Article Text

Abstract
Background Pancreatic ductular adenocarcinoma (PDAC) is among the most dreadful of malignancies, in part due to the lack of efficacious chemotherapy. Immune checkpoint inhibitors, including anti-programmed cell death 1 (anti-PD-1) antibodies, are novel promising forms of systemic immunotherapy. In the current study, we assessed whether gemcitabine (GEM) combined with anti-PD-1 antibody treatment was efficacious as immunochemotherapy for advanced PDAC using a murine model of liver metastasis.
Methods The murine model of PDAC liver metastasis was established by intrasplenically injecting the murine pancreatic cancer cell line PAN02 into immunocompetent C57BL/6J mice. The mice were treated with an anti-PD-1 antibody, GEM, or a combination of GEM plus anti-PD-1 antibody, and compared with no treatment (control); liver metastases, immune cell infiltration, gene expression, immune cell response phenotypes, and overall survival were investigated.
Results In the metastatic tumor tissues of mice treated with GEM plus anti-PD-1 antibody, we observed the increased infiltration of Th1 lymphocytes and M1 macrophages. Gene expression profile analysis of peripheral blood cells obtained from mice treated with GEM plus anti-PD-1 antibody clearly highlighted T cell and innate immune signaling pathways. Survival of PDAC liver metastasis mice was significantly prolonged by the combination therapy (median survival, 66 days) when compared with that of GEM alone treatment (median survival, 56 days). Expanded lymphocytes, which were isolated from the splenocytes of PDAC liver metastasis mice treated with GEM plus anti-PD-1 antibody, had an increased number of M1 macrophages.
Conclusion The combination of anti-PD-1 antibody immunotherapy with GEM was beneficial to treat a murine model of PDAC liver metastasis by enhancing the immune response mediated by Th1 lymphocytes and M1 macrophages and was associated with CD8+ T cells.
- drug therapy
- combination
- immunity
- immunotherapy
- macrophages
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Pancreatic ductular adenocarcinoma (PDAC) is among the most dreadful of malignancies with an extremely poor prognosis.1 Most cases are diagnosed in the advanced stages owing to the extreme difficulty of diagnosing PDAC in the early stages because of a lack of specific symptoms and efficient and convenient examination methods. As a consequence, liver metastasis develops in the majority of patients with PDAC.2 Furthermore, patients with PDAC in the advanced stages are unable to undergo radical surgical treatment, but they can receive chemotherapy; however, it is partially effective at best, and not curative. Thus, novel effective treatments for advanced patients with PDAC should be developed to improve their prognosis.
We have previously shown that human PDAC is associated with systemic and local immune responses.3 As a patient’s immune condition is important for cancer pathophysiology, modulation of the immune response in patients with cancer is an emerging therapeutic strategy. In fact, immune checkpoints have been shown to be promising targets for the development of immunotherapy against cancer. Considering that PDAC is a disease associated with the immune response,4 appropriate immune-modulation should be explored as a potential therapeutic strategy. However, the currently available immunotherapeutic agents have not been shown to be effective for PDAC.5
In this study, we used a murine model of PDAC liver metastasis to investigate the efficacy of gemcitabine hydrochloride (GEM) chemotherapy, which is one of the most standard chemotherapeutic drug treatments, in combination with anti-programmed cell death 1 (anti-PD-1) antibody (Ab) treatment. We observed that this combination induced a prominent anticancer immune response featuring an increase in the number of M1 macrophages, Th1 CD4+ cells, and cytotoxic CD8+ T cells in tumor tissue. We confirmed that GEM plus anti-PD-1 Ab treatment prolonged survival in the murine model of PDAC liver metastasis.
Methods
Cell line
The PDAC cell line (PAN02 cells; DCTD Tumor Repository, NCI-Frederick, Frederick, MD) was cultured and expanded in Dulbecco’s modified Eagle’s medium (DMEM; Nacalai Tesque, Kyoto, Japan) supplemented with 10% heat-inactivated fetal bovine serum and 100 µg/mL penicillin and streptomycin (P/S; Life Technologies, Carlsbad, California, USA). Cells were cultured in an incubator at 37°C/5% CO2.
Establishment of the murine model of PDAC liver metastasis and treatments
C57BL/6J male mice (Charles River Laboratories, Yokohama, Japan) were injected intraperitoneally with an anesthetic mixture of 0.3 mg/kg medetomidine (1 mg/mL Domitor; Nippon Zenyaku Kogyo, Koriyama, Japan), 4 mg/kg midazolam (10 mg/2 mL Dormicum; Astellas Pharma, Tokyo, Japan), and 5 mg/kg butorphanol (5 mg/mL Vetorphale; Meiji Seika Pharma, Tokyo, Japan), and then injected intrasplenically with 2.5×106 PAN02 cells to establish the murine model of PDAC liver metastasis, as described previously.6 The dissemination pattern within mice injected with PAN02 cells was verified at necropsy in order to test the stability of the liver metastasis model; we checked the spleen, liver, pancreas, lung and peritoneal cavity for metastatic lesions. Furthermore, we verified tumor dissemination of the liver metastasis model by single-photon emission CT/X-ray CT (SPECT/CT) using a VECTor+/CT (MILabs, Houten, The Netherlands); 5 MBq F-18 fluorodeoxyglucose (Kanazawa Advanced Medical Center, Kanazawa, Japan) was injected via the tail vein to each mouse and followed by real-time imaging. The PDAC mice were treated with 50 mg/kg GEM (G0367; TCI, Tokyo, Japan) by tail vein injection, and with or without intraperitoneal injection of anti-PD-1 Ab (200 µg; clone: J43; BD Biosciences, Franklin Lakes, New Jersey) or anti-CD8 Ab (200 µg; clone: 53–6.7; BD Biosciences). To evaluate the presence of tumor-infiltrating inflammatory cells (TICs), the mice received a single dose of phosphate-buffered saline (PBS), GEM, or GEM plus anti-PD-1 Ab at 28 days after tumor implantation; the mice were euthanized 2 days later. To assess the immune response in peripheral blood and for histological analysis, PDAC mice received PBS, GEM, or GEM plus anti-PD-1 Ab twice a week from day 7 to day 33, for a total of 8 treatments; blood was collected and tumor tissues were isolated on day 34. To determine the effect on survival, the mice were administered PBS, anti-PD-1 Ab, GEM, GEM plus anti-PD-1 Ab, or GEM plus anti-PD-1 Ab plus anti-CD8 Ab, twice a week from day 7 to day 39. All animal experiments were approved by our institutional review board.
