, these data show that tumor cells had become differently wired as a consequence of a non-curative T-cell attack, but the changes in the TGF-β and p53 pathways were not the underlying cause for therapy resistance.

Local recurrences display an altered transcriptome. (A–E) mice were injected with TC-1 tumor cells on day 0. Then, mice were vaccinated with prime SLP suboptimal vaccine on day 8 (regressed) or kept untreated (untreated) and were sacrificed on day 18. Another group of mice vaccinated with prime and boost SLP suboptimal vaccine on day 8 and 22 were sacrificed at the time of relapse on day 39 (relapsed). (A, B) RNA seq and gene set enrichment analysis of tumor cells sorted from untreated tumor-bearing mice (day 18) or mice with relapsed tumors (day 39). (C) TGFβ score of the rim of the tumor microenvironment from untreated and relapsed tumor-bearing mice. (D) Measurement of TGFβ production by ELISA from the cells present in the tumor microenvironment of the untreated mice or SLP suboptimal vaccinated mice at the time of the regression (day 18) or relapse (day 39). (E) RNA expression of mdm2, ccng1, ei24 and pidd genes by qPCR from tumor cells sorted from untreated mice (day 18) or SLP suboptimal vaccinated mice at the time of the relapse (day 39). (F–G) Survival of the mice treated with prime and boost SLP suboptimal vaccination alone or in combination with TGFβ-blocking antibody (F) and/or RG7112 (G). Mice were injected with TC-1 tumor cells on day 0. Then, mice were vaccinated with prime and boost SLP suboptimal vaccine on day 8 and 22. TGFβ neutralizing antibody and RG7112 were administered from day 20 till 32 as described in methods. data presented in C–E are mean±SEM, and statistical analysis was performed using Mann-Whitney U test. statistical analysis shown in G and F is determined by a log-rank (Mantel-Cox) test. *P<0.05. ns, not significant; TGFβ, transforming growth factor-β.
Non-responsiveness relates to impaired inflammatory myeloid cell infiltration in mice and man
Cure of TC-1-bearing mice requires the influx of tumor-specific CD8+ T cells and that of matured (MHC class II+) inflammatory (Ly6Chi) phagocytic myeloid cells,27 37 which kill tumor cells by TNF and phagocyte them.38 Whereas the percentage of myeloid cells was unaltered between regressed and relapsed tumors, the composition of these intratumoral cells, comprizing inflammatory (Ly6Chigh) MHC class II+ myeloid cells, tumor associated macrophages (Ly6Clow cells) and neutrophils (Ly6G+), was changed in favor of the non-inflammatory Ly6ClowF4/80hi/low cells (figure 3A–C and online supplementary file 15A), and coincided with higher levels of IL-6 (online supplementary file 15B), both of which may foster local immune suppression. To test this, we depleted Ly6Clow cells using the CSF1R inhibitor PLX3397 (PLX) from day 20, 4 days after the start of regression on day 16. As expected,27 this depleted intratumoral but not splenic macrophages and had no effect on the vaccine-induced CD8+ T-cell response (online supplementary file 15C,D). However, neither PLX (online supplementary file 15E,F), nor blockade of IL-6 in combination with TGF-β blocking antibodies (online supplementary file 15G,H) had an effect on tumor recurrence. This not only showed that the Ly6Clow myeloid cell population was not required for recurrence, but also implied that the inflammatory myeloid cell population, known to assist tumor killing and to be affected by PLX treatment,27 was not present. Thus, it appeared that acquired resistance was associated with a lack of inflammatory myeloid cell infiltration. To validate these findings in the human setting, we analyzed the immune microenvironment of patients with HPV16-induced vHSIL, after their systemic HPV16-specific T-cell response had been amplified by HPV16 SLP vaccination (n=24).20 Interestingly, clinical responders (defined as≥50% lesion regression 12 months after vaccination) displayed significantly higher infiltration with CD14+CD68+/-CD163- inflammatory macrophages after vaccination when compared with non-responders (figure 3D). This influx of CD14+ inflammatory macrophages coincided with significant T-cell infiltration, primarily type I cytokine-producing (Tbet+) T cells (figure 3D). Interestingly, several of the non-responding lesions are infiltrated with T cells postvaccination, but hardly with inflammatory myeloid cells. Thus, our data clearly showed that vaccine-resistance was associated with an impaired local inflammatory myeloid cell infiltration after vaccination, both in our mouse model and in patients.
