Article Text

Abstract
Background Immunotherapy represents the future of clinical cancer treatment. The type of cancer cell death determines the antitumor immune response and thereby contributes to the efficacy of anticancer therapy and long-term survival of patients. Induction of immunogenic apoptosis or necroptosis in cancer cells does activate antitumor immunity, but resistance to these cell death modalities is common. Therefore, it is of great importance to find other ways to kill tumor cells. Recently, ferroptosis has been identified as a novel, iron-dependent form of regulated cell death but whether ferroptotic cancer cells are immunogenic is unknown.
Methods Ferroptotic cell death in murine fibrosarcoma MCA205 or glioma GL261 cells was induced by RAS-selective lethal 3 and ferroptosis was analyzed by flow cytometry, atomic force and confocal microscopy. ATP and high-mobility group box 1 (HMGB1) release were detected by luminescence and ELISA assays, respectively. Immunogenicity in vitro was analyzed by coculturing of ferroptotic cancer cells with bone-marrow derived dendritic cells (BMDCs) and rate of phagocytosis and activation/maturation of BMDCs (CD11c+CD86+, CD11c+CD40+, CD11c+MHCII+, IL-6, RNAseq analysis). The tumor prophylactic vaccination model in immune-competent and immune compromised (Rag-2−/−) mice was used to analyze ferroptosis immunogenicity.
Results Ferroptosis can be induced in cancer cells by inhibition of glutathione peroxidase 4, as evidenced by confocal and atomic force microscopy and inhibitors’ analysis. We demonstrate for the first time that ferroptosis is immunogenic in vitro and in vivo. Early, but not late, ferroptotic cells promote the phenotypic maturation of BMDCs and elicit a vaccination-like effect in immune-competent mice but not in Rag-2−/− mice, suggesting that the mechanism of immunogenicity is very tightly regulated by the adaptive immune system and is time dependent. Also, ATP and HMGB1, the best-characterized damage-associated molecular patterns involved in immunogenic cell death, have proven to be passively released along the timeline of ferroptosis and act as immunogenic signal associated with the immunogenicity of early ferroptotic cancer cells.
Conclusions These results pave the way for the development of new therapeutic strategies for cancers based on induction of ferroptosis, and thus broadens the current concept of immunogenic cell death and opens the door for the development of new strategies in cancer immunotherapy.
- cytotoxicity
- immunologic
- immunogenicity
- vaccine
- immunotherapy
- phagocytosis
- alarmins
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immunotherapy has emerged as an independent and effective antineoplastic strategy alongside surgery, radiotherapy and chemotherapy.1 Immunogenic cell death (ICD) has recently been recognized as a critical determinant of the efficacy of cancer therapy.2–7 The ICD concept combines the ability to effectively kill cancer cells with the activation of an immune response specific for the cancer cells and leading to a strong and longlasting anticancer immunity. ICD-inducing agents elicit activation of a danger pathway involving the emission of ICD mediators known as damage-associated molecular patterns (DAMPs). DAMPs constitute a family of endogenous molecules that acquire immunostimulatory properties when exposed on the outer cell membrane or released in the extracellular matrix in a defined spatial–temporal manner. They include ATP, high-mobility group box 1 (HMGB1), calreticulin (CRT) and proinflammatory cytokines such as type I interferons (IFNs).8–12 When mobilized, DAMPs act as danger signals and as adjuvant molecules to activate the immune system.2 7 13
Initially, the ICD concept was described for cancer cells undergoing apoptosis.11 14–16 However, evasion of apoptosis is in itself a hallmark of cancer and can abolish the cell death response, which often leads to the development of apoptosis resistance.17 18 The discovery of other forms of regulated cell death paved the way for the development of alternative strategies for cell death induction. For example, necroptosis is transduced by receptor-interacting protein kinase-1 (RIPK-1), RIPK-3 and a mixed lineage kinase domain-like pseudokinase triggering necroptosis. Induction of necroptosis has become of utmost importance in experimental cancer therapy as an alternative strategy to apoptosis.19–21 It was recently reported that necroptotic cancer cells could be proinflammatory and immunogenic.22–24 Although the induction of immunogenic necroptosis in cancer cells seems to be promising in terms of activating antitumor immunity in experimental mouse models, it is important to stress that many cancers often develop necroptosis resistance.25–27 Therefore, triggering immunogenic apoptosis or necroptosis would not always be the optimal strategy. It is of great importance to find novel ways to kill tumor cells by triggering cell death modalities other than apoptosis and necroptosis. Ferroptosis is an iron-dependent cell death type and it can be selectively triggered in cells expressing oncogenic mutants of RAS.28 Oxidized phosphatidylethanolamines are crucial for ferroptosis execution, and ferroptosis can be induced by blocking the xc cystine/glutamate antiporter system or glutathione peroxidase 4 (GPX4), resulting in a defective GSH-redox system.28–30 Thus, in addition to apoptosis and necroptosis, ferroptosis might be another option to overcome cell death resistance and enhance the efficacy of anticancer therapy. Although the concept of ferroptosis-based cancer therapy is novel and provides potentially promising pharmacological strategy,31–35 whether ferroptotic cancer cells are immunogenic is currently unknown.