Isolation of TICs
Metastasized PDAC tumors were resected from the liver, washed in PBS, and minced. Tissue homogenates were digested in RPMI 1640 medium (Nacalai Tesque) with digestive enzyme mix (Tumor Dissociation Kit; Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions and incubated under constant rotation by using a gentleMACS Dissociator (Miltenyi Biotec). Dissociated cells were washed and passed through 100 µm and 40 µm cell strainers (BD Biosciences). Cells were centrifuged and resuspended in 5 mL of 40% Percoll PLUS (GE Healthcare, Uppsala, Sweden), which was gently applied over 3 mL Histopaque-1083 (Sigma-Aldrich, St. Louis, Missouri, USA). After centrifugation, TICs were collected from the interface between the Percoll and Histopaque layers.
Results
Histological features of PDAC liver metastasis tumors in mice treated with GEM plus anti-PD-1 Ab
We established a murine model of PDAC liver metastasis by injection of syngeneic PAN02 pancreatic cancer cells. We performed two independent experiments with 23 and 30 mice, respectively; 3 mice from the first experiment were macroscopically verified at necropsy on day 7, and metastases were not observed in any organ (online supplemental figure S1A). The remaining 50 mice were macroscopically observed at necropsy on day 34, and they all developed tumor foci in the liver, with a reproducibility of 100% (online supplemental figure S1A,B). We confirmed that the tumor metastases were localized only in the liver and not in other organs, such as the spleen, peritoneal cavity, pancreas, or lung, at necropsy (online supplemental figure S1A) and by SPECT/CT analysis (online supplemental figure S1C). The PDAC mouse model was treated with GEM with or without intraperitoneal anti-PD-1 Ab administration, twice a week up to day 33 for a total of 8 injections. On day 34, the mice were euthanized and assessed for treatment effect and efficacy (figure 1A). The number of metastatic foci in the liver was decreased in PDAC mice treated with GEM and GEM plus anti-PD-1 Ab, compared with untreated or anti-PD-1 Ab-treated PDAC mice; moreover, the average volume of liver nodules was the lowest following treatment with GEM plus anti-PD-1 Ab when compared with the other treatments (figure 1B,C). Histologically, the liver tumors of mice without any treatment contained immune-mediating cells including CD8a+, CD4+, and CD11b+ cells (figure 1D). The number of CD8a+ cells and CD4+ cells infiltrating the tumors was increased in mice treated with only GEM and GEM plus anti-PD-1 Ab when compared with no treatment or anti-PD-1 Ab alone. The increase in the number of infiltrating CD11b+ cells in tumor tissues was more prominent in mice treated with GEM plus anti-PD-1 Ab than in those treated with only GEM or anti-PD-1 Ab (figure 1D). The tumors of PDAC mice treated with GEM showed an increase in PD-1 expression when compared with control. When PDAC mice were treated with anti-PD-1 Ab or GEM plus anti-PD-1 Ab, PD-1 expression in tumor tissues disappeared (online supplemental figure S2).
Supplemental material
Supplemental material

The response in pancreatic ductular adenocarcinoma (PDAC) liver metastasis tumors of mice treated with gemcitabine (GEM) and anti-programmed cell death 1 (anti-PD-1) Ab. (A) Experimental schedule showing the treatments for PDAC liver metastasis mice: no treatment (phosphate-buffered saline), anti-PD-1 Ab, GEM, and GEM plus anti-PD-1 Ab. Treatment was administered twice a week from day 7 to day 33. Tumor tissues were obtained and stained immunohistochemically on day 34 (n=3 per group). (B) Number of liver metastasis tumor nodules and averaged nodule volumes and (C) macroscopic images of tumors are shown; (B) bars represent mean±SEM; Student’s t-test was performed as statistical analysis. (D) Immunohistochemical analysis of tumors for CD8a+, CD4+, and CD11b+ inflammatory cells. Magnification: ×200; bars: 100 µm. Quantification of cell infiltration by using ImageJ (each area analyzed=1.576 mm2, three different areas were analyzed for each sample). White bar: no treatment; light gray bar: anti-PD-1 Ab; dark gray bar: GEM; black bar: GEM plus anti-PD-1 Ab; bars represent mean±SEM; one-way analysis of variance followed by Tukey’s HSD post hoc test was performed as statistical analysis; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. DAB, 3,3'-diaminobenzidine; HSD, honestly significant difference.
Immunity-related gene expression of TICs treated with GEM and anti-PD-1 Ab
As we observed that GEM plus anti-PD-1 Ab treatment affected the immune response of PDAC tumors in the liver, we examined the gene expression of cytokines and chemokines in TICs isolated from PDAC liver metastasis tumors according to treatment. Expression of cytokines and chemokines related to anticancer immunity, namely, Il12a, Il12b, tumor necrosis factor (Tnf), and chemokine (C-C motif) ligand 2 (Ccl2), was significantly upregulated by GEM plus anti-PD-1 Ab treatment (figure 2). In addition, genes related to cancer-prone immunity, namely, arginase, liver (Arg1), Il10, transforming growth factor, beta 1 (Tgfb1), and matrix metallopeptidase 9 (Mmp9), were also significantly upregulated by the combination therapy (figure 2). Expression of Il6, Il1b, and chemokine (C-X-C motif) ligand 10 (Cxcl10) tended to increase with GEM plus anti-PD-1 Ab treatment; furthermore, there was a clear increase of programmed cell death 1 (Pdcd1) and cytotoxicity-related perforin 1 (Prf1) expression in the GEM plus anti-PD-1 Ab treatment group (figure 2). Taken together, although the expression of anticancer and cancer-prone immunity genes was mixed, GEM plus anti-PD-1 Ab treatment eventually induced a reactive immune condition in the tumor microenvironment.