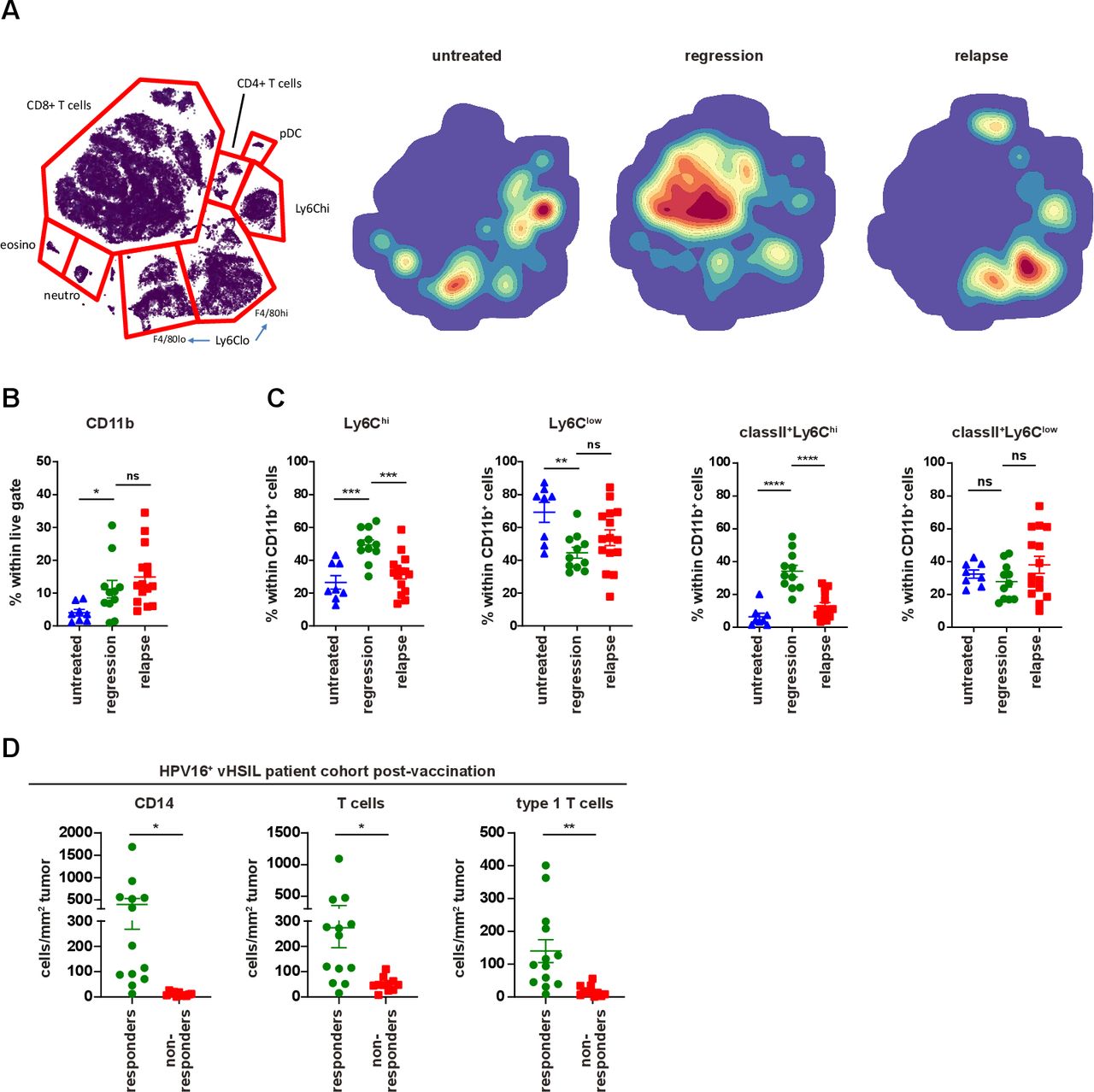
Response to vaccine is correlated with infiltration of inflammatory myeloid cells. Mice were injected with TC-1 tumor cells on day 0. Then, mice were vaccinated with prime SLP suboptimal vaccine on day 8 (regressed) or kept untreated (untreated) and were sacrificed on day 18. Another group of mice vaccinated with prime and boost SLP suboptimal vaccine on day 8 and 22 were sacrificed at the time of relapse on day 39 (relapsed). (A) H-SNE plots of total CD45+ immune infiltrates of TC-1 tumors of untreated, regressed or relapsed. Tumors were analyzed by mass cytometry followed by non-supervised clustering using Cytosplore as described in materials and methods. The relative contribution of major myeloid and lymphoid cell types is indicated in the H-SNE plot, as indicated. (B) The percentage of CD11b+ within live gate. (C) The percentage of Ly6Chi and Ly6Clow cells as well as classII+Ly6Chi and classII+Ly6Clow within CD11b+ cells on day 18 at the time of regression (regression) or at the time of relapse on day 39 (relapse). The scheme is shown in online supplementary file 7A. (D) Immune microenvironment of vHSIL patients 3 months after therapeutic vaccination by HPV16 SLP, separated for responders (n=13) and non-responders (n=11). Depicted are the number of infiltrating CD14+ macrophages (CD14+CD68-/+CD163-/+), CD3+ T cells (CD3+CD8-/+Foxp3-) and type one cytokine producing (Tbet+) T cells (CD3+CD8-/+Foxp3-Tbet+) per mm2 tumor post vaccination. data presented are mean±SEM, and statistical analysis was performed using unpaired t-test. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. ns, not significant; vHSIL, vulvar high-grade squamous intraepithelial lesions.
