Article Text

Abstract
Neoadjuvant chemo(radio)therapy is part of the established standard of care in cancer treatment; neoadjuvant application of immunotherapy, however, is only performed within recent trials. Combination of programmed cell death protein 1 and cytotoxic T lymphocyte antigen 4 blockade shows promising results with high pathologic response rates in the neoadjuvant setting and a very low relapse rate in the responding patients. In addition, neoadjuvant administration allows direct determination of treatment efficacy within the individual patient, and offers easy access to paired tumor material, both pretherapy and post-therapy, thus facilitates the rational development of new combinations driven by preclinical analyses. Patient-derived human tumor explant systems such as a recently developed human patient-derived tumor fragment platform can provide an additional tool to further rationalize the development of new treatment combinations. We will discuss neoadjuvant immunotherapy as a unique opportunity for rational trial design, the development of immune signatures for non-responding patients to steer clinical trial development, and the use of patient-derived ex vivo models to identify new personalized immunotherapy combinations. In this context, we propose the ‘Lombard Street Approach’, a back and forth approach of characterizing non-responders on neoadjuvant immunotherapy combinations, identifying promising new combinations for this group in the tumor fragment platform, and performing subsequently signature-driven small proof-of-concept combination trials. Repeating this approach with smaller and smaller groups of non-responders will step by step increase the percentage of patients benefiting from neoadjuvant immunotherapy in a rational and fast manner.
- melanoma
- immunotherapy
- translational medical research
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Neoadjuvant checkpoint inhibition - new aspects in an old concept
Neoadjuvant systemic therapy has an important role in cancer treatment, with the goal to improve local and distant tumor control, perform organ-sparing surgery and create the possibility of direct response evaluation.1 2 The response assessment allows to tailor postoperative treatment, as pathologic response has been associated with durable disease control and a favorable survival outcome in neoadjuvant chemotherapy in breast cancer.3 Recently, neoadjuvant immune checkpoint blockade has been proposed and tested in small conceptual trials. In preclinical models, T cell checkpoint blockade before surgery has been shown to induce a broader immune response by expanding a wider range of tumor-resident T cell clones compared with adjuvant administration, which also translates into long-term survival.4 This broader expansion of tumor-resident T cells has also been shown in patients with melanoma treated with neoadjuvant programmed cell death protein 1 (PD-1) plus cytotoxic T lymphocyte antigen 4 (CTLA-4) blockade.5
Pathologic response evaluation has been proposed as a strong outcome parameter and major pathologic response (MPR), defined as presence of 10% or less residual viable tumor cells after neoadjuvant therapy, is used regularly as surrogate outcome marker.6 7 While pathologic response rates are promising with neoadjuvant anti-PD-1 monotherapy in melanoma (30%–33% pathologic response rate),8 9 neoadjuvant combined CTLA-4 and PD-1 blockade induces high pathologic response rates of 74%–78% in melanoma,5 10 100% pathologic response in microsatellite instability-high colorectal carcinoma,11 and 44% MPR in bladder cancer.12 A more comprehensive overview of trials performed with different neoadjuvant combinations is provided in two recently published reviews.13 14
Strikingly, a very low relapse rate has been observed in patients achieving a pathologic response on neoadjuvant CTLA-4 and PD-1 blockade in patients with melanoma. So far, only 1 (1.4%) of 71 patients treated within the OpACIN and OpACIN-neo trials have relapsed (with a median follow-up of 36 and 24 months, respectively).15 16 Vice versa, patients not responding within the 6 weeks of neoadjuvant combination checkpoint inhibition have a very high risk of relapse (15 of 23 non-responders, 65%, have relapsed so far). A pooled analysis of six neoadjuvant melanoma trials showed that pathologic complete response (pCR) was associated with complete freedom from relapse within 10 months of follow-up after checkpoint inhibition, while this was not the case for patients with pCR after targeted therapy.7 Thus, pathological non-response on checkpoint inhibition seems a strong predictor for poor outcome. While pCR is an Food and Drug Administration accepted endpoint in neoadjuvant chemotherapy trials,3 this is yet to be established for pathologic response after neoadjuvant checkpoint inhibition. This is surprising, as the correlation of pathologic response with a favorable relapse-free survival (RFS) is much stronger for checkpoint inhibition than for targeted therapy and chemotherapy.7 17
Neoadjuvant therapy allows to fine-tune treatment for each patient, as treatment can be subsequently de-escalated in responding patients and escalated in non-responding patients. With the promise of pathologic response as good predictor for outcome, the PRADO trial was developed to answer if extensive surgery can be safely omitted in patients achieving a MPR after neoadjuvant combined CTLA-4 and PD-1 blockade, and whether RFS in non-responding patients can be improved by adding adjuvant therapy (radiotherapy and systemic therapy). The first results show again a high pathologic response rate of 71%, and patients with MPR in whom extensive surgery was omitted had reduced surgical morbidity and higher quality of life scores.18 The RFS data are still immature with a median follow-up of less than a year.