Gene expression analysis of tumor-infiltrating inflammatory cells (TICs) from pancreatic ductular adenocarcinoma (PDAC) liver metastasis model mice by qRT-PCR. PDAC liver metastasis mice received a single dose of the indicated treatment on day 28 and tumor tissues were obtained 2 days later for TIC isolation, followed by RNA extraction. Quantitative RT-PCR showing the expression of M1 macrophage-related cytokine genes (Il6, Il12a, Il12b, Il1b, and Tnf), M2 macrophage-related genes (Arg1, Il10, Tgfb1, and Mmp9), proinflammatory chemokines (Cxcl10 and Ccl2), and T-cell activation markers (Pdcd1 and Prf1); n=3; bars represent mean±SEM; Student’s t-test was used for statistical analysis; *p<0.05, **p<0.01. GEM, gemcitabine; PD-1, programmed cell death 1; qRT-PCR, quantitative real-time polymerase chain reaction.
Characteristics of lymphoid-lineage cells of TICs from PDAC tissues in mice according to treatment
Expression of anticancer immunity-related genes was significantly upregulated in the TICs of PDAC liver metastasis tumors, and histological analysis of PDAC liver metastasis tumors suggested an increase in the number of infiltrating lymphocytes and CD11b+ myeloid-lineage cells following GEM plus anti-PD-1 Ab treatment. To further assess the characteristics of the affected immune-mediating inflammatory cells of metastatic tumors, we characterized tumor-infiltrating lymphoid-lineage cells by flow cytometry (FCM). We observed that the populations of CD4+ cells and CD8a+ cells within TICs were increased in mice treated with only GEM and GEM plus anti-PD-1 Ab when compared with mice with no treatment or anti-PD-1 Ab alone (figure 3A; online supplemental figure S3A,B). We also performed intracellular staining of cytokine expression in TICs and assessed their expression by FCM. The population of Th1 CD4+ cells expressing T-bet and interferon (IFN)-γ, which induces cytotoxic T cells, was the most prevalent in the group treated with GEM plus anti-PD-1 Ab (figure 3B; online supplemental figure S3C). As for CD8+ cells within TICs, IFN-γ-secreting cells were prominent in the GEM and GEM plus anti-PD-1 Ab treatment groups (figure 3B; online supplemental figure S3D). To further confirm the effect of treatment on the capability of CD8+ TICs to secrete IFN-γ, we cocultured the TICs with PAN02 cells supplemented with IL-2 and anti-CD3/CD28 Abs, followed by an IFN-γ secretion assay. We observed that the frequency of IFN-γ-secreting cells within the CD8+ cells was increased when compared with the no treatment and anti-PD-1 Ab groups. A slight increase of IFN-γ-secreting cells was also observed in the GEM plus anti-PD-1 Ab group when compared with the GEM only group, despite not reaching statistical significance (online supplemental figure S4). Interestingly, TICs in liver metastasis tumors of mice treated with GEM alone contained more cells with a regulatory T cell phenotype (CD4+CD25+FoxP3+), than did those in mice without any treatment. However, this increase was not observed in TICs of mice when they were treated with GEM plus anti-PD-1 Ab. Furthermore, the cells with the CD4+CD25+FoxP3+ regulatory phenotype in TICs of mice treated with GEM plus anti-PD-1 Ab produced less IL-10, which is generally considered to be a cancer-prone cytokine (figure 3C; online supplemental figure S3E). Thus, the lymphocyte population in TICs of PDAC liver metastasis tumors in mice treated with GEM plus anti-PD-1 Ab were shown to be in an anticancer immune condition.
Supplemental material
Supplemental material
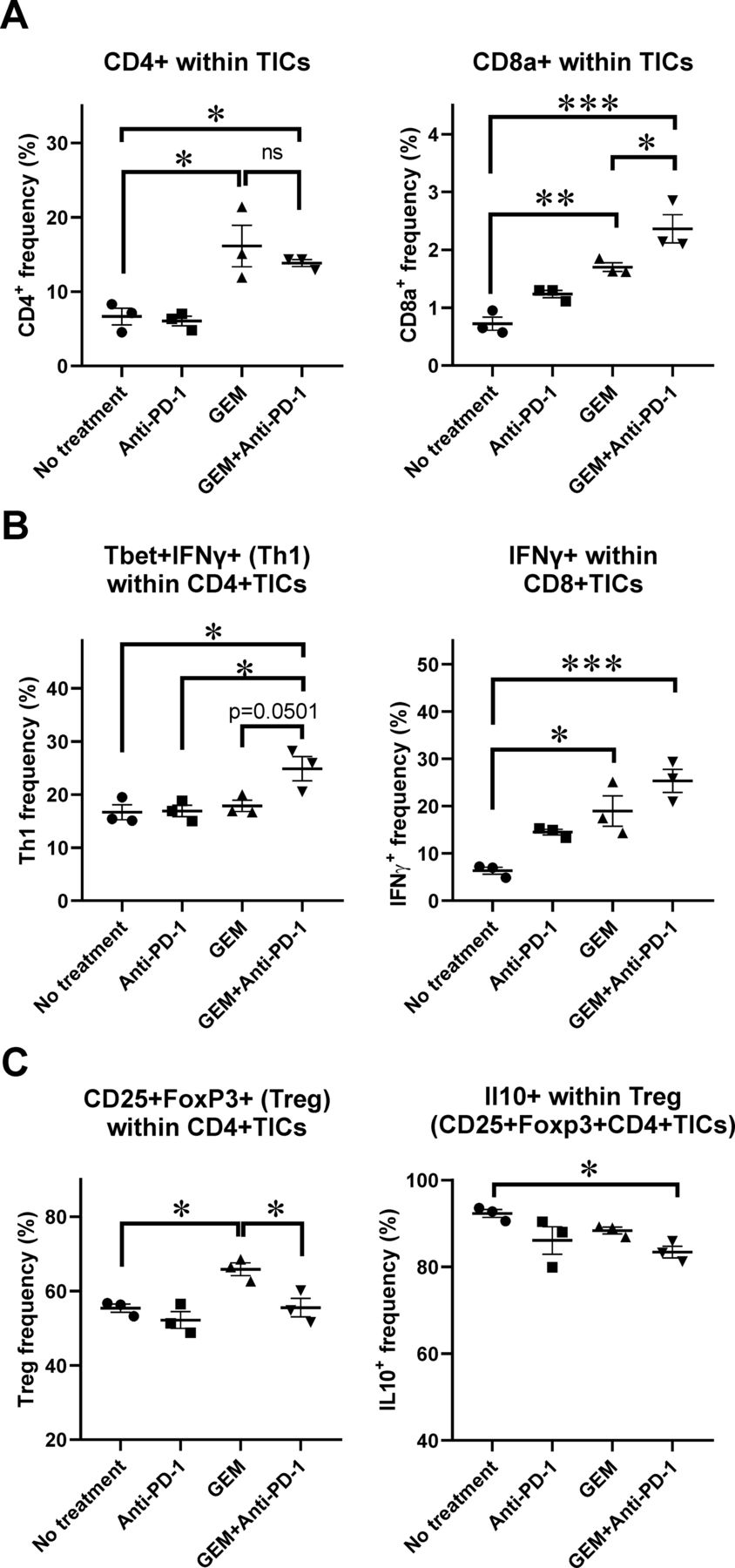
Flow cytometry (FCM) analysis of tumor-infiltrating inflammatory cells (TICs) for lymphoid-lineage cells. Pancreatic ductular adenocarcinoma liver metastasis mice received a single dose of the indicated treatment on day 28 and tumor tissues were obtained 2 days later for TIC isolation followed by FCM analysis; n=3. (A) CD4+ and CD8a+ cells within TICs. (B) Th1 CD4+ cells expressing T-bet and interferon (IFN)-γ; IFN-γ-secreting cells within CD8+ TICs. (C) Regulatory T cell phenotype, CD4+CD25+FoxP3+ cells, and interleukin (IL)-10+-producing cells within Treg cells. (A–C) Bars represent mean±SEM; one-way analysis of variance followed by Tukey’s HSD post hoc test was performed as statistical analysis; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. GEM, gemcitabine. PD-1, programmed cell death 1.