Secondary immune resistance is caused by a tumor cell-imprinted failure to attract myeloid effector cells
Until now, we had investigated secondary resistance to vaccination in a setting of local recurrence, the actual cause which may be obscured due to several different biological processes such as immune cell infiltration and tumor regression, wound healing and oncogenic pathway rewiring in tumor cells, all acting in a relatively small time window and in the same anatomical location during the regrowth of tumors. To interrogate the major mechanism underlying therapy-induced secondary resistance, we tested if TC-1 tumor cells isolated at the time of the relapse respond to the same vaccination protocol when injected in a naïve host. Flow cytometry-sorted live TC-1 tumor cells were reinjected in two groups of naïve mice, one of which received the vaccine on day 8 and 22 while the other was left untreated (figure 4A). The reinjected tumor cells from untreated mice regressed on SLP vaccination, whereas reinjected recurrent tumor cells did not respond at all (figure 4B). This implied that the relapsed phenotype was imprinted into the cancer cells. Analysis of the tumor immune microenvironment at day 15 revealed that the tumors originating from reinjected recurrent tumor cells displayed a strong reduction in the number of CD45+ tumor-infiltrating immune cells (figure 4C). Remarkably, this was not due to a lack of CD8+ T-cell infiltration as their number, type 1 cytokine production, Ki-67 expression and intratumoral distribution, was similar to what is seen in the vaccine-responsive tumors originating from TC-1 tumor cells (figure 4D–G). Instead, the numbers of tumor-infiltrating CD11b+, matured (MHC class II+) and inflammatory (Ly6Chigh) myeloid cells were decreased (figure 5A). This included a decrease in CD11b+F4/80+iNos+ (M1-type) macrophages CD11b+F4/80-CD11c+ cells and plasmacytoid dendritic cells (pDCs) (figure 5B). In addition, the percentage of intratumoral CD11b+F4/80+Egr2+ (M2-type) macrophages and Ly6G+ neutrophils were somewhat increased in the resistant tumors (figure 5C). Consistent with this change, an