Neoadjuvant therapy also provides an attractive setting to develop new treatment regimens. Earlier stage disease is thought to be more responsive to immunotherapy due to a lower tumor burden, less immune escape mutations developed and less systemic immune suppression.19–21 In addition, early stage patients are more homogenous patient populations (eg, all having normal lactate dehydrogenase and C-reactive protein levels and lymphocyte counts) allowing to generate meaningful results in smaller trials. As patients commonly receive only one or two cycles of therapy, treatment time is short, which leads to faster completion of clinical trials. As a unique feature of neoadjuvant therapy, treatment efficacy can be directly assessed within the individual patient. Due to the subsequent surgery, tumor tissue can easily be collected at pretherapy and post-therapy time points. This offers an attractive opportunity for translational research to develop both predictive biomarkers and new treatment strategies, as well as to understand resistance mechanisms in non-responding tumors. Therefore, the neoadjuvant therapy setting provides the ideal framework to establish new personalized immunotherapy combinations that are driven by preclinical analyses.
For example, the interferon-gamma (IFN-γ) signature22 was identified as strong biomarker for response in the OpACIN-neo trial, associated with a higher response rate of 78% versus 58% for high versus low IFN-γ signature tumors at baseline, which resulted in an impressive lower relapse rate of 6% versus 25%, respectively.23 Combining the IFN-γ signature with tumor mutational burden (TMB) improved the baseline predictive value further; patients with both high IFN-γ and TMB had a 100% response rate to neoadjuvant CTLA-4 and PD-1 blockade, while only 37% responded in patients who scored low on both IFN-γ and TMB.23 Thus, the combination of IFN-γ signature and TMB can be used to upfront identify patients with a very high likelihood to respond to neoadjuvant CTLA-4 plus PD-1 blockade. Other examples of signatures are the T cell signature24 and the Batf3+ dendritic cell signature25; both have been shown to correlate with good clinical outcome after neoadjuvant CTLA-4 and PD-1 blockade.26
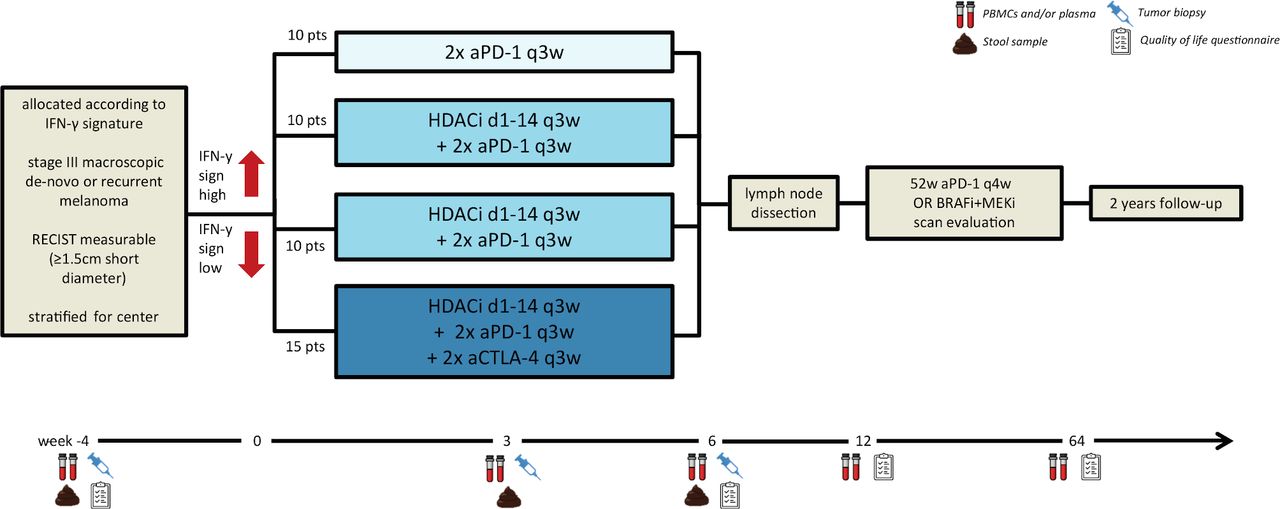
The first of such a personalized neoadjuvant immunotherapy trial is currently recruiting. The DONIMI trial is testing single PD-1 blockade, the combination of histone deacetylase inhibition in combination with PD-1 blockade, and in combination with PD-1 plus CTLA-4 blockade, allocating patients according to IFN-γ signature in their tumor to less or more extensive (combination) therapy (figure 1).27 In preclinical models and early clinical trial data it has been shown that class I-selective histone deacetylase inhibitor (HDACi) can increase antigen presentation, tumor T cell infiltration, and induces the IFN-γ signature, a signature previously shown to be associated with response to PD-1 blockade in stage IV melanoma and to combined neoadjuvant CTLA-4 plus PD-1 blockade.5 22 28