Characteristics of the myeloid-lineage population of TICs of PDAC liver metastasis tissues in mice according to treatment
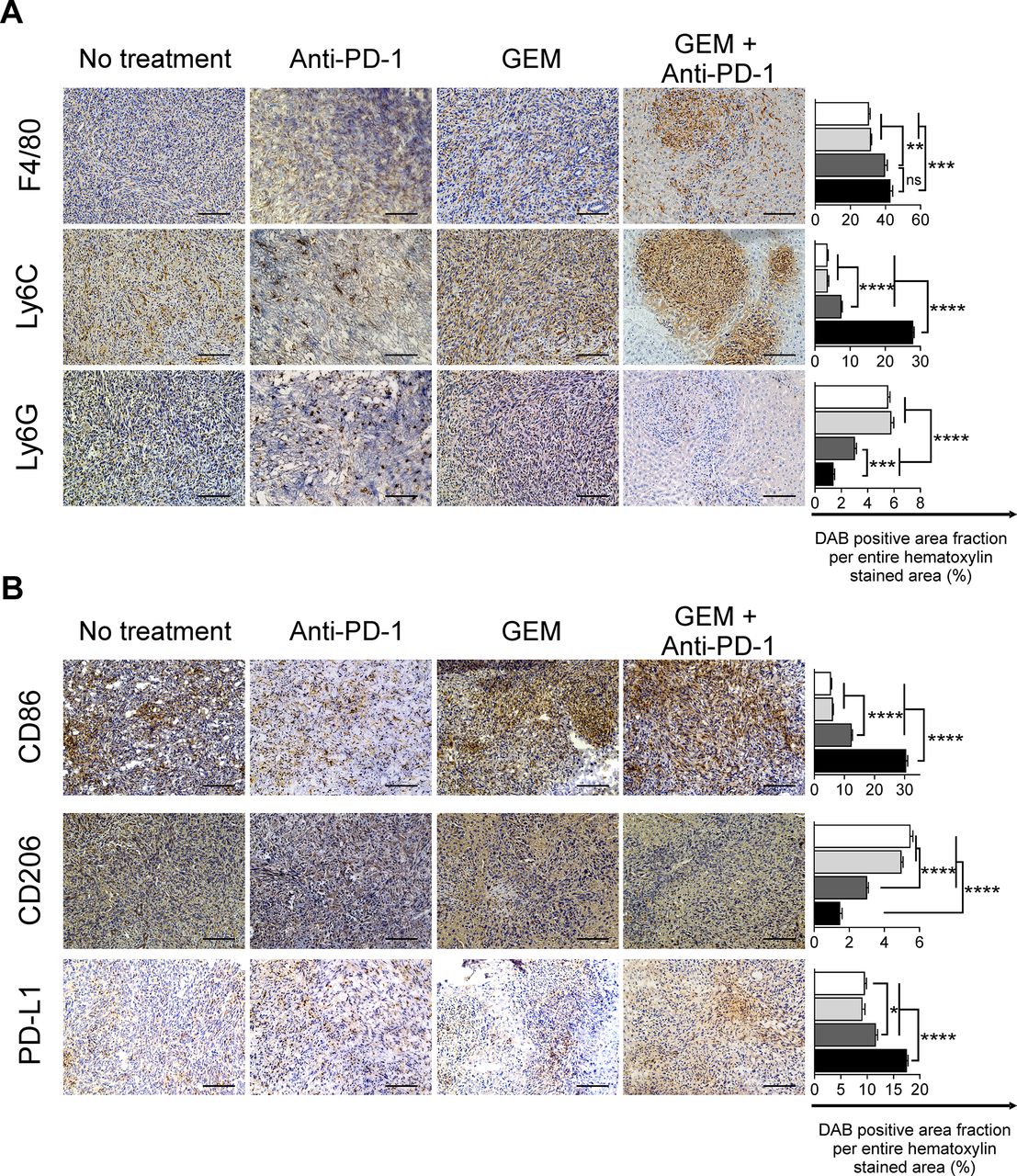
We observed that the lymphoid-lineage inflammatory cells had increased anticancer features after GEM plus anti-PD-1 Ab treatment. We next analyzed the infiltration of myeloid-lineage inflammatory cells, which also play an important role in cancer immunity,7 8 in PDAC liver metastasis tumors. We observed a substantial infiltration of F4/80+ cells in tumor tissues treated with GEM or GEM plus anti-PD-1 Ab. As for monocytic myeloid cells expressing the Ly-6C antigen, their infiltration of liver metastasis tumors was significantly increased after treatment with GEM plus anti-PD-1 Ab, compared with the control, anti-PD-1 Ab, and GEM alone treatment groups (figure 4A). In contrast, the population of infiltrating granulocytic myeloid-lineage cells expressing Ly-6G, which were prominent in tumor tissues of mice without any treatment, were decreased by treatment with GEM alone, and this decrease was further enhanced by GEM plus anti-PD-1 Ab treatment (figure 4A). We also confirmed that the frequency of CD11b+Ly6G+ granulocytic-myeloid-derived suppressor cells (G-MDSCs) was decreased within TICs isolated from mice treated with GEM plus anti-PD-1 Ab when compared with mice receiving the other treatments. We performed cell sorting of CD11b+Ly6G+ TICs and analyzed arginase activity, a surrogate marker of suppressive capability. We observed that there was no difference in arginase activity between the G-MDSCs of mice receiving different treatments (online supplemental figure S5). We next characterized the subtypes of infiltrating monocytic cells in detail by staining for CD86 and CD206 antigens, which indicate M1-immune-activating and M2-immune-suppressive types, respectively. We observed a significant increase of CD86+ cells and decrease of CD206+ cells in the tumors of mice treated with GEM plus anti-PD-1 Ab (figure 4B). As for the expression of the immune checkpoint ligand PD-L1, the number of inflammatory cells expressing PD-L1 was slightly increased by GEM plus anti-PD-1 Ab treatment (figure 4B).