overall reduction in the expression of several IRF was found in the microenvironment (online supplementary file 16A). Furthermore, the expression of IRF-1 in CD11b+ myeloid cells, important for the commitment of M1 macrophages,39 was reduced (figure 5D). Moreover, in spite of the presence of similar numbers of intratumoral type 1 cytokine-producing CD8+ T cells, the expression level of TNF was also strongly decreased in the reinjected recurrent tumors (figure 5E), suggesting that this was produced by the myeloid effector cells and potentially explaining the observed reduction in TNF-signaling in the transcriptome of relapsed tumor cells (figure 2A,B). Thus, secondary immune resistance is not due to a lack of intratumoral functionally active CD8+ T cells but is associated with an impaired influx of inflammatory mature myeloid effector cells after vaccination.

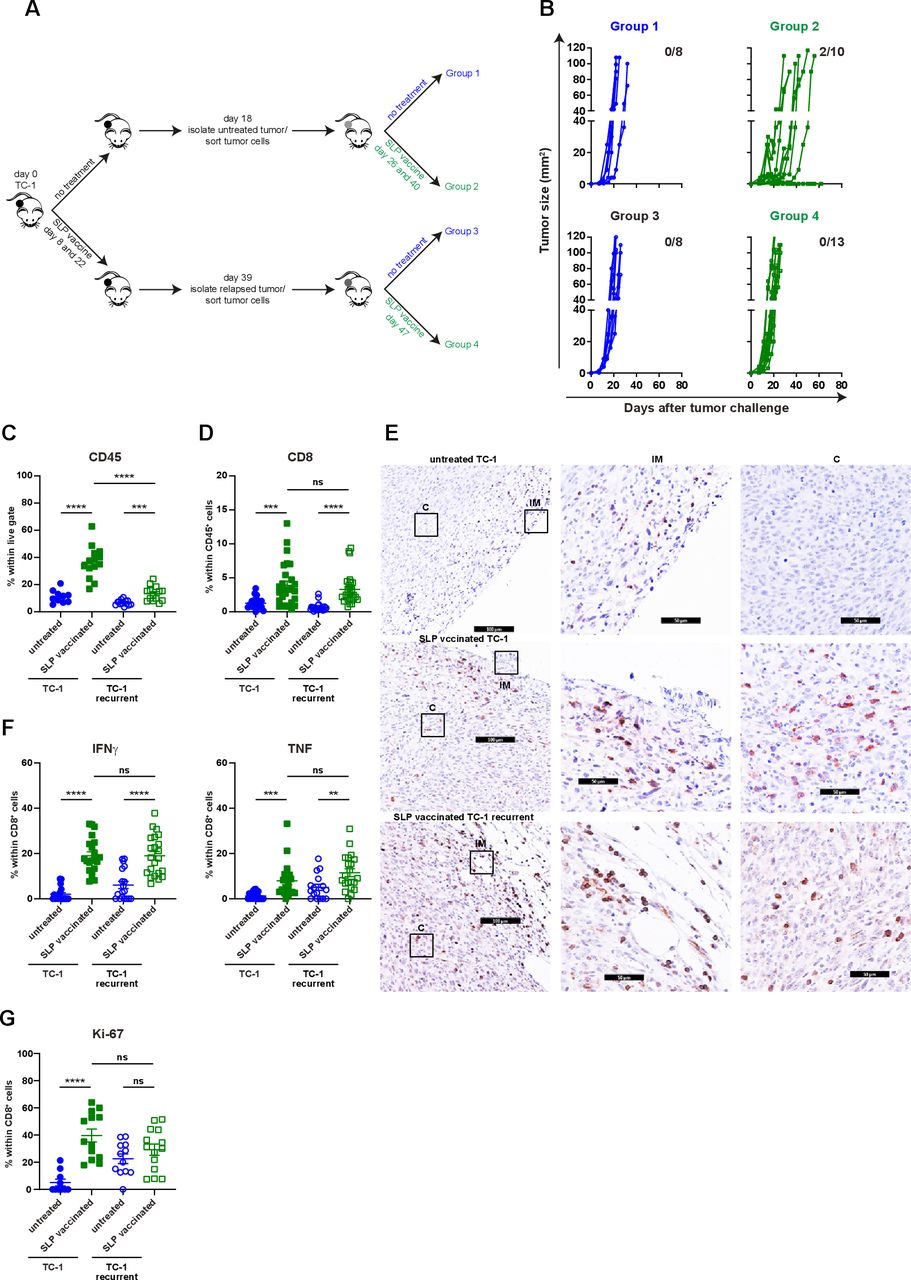
Non-Curative immunotherapy drives the development of immune resistant tumors. (A) Scheme of the experimental procedure. In brief, mice were injected with TC-1 tumor cells on day 0. Then, mice were suboptimal vaccinated with SLP vaccine on days 8 and 22 or kept untreated. Untreated mice were sacrificed on day 18. Tumor cells were sorted and injected back into naïve mice. these mice received suboptimal vaccination on days 26 and 40 or kept untreated. Similarly, suboptimal vaccinated mice were sacrificed on day 39, tumor cells were sorted and injected back into naïve mice, which were suboptimal vaccinated on days 47 or left untreated. (B) Tumor outgrowth of the mice shown in (A). The number of tumor-free mice from total is indicated. Data are pooled of two experiments. (C–G) The scheme of the experiment is shown in (A). The mice (group 1–4) were sacrificed on day 15 post tumor challenge. (C) The percentage of CD45+ cells within live gate in the tumor microenvironment. (D) The percentage of intratumoral CD8+ T cells within CD45+ cells. (E) The representative immunohistochemistry images of CD3+ T cells in tumors of untreated TC-1, suboptimal SLP-vaccinated TC-1 and suboptimal SLP-vaccinated TC-1 recurrent tumor-bearing