Study design of the DONIMI trial. aCTLA-4, anti-cytotoxic T lymphocyte antigen 4; aPD-1, anti-programmed cell death protein 1; BRAFi, BRAF inhibition; d, day; HDACi, histone deacetylase inhibitor; IFN-γ, interferon-gamma; MEKi, MEK inhibition; PBMCs, peripheral blood mononuclear cells; q3w, 3 weekly; sign, signature; w, week.
Signature-driven patient selection for personalized neoadjuvant combination therapy
Rational development of immunotherapy combinations could be most efficient when identifying baseline biomarkers, defining patient groups benefiting from this treatment combination, using early read-outs of response, and identification of other combinations overcoming resistance to the current therapies. In stage IV disease many combinations are currently tested without such rational approach, resulting regularly in disappointing results of large phase 3 trials, for example, the KEYNOTE-252 trial, in which the indoleamine 2,3-dioxygenase inhibitor epacadostat combined with anti-PD-1 did not contribute to any clinical benefit compared with anti-PD-1 monotherapy for patients with melanoma.29 Both the IMmotion151 trial, in which first-line atezolizumab plus bevacizumab was compared with sunitinib in patients with renal cell cancer,30 and the START trial, comparing tecemotide versus placebo in patients with stage III non-small cell lung cancer, did not result in an overall survival (OS) difference.31 This illustrates the need for early optimization of combination trial design by preclinical risk reduction and a better definition of patients who are not likely to respond to available treatments. These patients are in great need for alternative combination treatments, which should be tested based on baseline biomarker definition and a strong preclinical rationale.
Also in the neoadjuvant setting, a plethora of new potential combination partners for checkpoint inhibitors are currently tested. The most frequently used neoadjuvant combination approach is the addition of PD-1 blockade to standard chemotherapies or to another checkpoint inhibitor that often is chosen based on availability within the individual pharmaceutical company. Such an irrational approach bears high risk of failure, as for example, seen in the recent combination trial of chemotherapy and PD-1 blockade in first-line treatment of bladder cancer, where the chemotherapy addition to PD-1 blockade was even contra productive in patients with high programmed death-ligand 1 (PD-L1) expression.32
In melanoma such an approach is not followed, making the trials in melanoma possible template trials for neoadjuvant (combination) immunotherapy. An overview of current neoadjuvant trials in melanoma is shown in table 1. But also for melanoma, preselection of patients likely to respond to the combination is currently not common. Such unselective approaches are, as said, associated with a high risk of failure. Thus, novel strategies need to be developed that can provide a clear rationale for new combinations and allow to specifically preselect patients for the compound tested.