Supplemental material

Immunohistochemical analysis of pancreatic ductular adenocarcinoma liver metastasis tumors for myeloid-lineage cells. Treatments were conducted twice a week from day 7 to day 33; tumors were obtained on day 34 and immunohistochemically analyzed for (A) F4/80+, Ly6C+, and Ly6G+, and (B) CD86+, CD206+ infiltrating cells, and programmed cell death-ligand 1 (PD-L1)+ for the indicated treatment. Magnification: ×200; bars: 100 µm. (A, B) Quantification of cell infiltration by using ImageJ (each area analyzed=1.576 mm2, 3 different areas were analyzed for each sample). White bar: no treatment; light gray bar: anti-PD-1 Ab; dark gray bar: gemcitabine (GEM); black bar: GEM plus anti-PD-1 Ab. Bars represent mean±SEM; one-way analysis of variance followed by Tukey’s HSD post hoc test was performed as statistical analysis; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
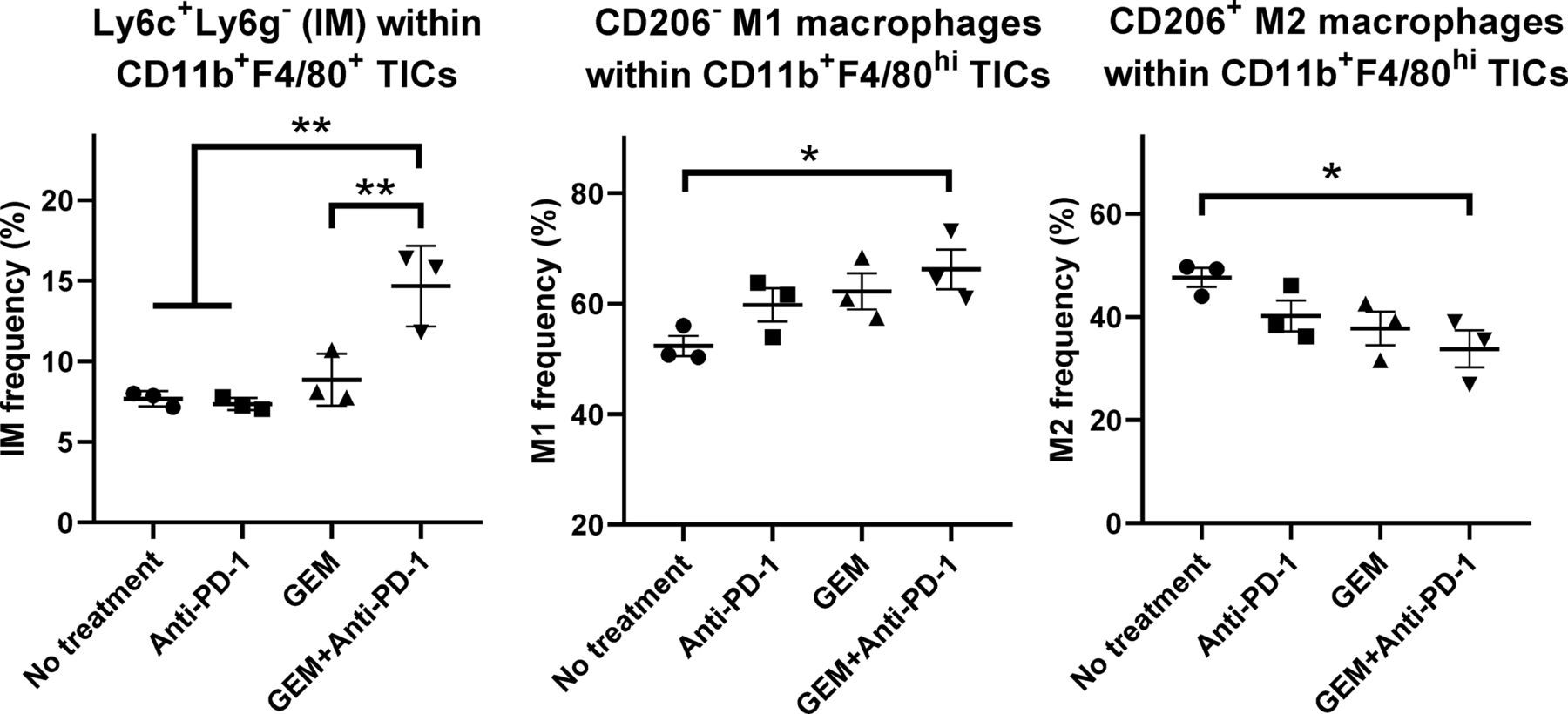
We also characterized CD11b+F4/80+ TICs from PDAC liver metastasis tumors in mice by FCM. The population of CD11b+F4/80+Ly6C+Ly6G− inflammatory monocytes with proinflammatory/antitumor properties9 10 was significantly increased by GEM plus anti-PD-1 Ab treatment (figure 5; online supplemental figure S6A). The CD206− M1 macrophage population within CD11b+F4/80high TICs was increased by GEM plus anti-PD-1 Ab treatment, whereas the CD206+ M2 macrophage population within CD11b+F4/80high TICs was decreased by GEM plus anti-PD-1 Ab treatment (figure 5; online supplemental figure S6B).