mice (C: center, IM: invasion margin). (F) IFNγ and TNF production of intratumoral CD8+ T cells. (G) The percentage of Ki67+ intratumoral CD8+ T cells. data presented in C, D, F and G are mean±SEM, and statistical analysis was performed using unpaired t-test. **P<0.01; ***P<0.001; ****P<0.0001. IFNγ, interferon-γ; ns, not significant; TNF, tumor necrosis factor.
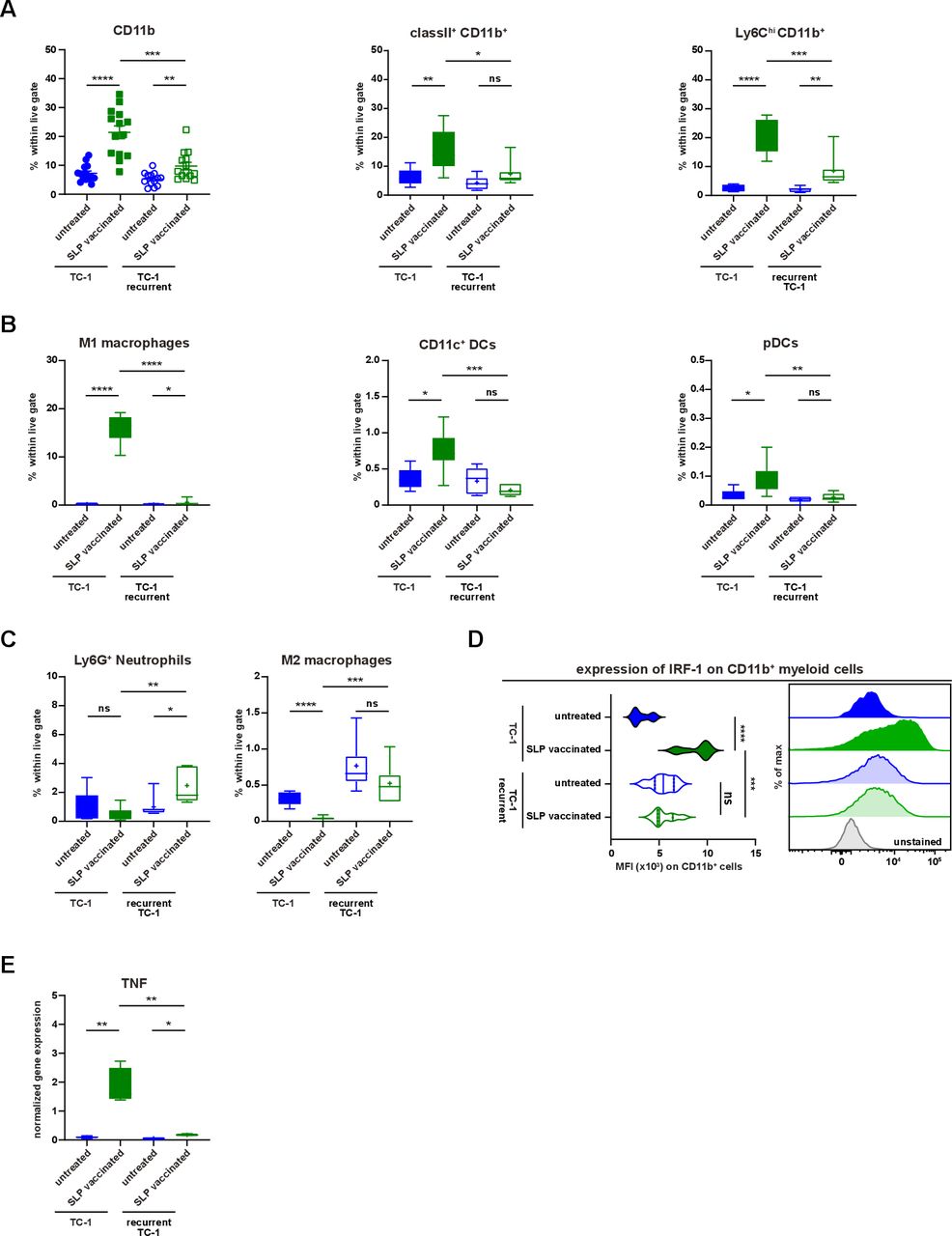
Recurrent tumors display impaired inflammatory myeloid cell infiltration. (A–E) The scheme of the experiment is shown in figure 4A. The mice (group 1–4) were sacrificed on day 15 post-tumor challenge. (A) The percentage of CD11b+, classII+CD11b+ and Ly6ChiCD11b+ cells within live gate in the tumor microenvironment of untreated and suboptimal SLP-vaccinated TC-1 and TC-1 recurrent tumor-bearing mice. (B) The percentage of M1-type macrophages, CD11c+ DCs and pDCs within live gate. (C) The percentage of intratumoral Ly6G+ neutrophils and M2-type macrophages within live gate. (D) The expression level of IRF-1 on intratumoral CD11b+ myeloid cells. The representative histogram is shown on the right. (E) The normalized gene expression level of TNF in the tumor microenvironment of untreated and SLP-vaccinated TC-1 and TC-1 recurrent tumor-bearing mice. Data presented are mean±SEM in the scatter plot and min to max (mean shown as +) in box and whiskers plots. Statistical analysis was performed using unpaired t-test. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. ns, not significant; TNF, tumor necrosis factor.
Reprogramming the microenvironment sensitizes escaped tumors to immunotherapy
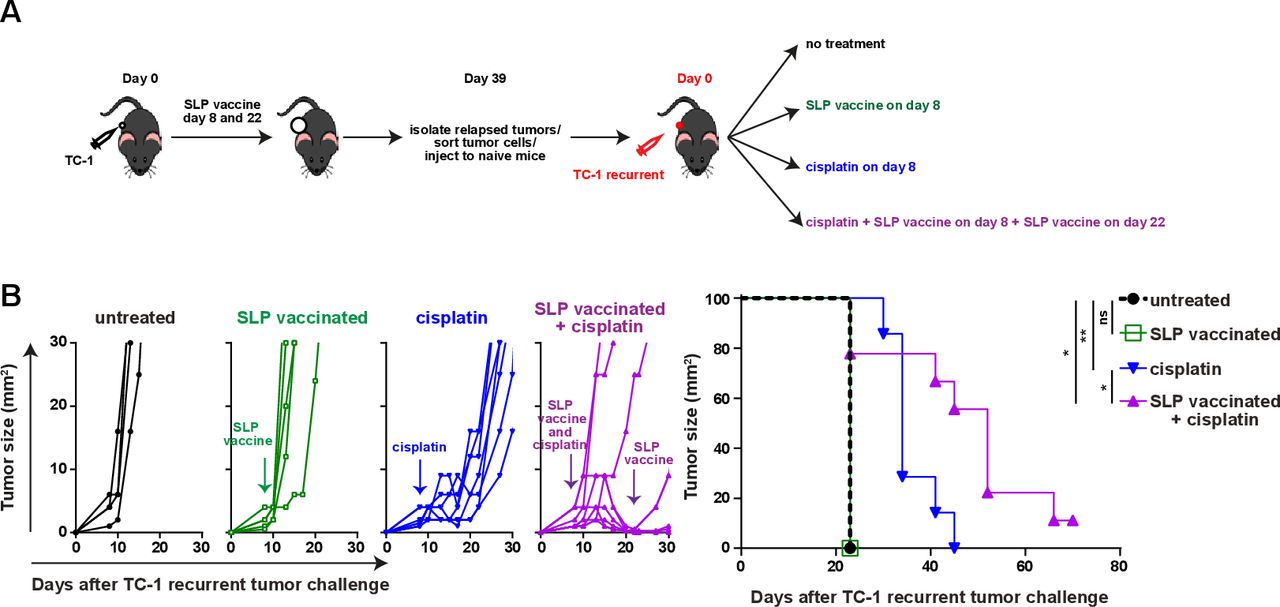
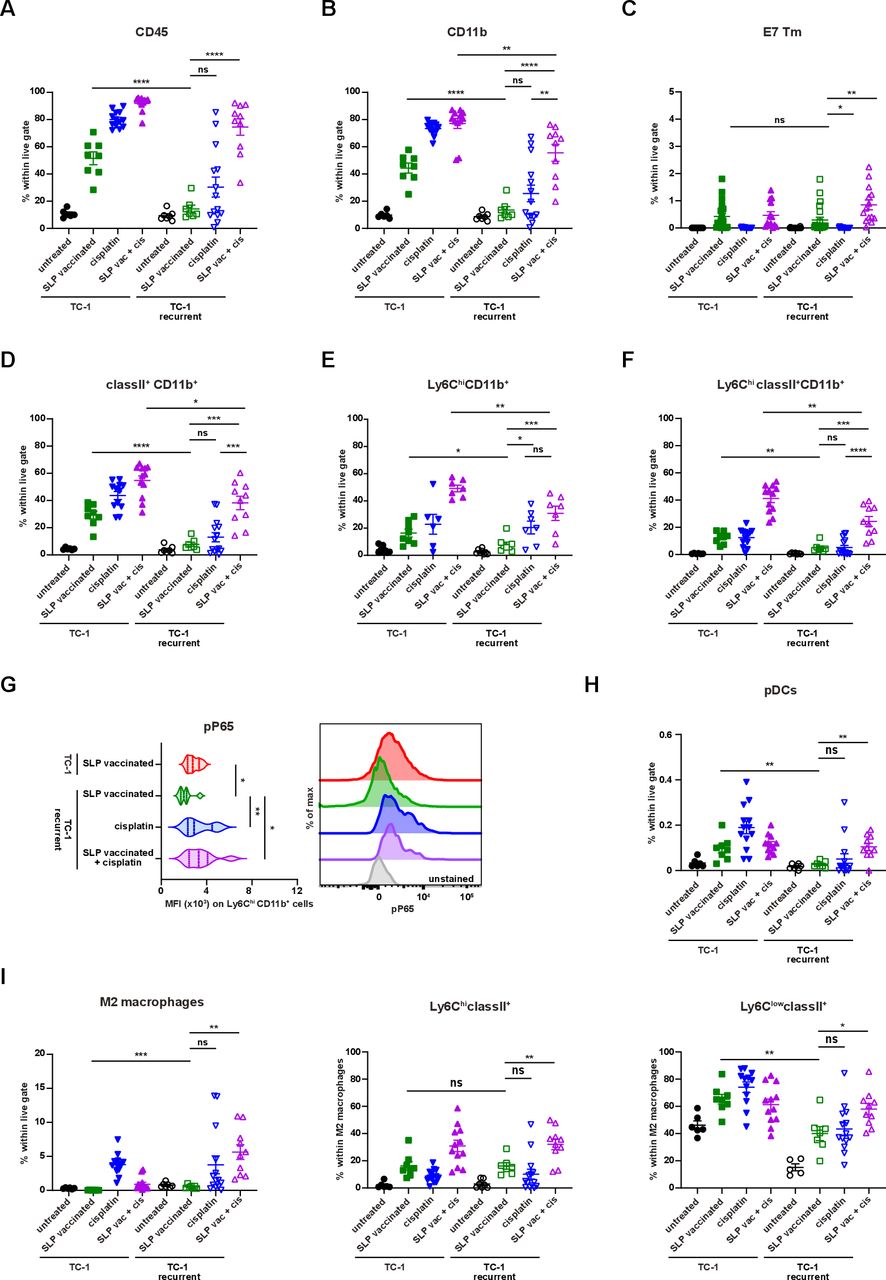
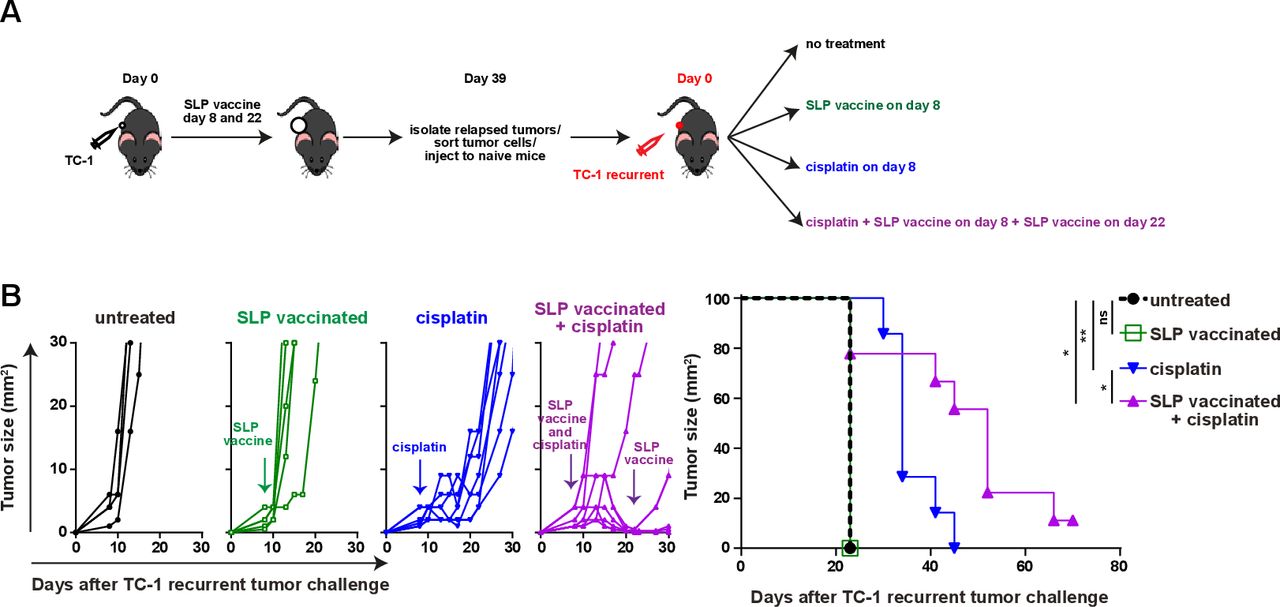
To test if a lack of tumor-infiltrating inflammatory myeloid cells facilitated vaccine resistance in this setting, we made use of cisplatin as it induces an influx of matured (MHC class II+) inflammatory (Ly6Chi) phagocytic myeloid cells27 37 (online supplementary file 17A). Especially, the combination of cisplatin and vaccination stimulated an increased CD45+ immune cell infiltration in the reinjected TC-1 recurrent