Overview on neoadjuvant trials currently recruiting in melanoma
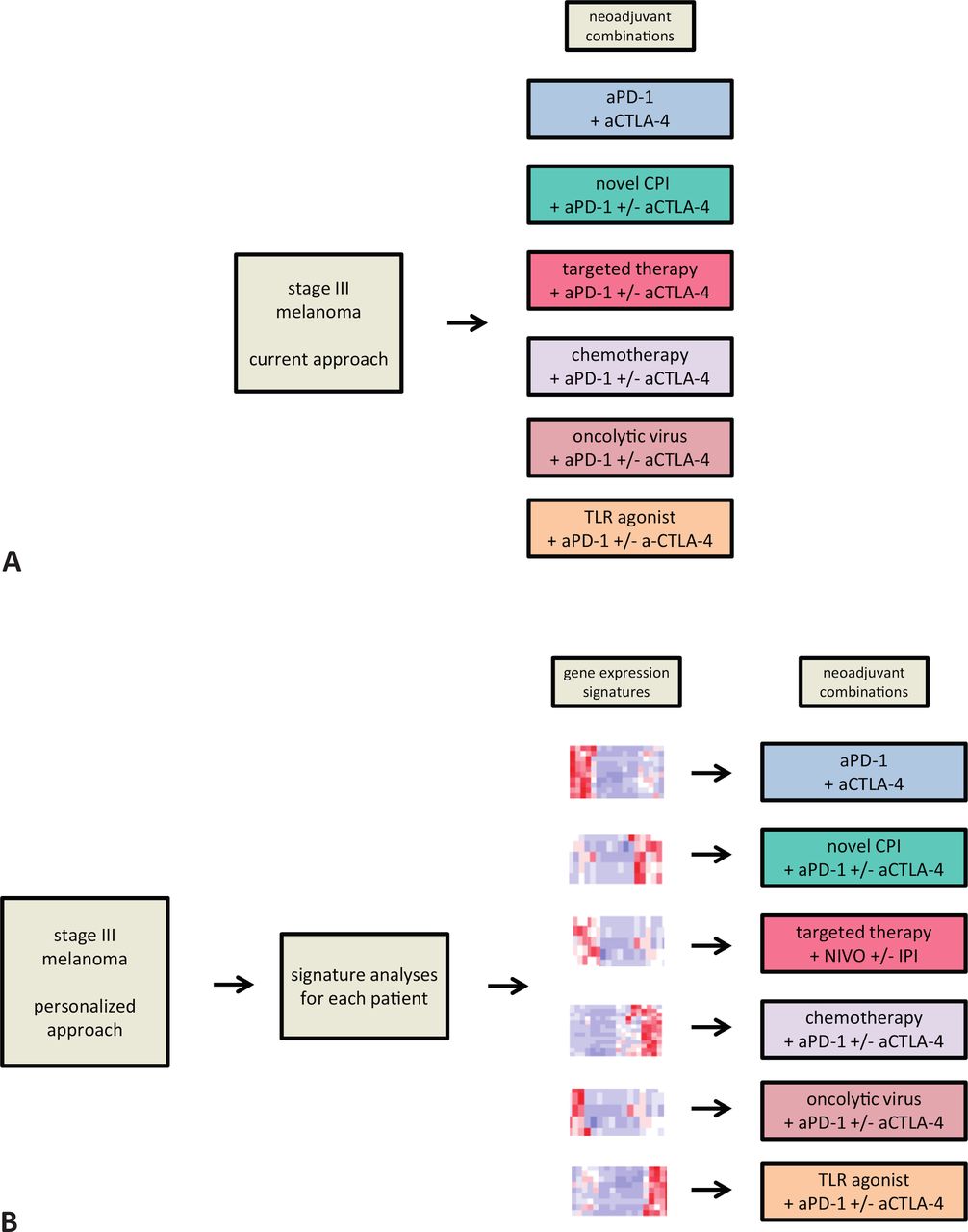
Furthermore, current neoadjuvant immunotherapy combination treatments that mostly follow an one-fits-all strategy, without prior patient selection, lead to a substantial number of patients without benefit (figure 2A). As pathologic non-response is a strong predictor of poor RFS and OS outcome, this early marker could be used in the search for baseline biomarkers. In addition, the homogeneity of the stage III patient population increases the reproducibility of efficacy data, even in small patient cohorts and especially when neoadjuvant trials are standardized, as advised by the International Neoadjuvant Melanoma Consortium.33

One-fits-all approach: patients with stage III melanoma get a neoadjuvant combination therapy within an available trial (A). Personalized neoadjuvant approach: the use of identified immune signatures could lead to selection of the combination with the highest chance of response for each individual patient (B). aCTLA-4, anti-cytotoxic T lymphocyte antigen 4; aPD-1, anti-programmed cell death protein 1; CPI, checkpoint inhibitor; TLR, toll-like receptor.
With more and more data emerging from stage IV disease and other cancer types, it is becoming clear that single biomarkers such as PD-L1 or TMB cannot sufficiently predict response to immunotherapy.34 35 In contrast, more complex biomarkers have been identified, such as immune signatures generated by mapping the network of antitumor immune responses, for example, the IFN-γ signature (see Neoadjuvant checkpoint inhibition - new aspects in an old concept), that may allow to better distinguish responding and non-responding patients. By identifying additional tumor and immune cell RNA signatures, single cell cytokine and (single cell) RNA signatures from peripheral blood, and serum markers, one will eventually predict with a high accuracy the responders versus non-responders for a certain neoadjuvant immunotherapy combination. This concept of signature-driven neoadjuvant combination therapy can be expanded to different treatment combinations, as outlined in figure 2B, and also be used to perform new trials in prespecified patient groups. We envision that this will eventually result in personalized early stage neoadjuvant treatment.