Supplemental material

Flow cytometry (FCM) analysis of tumor-infiltrating inflammatory cells (TICs) for myeloid-lineage cells. Pancreatic ductular adenocarcinoma liver metastasis mice received the following treatments: no treatment, anti-programmed cell death 1 (anti-PD-1) Ab, gemcitabine (GEM), or GEM plus anti-PD-1 Ab on day 28 and tumor tissues were obtained 2 days later for TIC isolation and FCM; n=3, bars represent mean±SEM. The following cell populations were analyzed: Ly6C+Ly6G− inflammatory monocytes within CD11b+F4/80+ TICs, CD206− M1 macrophages within CD11b+F4/80high TICs, and CD206+ M2 macrophages within CD11b+F4/80high TICs. One-way analysis of variance followed by Tukey’s HSD post hoc test was performed as statistical analysis; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Gene expression profile features of peripheral blood cells affected by GEM and anti-PD-1 Ab treatment
Peripheral blood cells circulate continuously throughout the body; they are affected by the host’s immune condition, which is altered by cancer.3 11 12 Therefore, in addition to analysis of the immunological features in PDAC liver metastasis tumors described above, we examined how peripheral blood cells were affected in the murine model of PDAC liver metastasis by treatment with GEM and anti-PD-1 Ab using gene expression profile analysis with a DNA microarray. Unsupervised cluster analysis using all 5710 filtered gene probes formed two clusters: one was consistent with no treatment or anti-PD-1 Ab treatment, and the other was consistent with GEM or GEM plus anti-PD-1 Ab treatment (online supplemental figure S7A). Cluster analysis of the gene expression profiles for GEM or GEM plus anti-PD-1 Ab, when compared with no treatment, revealed distinct clusters between them (online supplemental figure S7B,C). In contrast, cluster analysis of peripheral blood gene expression for anti-PD-1 Ab treatment alone and no treatment, as well as GEM and GEM plus anti-PD-1 Ab treatment, did not show any apparent clusters (online supplemental figure S7D,E), suggesting that anti-PD-1 Ab treatment alone did not drastically affect the gene expression profile of whole peripheral blood cells.
Supplemental material
To assess further how the effect of anti-PD-1 treatment plus GEM treatment could be observed in the features of the gene expression profiles of peripheral blood cells, we performed pathway map analysis of the differentially expressed genes between no treatment and GEM or GEM plus anti-PD-1 Ab. We observed that 5706 genes were upregulated (parametric p value<0.05) for GEM treatment when compared with the no treatment group. Analysis to identify the pathway maps related to the 5706 upregulated genes included the immune response pathway maps ‘IFN-alpha/beta signaling via MAPKs’ and ‘TCR alpha/beta signaling pathway’, which were suggestive of anticancer immunity, as well as immune checkpoint-related pathway maps such as ‘Inhibitory PD-1 signaling in T cells’ and ‘Regulation of T cell function by CTLA-4’, which inhibit anticancer immunity (table 1). We found that 2532 genes were upregulated by GEM plus anti-PD-1 Ab treatment when compared with the no treatment group. The highlighted pathway maps for these genes were related to anticancer immune responses, but did not include the immune checkpoint-related pathway maps ‘Inhibitory PD-1 signaling in T cells’ and ‘Regulation of T cell function by CTLA-4’ (table 1).
List of pathway maps generated by MetaCore enrichment analysis of differentially upregulated genes defined by BrB class comparison analysis
In addition, the immune response pathway maps ‘IFN-alpha/beta signaling via JAK/STAT’, ‘HMGB1 release from the cell’, and ‘Apoptosis and survival map Granzyme A signaling’ were also featured only for GEM plus anti-PD-1 Ab treatment. Thus, analysis of the gene expression profile features of peripheral blood cells implied that the combination of GEM plus anti-PD-1 Ab treatment induced an enhanced immune response against cancer.
Anti-PD-1 Ab plus GEM treatment has a therapeutic effect on PDAC
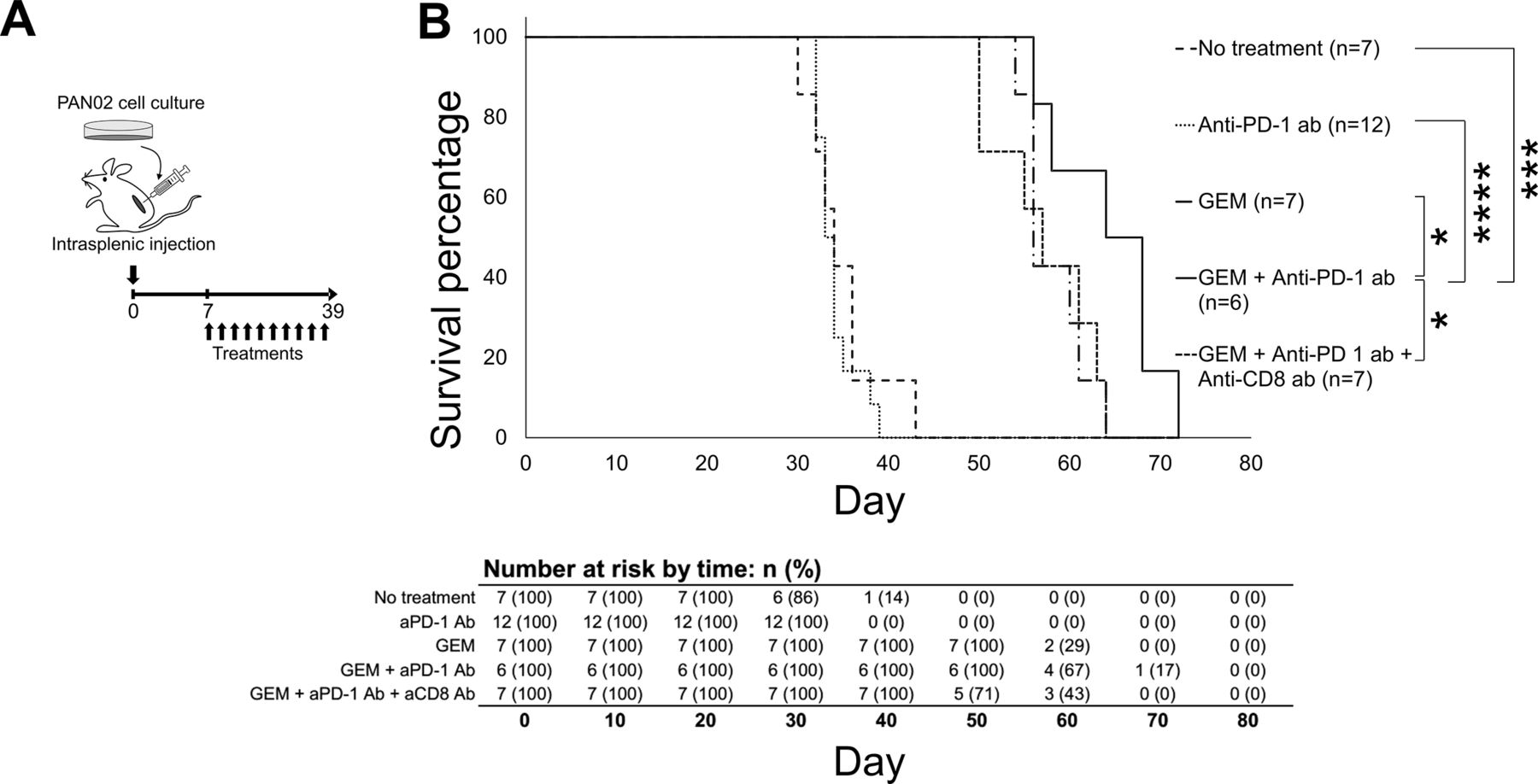