tumors. Due to an influx of CD11b+ and CD11b-CD11c+ myeloid cells expressing MHC class II and/or Ly6C (figure 6A–F; online supplementary file 17B-D), the percentages of the inflammatory mature CD11b+ myeloid cells in the reinjected TC-1 recurrent tumors reached similar levels as observed in normal TC-1 tumors after vaccination without cisplatin treatment. The level of phosphorylated p65, as a witness of proinflammatory NF-Kβ pathway activation, was also increased in the reinjected TC-1 recurrent tumor-infiltrating Ly6ChiCD11b+ myeloid cells, to the same level as detected in these cells from normal TC-1 tumors after vaccination (figure 6G). Detailed analysis of the myeloid cell composition revealed that the combination treatment drove the influx of predominantly CD11b-CD11c+ pDCs and MHC class II+ M2-type macrophages and not CD11b-CD11c+MHCclasssII+CD103+ (cDC1) and CD11b+CD11c+F4/80- (cDC2) (figure 6H,I; online supplementary file 17E). In addition, the combination treatment restored the percentage of tumor-resident (Ly6Clow) matured M2-type macrophages expressing MHC class II in reinjected TC-1 recurrent tumor, to the levels seen in normal TC-1 tumors after vaccination (figure 6I). The combination treatment, but not cisplatin alone, also resulted in an increased infiltration with CD4+ and CD8+ T cells in TC-1 recurrent tumors (online supplementary file 17F), the percentage of which exceeded the number of HPV-specific T cells (figure 6C). Based on the fact that cisplatin does not induce immunogenic cell death40 and did not drive overt CD4+ and CD8+ T cell infiltration, it is likely that the inflamed tumors attracted many bystander CD8+T cells,41 these non-HPV-specific CD8+T cells are unlikely to have a major contribution to the clinical response in the combination treatment of TC-1 recurrent tumors. Finally, we analyzed if this apparent restoration of the mature and inflammatory intratumoral myeloid cell population also reinstalled the sensitivity of the reinjected TC-1 recurrent tumors to T-cell therapy. Indeed, while cisplatin only delayed tumor outgrowth, combination of cisplatin with SLP vaccination induce