In addition, such immune signatures could also be of relevance in case of relapse. A patient with an early relapse and therefore no sufficient response to the neoadjuvant signature-driven therapy should be treated with a different scheme. Reanalyzing tumor immune signatures of this patient and treating her/him with a combination that fits best with another signature coupled to this treatment combination will also guide late-stage therapy. Vice versa, one could imagine that a patient with a very late relapse (eg, several years after response on neoadjuvant treatment), and still having the same tumor signature, might respond to reintroduction of the same combination therapy that she/he received as neoadjuvant therapy (figure 3). In that way, efforts identifying the optimal neoadjuvant therapy according to the patient’s tumor signature will also give guidance for optimal late-stage therapy, that hopefully will be needed very infrequently.

The future of personalized cancer immunotherapy in the neoadjuvant setting and late-stage disease setting. In stage III disease the tumor of the patient will be analyzed for previously identified signatures associated with a higher chance of response on a certain neoadjuvant treatment combination. The neoadjuvant therapy will be based on such signatures. Most of the patients will then have a pathologic response, associated with RFS. Patients relapsing will be reanalyzed for their gene expression signature, and in combination with the initial result of the neoadjuvant therapy, the time to relapse and the new signature the physician can decide to retreat the patient with the same combination (in case of detection of the same signature, initially MPR, long time to relapse) or choose an alternative combination fitting best to the newly detected signature (in case of detection of a deviating signature, early relapse after non-response to the neoadjuvant therapy). In that way, personalized neoadjuvant therapy will also deliver support for treatment decisions in the late-stage situation. aCTLA-4, anti-cytotoxic T lymphocyte antigen 4; aPD-1, anti-programmed cell death protein 1; CPI, checkpoint inhibitor; MPR, major pathologic response; RFS, relapse-free survival; TLR, toll-like receptor.
We envision that in the future for every combination therapy tested, a response-predicting signature can be developed in parallel. This should lead to a set of immune signatures that allow preselection of patients with stage III melanoma for different neoadjuvant combination treatments, ultimately leading to a true personalized treatment approach.
Rationalizing neoadjuvant combination therapies - the Lombard Street Approach
Signature-driven neoadjuvant immunotherapy that is employed at the level of trial design, should accelerate the development of new treatments to overcome ineffective antitumor response. To this end, instead of performing trials using the current one-fits-all strategy, we propose an approach that we termed the ‘Lombard Street Approach’. Such as this famous street in San Francisco winds back and forth down the hill, we envision that trials evaluating new neoadjuvant immunotherapy combinations should be performed in a stepwise manner, accompanied by in-depth (dynamic) preclinical analyses of pretreatment and post-treatment tumor tissue. The Lombard Street Approach consists of three major components: (1) adaptive small neoadjuvant trials in pre-specified patient populations, (2) immune signature-driven patient selection, and (3) design of new trials based on preclinical testing of immunotherapy combinations in human tumor explants of non-responding patients (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The Lombard Street Approach as an approach to test new treatment combinations in subgroups of patients with unfavorable tumor characteristics. aCTLA-4, anti-cytotoxic T lymphocyte antigen 4; aPD-1, anti-programmed cell death protein 1; NR, non-responder; PDTFs, patient-derived tumor fragments.
Trials with adaptive designs have been performed previously. Since its opening in 2005, the STAMPEDE trial has tested several approaches treating high-risk prostate cancer, where each new alternative in treatment is added as an additional treatment arm and compared with the same standard-of-care treatment arm.36 Another platform trial, I-SPY 2, focuses on neoadjuvant treatment in high-risk breast cancer subtypes, comparing multiple treatment arms to a standard backbone.37 A baseline biomarker assessment, based on HER2 receptor and hormone receptor expression, classifies the patients in predefined groups.
The crucial difference to the proposed Lombard Street Approach is that these platform trials include always the same patient population and therefore do not identify a new treatment option for patients not responding to the previously tested combination. Using the Lombard Street Approach, for each subsequent combination tested, only patients with tumors lacking the responder signature (or displaying a specific non-responder signature) will be included, as for the responder group already an efficient treatment has been found. Repeating this approach will reduce the group of non-responders with every trial (figure 4).