The immune condition of the murine model of PDAC liver metastasis was confirmed to be in an anticancer response state in PDAC liver metastasis tumors and peripheral blood following GEM plus anti-PD-1 Ab treatment. To assess whether this affected condition is therapeutically beneficial in the model mice, we assessed overall survival following treatment with GEM plus anti-PD-1 Ab (figure 6A). The PDAC liver metastasis model was sufficiently reproducible and stable, with an expected survival period of 30–45 days in the absence of treatment. In order to avoid any bias, the mice were assigned randomly to each group (no treatment, anti-PD-1, GEM, and GEM plus anti-PD-1 Ab). We observed that GEM treatment prolonged the survival of PDAC mice, compared with mice without any treatment or with anti-PD-1 Ab administration (figure 6B); by day 45, none of the mice in the last two groups had survived, while all of the mice in the GEM alone or GEM plus anti-PD-1 Ab groups were alive. Furthermore, the overall survival of mice treated with GEM plus anti-PD-1 Ab was significantly improved; by day 65, 50% of the mice in this group were still alive, whereas all of the mice in the other groups had died. The median survival values were 34 days for the no treatment group, 34 days for the anti-PD-1 Ab group, 56 days for the GEM group, 66 days for the GEM plus anti-PD-1 Ab group, and 57 days for the GEM plus anti-PD-1 Ab and anti-CD8 Ab group (figure 6B); the Kaplan-Meier survival probability estimates are shown in online supplemental table S1. Thus, the survival of PDAC liver metastasis mice was enhanced, as expected, by GEM plus anti-PD-1 Ab treatment compared with GEM alone treatment.
Supplemental material

Survival of pancreatic ductular adenocarcinoma (PDAC) liver metastasis model mice that underwent treatment. (A) Experimental schedule showing treatment timing: the PDAC liver metastasis mice received treatment 10 times, twice a week from day 7 to day 39, and survival percentage was monitored until day 72. (B) Survival curves of PDAC liver metastasis mice that received: no treatment (only phosphate-buffered saline; n=7), anti-programmed cell death 1 (anti-PD-1) Ab (n=12), gemcitabine (GEM) (n=7), GEM plus anti-PD-1 Ab (n=6), or GEM plus anti-PD-1 Ab plus anti-CD8 Ab (n=7). The log-rank test was performed to obtain the p values: no treatment/anti-PD-1 Ab=0.492723; no treatment/GEM=0.000172; no treatment/GEM plus anti-PD-1 Ab=0.000446; anti-PD-1 Ab/GEM=0.000034; anti-PD-1 Ab/GEM plus anti-PD-1 Ab=0.000098; GEM/GEM plus anti-PD-1 Ab=0.034672; and GEM plus anti-PD-1 Ab/GEM plus anti-PD-1 Ab plus anti-CD8 Ab=0.0345.
Lymphocytes of PDAC mice treated with GEM plus anti-PD-1 Ab cause macrophages to be shifted to an M1 antitumor macrophage type
The results shown above suggested that lymphoid-lineage cells and myeloid-lineage cells in PDAC liver metastasis tissues had anticancer immunity features, with Th1-mediated and M1-mediated responses, leading to a therapeutic effect. To confirm this immune response further, we isolated splenocytes from the treated mice, followed by T lymphocyte activation and expansion in vitro. Then, resting (M0) bone marrow-derived macrophages were cocultured with the expanded lymphocytes, followed by the selective recollection of macrophages for FCM and gene expression analyses. Macrophages cocultured with lymphocytes, which were isolated and expanded from the spleen of PDAC liver metastasis mice treated with GEM plus anti-PD-1 Ab, included a higher number of CD86+ cells (M1) and lower number of CD206+ cells (M2) among the CD11b+ population than those treated with GEM alone (figure 7A), resulting in a higher M1/M2 ratio. Similarly, the expression of anticancer immunity-related cytokine genes in macrophages, namely, Il6, Il12a, Il1b, and Tnf, was also significantly upregulated (figure 7B). In addition, genes related to the cancer-prone immune condition, namely, Il10, Tgfb1, and Mmp9, were slightly upregulated by treatment with GEM, but not by GEM plus anti-PD-1 Ab (figure 7C), suggesting that the immune response related to GEM treatment was further modified by the addition of anti-PD-1 Ab to induce an anticancer immune response. In contrast, Arg1 expression was slightly upregulated by GEM and GEM plus anti-PD-1 Ab treatment; however, Arg1 is known to be partially induced in M1 macrophages after shifting from resting M0 macrophages.13

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vitro macrophage polarization assay. Pancreatic ductular adenocarcinoma liver metastasis mice received a single dose of the indicated treatment on day 28 and splenocytes were obtained on day 30. The splenocytes were activated by coculture with irradiated PAN02 cells; after 7 days, activated splenocytes were harvested and cocultured with bone marrow-derived macrophages. After 48 hours, macrophages were collected for (A) flow cytometry analysis of CD11b, CD86, and CD206; and total RNA was isolated for qRT-PCR analysis of (B) Il6, Il12a, Il1b, and Tnf (M1 macrophage-related genes) or (C) Arg1, Il10, Tgfb1, and Mmp9 (M2 macrophage-related genes). (A–C) n=3; bars represent mean±SEM; Student’s t-test was performed for statistical analysis; *p<0.05, **p<0.01, ***p<0.001. GEM, gemcitabine; PD-1, programmed cell death 1.