full tumor regression in the majority of mice (figure 7A,B), similar to what was seen with the original TC-1 tumor (figure 1B), and significantly improved the survival in reinjected TC-1 recurrent tumor-bearing mice. Thus, the major mechanism underlying secondary immune resistance after vaccination was the lack of sufficient intratumoral mature myeloid effector cells after vaccination.

Reprogramming the microenvironment enhances myeloid cell infiltration. (A–I) The scheme of the experiment is shown in online supplementary file 17A. (A–F) The percentage of intratumoral CD45+ cells (A), CD11b+ (B), E7 Tm+CD8+ T cells (C) classII+CD11b+ (D), Ly6ChiCD11b+ (E) and Ly6ChiclassII+CD11b+ (F) within live gate of untreated, suboptimal SLP-vaccinated, cisplatin treated and combined cisplatin and suboptimal SLP-vaccination treated TC-1 and TC-1 recurrent tumor-bearing mice. (G) The expression level of pp65 on Ly6ChiCD11b+ myeloid cells. The representative histogram is shown on the right. (H–I) The percentage of intratumoral pDCs (H) and M2-type macrophages within live gate, Ly6ChiclassII+ and Ly6ClowclassII+ within M2 macrophages (I) of untreated, SLP-vaccinated, cisplatin treated and combined cisplatin and SLP-vaccination treated TC-1 and TC-1 recurrent tumor-bearing mice. data presented are mean±SEM, and statistical analysis was performed using unpaired t-test. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. ns, not significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reprogramming the microenvironment overcomes acquired resistance. (A) Scheme of the experiment. In brief, mice were injected with TC-1 tumor cells on day 0 and suboptimal vaccinated with SLP vaccine on days 8 and 22. when the tumor relapsed on day 39, mice were sacrificed and tumors were harvested and tumor cells were isolated. sorted tumor cells (TC-1 recurrent) were injected into naïve mice and treated with SLP vaccine, cisplatin, the combination or kept untreated. (B) Tumor outgrowth and survival of the mice as shown in (A). Differences in survival was determined by a log-rank (Mantel-Cox) test. *P<0.05; **P<0.01;. ns, not significant.