To illustrate the Lombard Street Approach, the prior trials testing neoadjuvant ipilimumab and nivolumab in melanoma would reflect the first step. Using the IFN-γ signature plus TMB, patients responding to or less likely to neoadjuvant CTLA-4 and PD-1 blockade have been identified.10 22 While for the patients with IFN-γ signature-high plus TMB-high an effective treatment option has been found, patients with low IFN-γ signature ± low TMB should possibly not be treated with this combination. In a subsequent trial, an alternative (combination) treatment can be evaluated in patients with low IFN-γ signature plus low TMB. In parallel, new immune signatures for responders and non-responders to the alternative treatment can be identified, resulting in a new well-defined patient group with an effective treatment. For patients with both unfavorable signatures, another treatment option needs to be identified based on preclinical analyses and so on. By following this approach, and in contrast to the STAMPEDE or I-SPY trials, the group of patients with melanoma for whom another therapy option needs to be found becomes smaller and smaller (figure 4).
To further support the development of rational neoadjuvant trials, new treatment combinations should be driven by dynamic preclinical assessment of tumors from non-responders. Human tumor explant systems, such as the patient-derived tumor fragment (PDTF) platform, we have recently developed (Voabil et al, manuscript in revision) can provide such a tool to rationalize the development of new treatment combinations for patients with ‘unfavorable’ tumors. Derived from freshly resected tumor specimens, PDTFs are tumor tissue fragments of ~1 mm3 size that, in contrast to other organotypic models such as tumor spheroids or organoids, are not dissociated and regrown, but microdissected and directly exposed to short-term culture. Therefore, PDTFs largely maintain the tumor microenvironment (especially the immune infiltrate) and architecture as found in the patient, and allow to not only test new immunotherapy treatments ex vivo, but to compare multiple treatments within samples from one patient. Ex vivo treatment of PDTFs can be combined with multiple RNA and protein readouts, allowing to assess immunological responses early on after treatment and to couple these response dynamics to baseline properties of the tumor. One potential limitation is that PDTFs only reflect the intratumoral cellular ecosystem and do not contain the systemic immune compartment. However, we recently found that in the case of ex vivo anti-PD-1 treatment, intratumoral immune reactivation in PDTFs correlated with clinical response in all patients assessed (Voabil et al, manuscript in revision). In addition, the comparison of ex vivo treated PDTFs with changes in on-treatment biopsies may allow to detect the effect caused by systemic immune reactivation.
In-depth analysis of post-treatment tumor material from non-responding patients can furthermore provide insights into potential resistance mechanisms. Based on such findings, new treatment combinations can be developed and directly tested ex vivo in the same tumor material, generating a rationale for new immunotherapy combination trials. As predictive immune signatures for identification of these patients can be developed in parallel, these new trials can be tailored to pre-specified patient groups.
For the Lombard Street Approach to work efficiently, a close and streamlined interaction is necessary between oncologists, surgeons, pathologists and translational researchers to align treatments, tumor material collection, trial design and patient selection. In addition, after the initially small trials, identified signatures and their correlated drug combinations will need to be validated in larger cohorts.
Concluding, we here plead for a more rational approach in testing neoadjuvant combination therapies. First, by identifying signatures associated with pathologic response on current immunotherapy combinations, the response chance for patients can be increased, and patients unlikely to respond can be spared adverse events without clinical benefit. Second, by identifying non-responders upfront, novel combinations should be preferentially offered to these patients. Third, such new combinations should be driven by preclinical analysis, for instance using human ex vivo platforms, thereby reducing the patient groups in need for an alternative treatment combination in a back-and-forth approach - a strategy that we introduced here as the ‘Lombard Street Approach’. These three steps should lead towards fast development of personalized neoadjuvant immunotherapy.
References
Footnotes
Contributors JMV wrote the first draft of the manuscript and processed writing and suggestions of the coauthors. DST and CUB contributed both in writing. All authors read and approved the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests All authors declare no direct conflicts with this work. For unrelated conflicts, DST reports research funding from BMS. CUB has received research funding from BMS, NanoString and Novartis; has an advisory role for AZ, BMS, GenMab, GSK, Lilly, MSD, Novartis, Pierre Fabre, Pfizer, Roche and Third Rock Ventures; is stockowner of Uniti Cars; and cofounder of Immagene BV.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.