Discussion
In this study, we assessed whether anti-PD-1 Ab treatment has a positive effect on the GEM treatment-associated immune response in a murine model of PDAC liver metastasis. We found that anti-PD-1 Ab plus GEM treatment provided a therapeutic benefit with anticancer immunity, leading to the prolonged survival of PDAC liver metastasis mice. The anticancer immunity induced by GEM plus anti-PD-1 Ab treatment was mediated by the Th1 and cytotoxic T cell response with M1 macrophages.
PD-1 is an immune checkpoint molecule14 that interacts with its ligands PD-L1 and PD-L2,15 and is principally involved in the suppression of activated T cells. Blockade of the PD-1 and PD-L1 or PD-L2 axis is efficacious for enhancing the anticancer immune response via the augmentation of T cell responses in some malignancies, such as non-small cell lung cancer, renal cell carcinoma, and malignant melanoma.16 Despite some clinical benefits for patients with cancer, details of how this axis can be modulated to enhance anticancer immunity have yet to be disclosed. PD-L1 or PD-L2 expression is observed in miscellaneous cell types in tumor tissues, such as, myeloid-lineage cells and tumor cells themselves.17 Therefore, the tumor microenvironment and its participation with miscellaneous cells, including immune-mediating cells, should be examined, especially for cancers that are resistant to conventional chemotherapy such as PDAC.
PDAC is the most difficult cancer to treat and is not curable by the currently available chemotherapeutic reagents, such as FOLFIRINOX and GEM.18 Although immune checkpoint inhibitors alone are not therapeutic for PDAC,16 the chemotherapy-related immune response of the host is considered to be important for the enhancement of the anticancer effect of treatment. In addition, effective checkpoint inhibition requires a modified tumor microenvironment that can induce a proinflammatory type 1 cytokine profile as well as the decreased suppression of T cell activity.19 We previously reported that MDSCs potentially diminished the effect of GEM treatment, and the ablation of myeloid-derived cells augmented the anti-cancer effect of GEM, which was mediated by CD8+ T cells.8 From the current results, anti-PD-1 Ab treatment appears to be a promising choice as a combinatorial agent for PDAC chemotherapy. Macrophages are reportedly affected by anti-PD-1 Ab treatment20 or can be directed toward M1 macrophages in the PD-1 knock-out condition.21
The current study also shows that the T cell population induced by GEM plus anti-PD-1 Ab treatment has an effect on the Th1 and cytotoxic T cells within the tumor environment associated with the M1 macrophage response. A possible mechanism was suggested by the gene expression profile of peripheral blood cells, which indicated a reduction of the immune-inhibitory effect by immune checkpoint inhibition. In fact, expanded T lymphocytes obtained from PDAC liver metastasis mice treated with GEM plus anti-PD-1 Ab shifted resting macrophages toward the M1 phenotype, which is important for anticancer immunity. Furthermore, there was a significant increase in the overall number of CD8+ T cells within the tumor microenvironment of mice treated with GEM plus anti-PD-1 Ab when compared with the other treatments; IFN-γ expression was also enhanced within CD8+ cells. In addition, a decrease in therapeutic effectiveness was observed when anti-CD8 Ab was administered together with GEM plus anti-PD-1 Ab, confirming that CD8+ T cells contribute, at least partially, to the therapeutic effect of GEM plus anti-PD-1 Ab. Previously, we observed changes of the host immune response by GEM treatment; specifically, there was a decreased number of MDSCs in tumor tissues and peripheral blood, an increase in the frequency of CD4+ and CD8+ T cells, and enhanced functional activity of CD8+ T cells for killing tumor cells.8 We also observed that the tumors of PDAC mice treated with GEM showed an increase in PD-1 expression, which was abolished by anti-PD-1 Ab treatment (online supplemental figure S2). Thus, GEM plus anti-PD-1 Ab treatment enhanced the anticancer immune effect compared with single GEM treatment.
In this study, we used a PDAC murine model created with a single cell line, PAN02, and demonstrated the efficacy of GEM plus anti-PD-1 treatment and characterized the immune response features. Despite these findings, the heterogeneity of pancreatic cancer in humans has to be taken into account. Other PDAC models created with different cell lines or xenografts of human-derived pancreatic cancer cells should also be used in future studies related to the immune-modulating anticancer effect of combination therapies.
Immune checkpoint inhibitor treatment is effective for only some patients with cancer,22–25 and a substantial number of patients cannot benefit from this therapeutic approach.26 These new combination therapies could be an alternative promising approach and obtain an improved therapeutic effect, especially for those patients with PDAC who are in an unsatisfactory condition with conventional chemotherapy or immune-modulatory therapy.
Despite these considerations, the mechanism by which inhibiting PD-1 and its interactions with its ligands enhances the immune response has yet to be disclosed, especially in combination with chemotherapy. One area of interest is the modulation of regulatory T cells,19 since we observed that the frequency of regulatory T cells was induced by GEM monotherapy when compared with no treatment, and not by GEM plus anti-PD-1 Ab treatment.
Conclusion
In conclusion, we observed that immune modulation with anti-PD-1 Ab is a promising approach for enhancing the efficacy of treatment for PDAC when used with GEM chemotherapy in a murine model of PDAC liver metastasis. Combination of immunotherapy with conventional chemotherapy is worthy of intense investigation for improving the treatment of patients with advanced PDAC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
TTBH and AN contributed equally.
Contributors TTBH, AN, AS, TK, SK, and YS conceived the research study; TTBH, AN, and HI conducted the experiments and acquired the data; TTBH, AN, AS, KK, and YS analyzed the data; TK, YK, and KS performed the radioisotope experiments in vivo; TTBH, AN, TarY, TatY, EM, TW, MH, SK, and YS wrote and revised the manuscript. All authors have agreed on the final manuscript and approved of its submission.
Funding This work was supported in part by a subsidy from the Japanese Ministry of Education, Culture, Sports, Science, and Technology.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The Institutional Review Board of Kanazawa University approved the procedures for the care and use of laboratory animals in this study (approval no. AP-173850).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.