Discussion
In our experimental model for acquired resistance, the induction of suboptimal tumor-specific T-cell response still induces regression of growing tumors but fails to wipe out all tumor cells, leading to immune escape and tumor relapse in accordance with the principles of cancer immunoediting.42 We excluded extrinsic mechanisms known to confer primary or secondary resistance to immunotherapy, such as Tregs, suppressive macrophages, TGF-β or IL-6 and showed that the intratumoral CD8+ T cells were still functional. Moreover, we excluded a role for the well-known intrinsic mechanisms associated with tumor antigen expression, alterations in the antigen processing pathway, checkpoint expression, resistance to killing or to IFNγ and TNF-signaling, all of which are required for activated CD8+ T cells to exert their anti-tumor function. The lowered CD8+ T cell infiltration observed in relapsed tumors did not form the underlying mechanism for escape, as reinjection of escaped tumor cells into naïve mice revealed that these tumors were still therapy resistant despite a strong influx with functionally active CD8+ T cells. Instead, the escape mechanism fostering resistance to immunotherapy was associated with a tumor cell imprinted incapacity to attract matured MHC class II+ myeloid cells, in particular intratumoral pDCs, and macrophages, which was observed both during relapse and in the reinjected escape tumors. Although we are unable to pinpoint the molecular signal of this deficit at this time, potentially because it may involve an altered cross talk between cancer and stromal cells, our data show that reinstating intratumoral inflammation restored their influx and sensitized escaped tumors to vaccine-induced tumor regression. This newly identified acquired immune escape mechanism may play a role in many types of tumors, because not only the immunotherapy-mediated control of TC-1 tumors27 38 but also that of YUMMER1.7 and B16/F10 melanoma,43 44 CT26 colon cancer, 4T1 breast carcinoma and mSCC1 squamous cell carcinoma45 requires the presence of CD8+ T cells and inflammatory matured MHC class II+ myeloid cells. Similarly, a combination of tumor-specific T cells and inflammatory MHC class II+ macrophages is required to prevent the development of hepatocellular cancer in mice.46 The requirement of both cell types in the tumor microenvironment fits well with the concept of programmed cell removal. This process is known to regulate cancer cell survival and is a key step in the removal of cells undergoing programmed cell death, executed by phagocytic innate cells.47 It can be envisioned that the tumor-specific CD8+T cells deliver the first hit which is then complemented by inflammatory myeloid effector cells, as they possess in vivo phagocytic functions.38 45 Additionally, myeloid effector cells may aid in this process via their production of TNF and nitric oxide, which also delivers a cytotoxic hit to tumor cells.38 44 45 The influx of M2 rather than M1 macrophages in the reinjected cisplatin plus vaccine treated tumors does not contradict this concept, as M2 macrophages alike to their M1 counterparts can phagocytose cancer cells, although at a lower rate.48 Our study outcomes are also relevant for human cancers, the clinical outcome of which often is better when they are infiltrated with type 1 cytokine-producing T cells and myeloid cells.49 This includes HPV16-driven cervical cancer.50 Indeed, we observed that the regression of HPV16-induced pre-malignant lesions after therapeutic vaccination is associated with the influx of both T cells and inflammatory (CD14+) myeloid cells. Although T cells infiltrated the lesions of clinical non-responders at lower numbers than in clinical responders, the numbers of inflammatory myeloid cells remained very low. The absence of a coordinated CD8+ T cell and inflammatory myeloid cell infiltration was also found to act as primary resistance mechanism in patients with HPV16-induced premalignant lesions.20
In conclusion, we propose that loss of the capacity to attract innate myeloid effector cells into the tumor microenvironment is a mechanism leading to primary or secondary therapy-resistance. The recognition that also tumor-infiltrating myeloid cells are required for tumor regression during immunotherapy asks for a reappraisal of their tumoricidal and phagocytic activities and new studies on how to harness this.
Acknowledgments
The authors thank LJAC Hawinkels from the department of Gastroenterology and Hepatology, Leiden University Medical Center, Leiden, The Netherland for his input on the TGFβ measurement. The authors appreciate the help of Guillaume Beyrend from the Immunohematology and Blood Transfusion department at Leiden University Medical Center, Leiden, The Netherland for mass cytometry panel design and analysis.
References
Footnotes
CL and ZA contributed equally.
Contributors Conception and design: EBN, TVH, RA and SHvdB. Experiments in mice: EBN, CL, MJvE, JWK, TCvdS and SvD. Collection and analyses of human materials: ZA and SHvdB. Molecular and transcriptomic analyses: AFAST, AGJ, JO and NFCCdM. Mass cytometry: EBN, CL, ZA, MJvE, JWK, TCvdS, SvD, AFAST, AGJ, JO, NFCCdM, TVH, RA and SHvdB. Immunohistochemistry: CL, ER, NFCCdM. Flow cytometry: EBN, CL and MJvE. Analysis and interpretation of data: EBN, CL, ZA, MJvE, JWK, TCvdS, AGJ, JO, NFCCdM, TVH, RA and SHvdB. Writing, review, and/or revision of the manuscript: EBN, TVH, RA and SHvdB. Study supervision: TVH, RA and SHvdB.
Funding This work was supported by grant 2015-7824 from the Dutch Cancer Society (SHvdB, TVH and RA) and a Biomedical Sciences Plus PhD grant from Leiden University Medical Center (EBN).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All animal experiments were approved by the Animal Experiments Committee of LUMC and were executed according to the animal experimentation guidelines of LUMC in compliance with the guidelines of Dutch and European committees.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.