In this study, we assessed the immunogenicity of ferroptotic cancer cells in vitro and in vivo and tested their potential as an alternative approach to cancer immunotherapy. We found that early ferroptotic cancer cells were efficiently engulfed by bone marrow-derived dendritic cells (BMDCs) and were able to promote BMDCs maturation and activation. However, late ferroptotic cancer cells were not immunogenic, as indicated by the percentage of BMDCs expressing major histocompatibility complex class II (MHCII) and CD80 and CD86 markers and by the levels of IL-6 as well as by RNAseq analysis. Moreover, in vitro data were confirmed in vivo by showing the effective vaccination activity of early ferroptotic cancer cells in a tumor prophylactic vaccination model in immune-competent mice but not in immune compromised Rag-2−/− mice. Importantly, the protective vaccination effect of the early ferroptotic cells was maximal but was absent in the late ferroptotic cancer cells. Moreover, ATP and HMGB1, the best-characterized DAMPs involved in ICD, have proven to be passively released along the timeline of ferroptosis and act as immunogenic signals associated with the immunogenicity of early ferroptotic cancer cells. These findings identify ferroptotic cancer cells in the early death stages as effective inducers of an adaptive immune response and open the door for the development of new strategies based on ferroptotic cell vaccination for cancer immunotherapy.
Materials and methods
Cell lines and cell culture
Murine fibrosarcoma MCA205 and murine glioma GL261 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 (Gibco) containing 2 g/L glucose. In addition, the RPMI medium was supplemented with L-glutamine (Gibco, 2 mM), heat-inactivated fetal bovine serum (FBS, Fisher Scientific, 10%), penicillin (100 U/mL) and streptomycin (Gibco, 100 µg/L). Murine glioma GL261 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Gibco) containing glucose (4.5 g/L) and supplemented with 10% heat-inactivated FBS, sodium pyruvate (Sigma Aldrich, 100 µM), penicillin (100 U/mL) and streptomycin (100 µg/L). All cells were maintained at 37°C under 5% CO2.
Cell death assay and inhibitor analysis by multimode microplate reader
MCA205 or GL261 cells were seeded at 105 cells/well in a 96-well plate. After incubation overnight, the cells were stained with 3.3 µM Sytox Green nucleic acid stain (molecular probes) and stimulated with 2.5 µM RAS-selective lethal 3 (RSL3) (Sigma Aldrich) for different durations (1, 3, 6 and 24 hours). Cell death was analyzed as described in Demuynck et al.36 Fluorescence was measured on a Tecan Spark 20M multimode microplate reader.
The following inhibitors were used to block different cell death modalities36 37: the pan-caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-(O-methyl)-fluoromethylketone (zVAD-fmk, 25 µM, apoptosis, Bachem), the inhibitor of ROS and lipid peroxidation ferrostatin-1 (Fer-1, 1 µM, ferroptosis, Sigma Aldrich), the iron chelator deferoxamine (DFO, 10 µM, ferroptosis, Sigma Aldrich), the nonspecific lipophilic antioxidant α-tocopherol (α-toc, 100 µM, ferroptosis, Sigma Aldrich) and the RIPK-1 inhibitor necrostatin-1s (Nec-1s, 20 µM, necroptosis, Abcam). The cell death inhibitors were added 1 hour before RSL3 stimulation.
For recovery experiments on both MCA205 and GL261 cells, after treatment for 1, 3 or 6 hours, RSL3-conditioned medium (2.5 µM RSL3) was replaced with drug-free medium; cell death was determined 24 hours after RSL3 stimulation as described above.
Cell death assay by flow cytometry
The cells were washed in Annexin-V binding buffer (10 mM Hepes, pH 7.4, 0.14 mM NaCl and 2.5 mM CaCl2), followed by staining with Sytox Blue Nucleic Acid Stain (molecular probes, S11348) and Annexin-V, Alexa Fluor 488 conjugate (molecular probes, A13201). The cells were run on a Becton Dickinson Fluorescence - Activated Cell Sorting (BD FACS) Canto flow cytometer, and the data were analyzed by using FlowJo software (V.10.0.8).
Live-cell imaging
Cells were seeded at 5.5×104 cells/mL of complete growth medium (DMEM) in a surface-treated 35 mm confocal dish (Van Waters and Rogers (VWR), cat-no: 75 856–740) 1 day before the experiments. Cell death was induced just before imaging, nuclei were visualized using Hoechst 33 342 (1 µM, Thermo Fisher) and cell death was monitored by propidium iodide (PI) exclusion (1 µM, Thermo Fisher). Live-cell imaging was performed on a Nikon A1R confocal microscope (Nikon) using a Chromatic Aberration Free Infinity (CFI) S Plan Fluor Extra-Long Working Distance (ELWD) 40XC air objective. Images were acquired sequentially every 2 min. From each image stack, montages of the multi-tiff time series and three-channel overlays (brightfield, Hoechst 33342, PI) were made using ImageJ 1.49 m, a public domain imaging software.
Morphological analysis by atomic force microscopy
The MCA205 cells were seeded 1 day before the experiments in glass-bottom Petri dishes (VWR, cat-no: 75 856–740) at a density of 5.5×104 cells/mL. The atomic force microscopy (AFM) instrument used to acquire the images was NanoWizard 4 (Bio-AFM, JPK/Bruker). Topographical images were acquired in the JPK QI mode using ATEC-CONT cantilevers. These sharp-tipped cantilevers (radius of curvature <10 nm) have a larger tip height (15 µm) to avoid contact of the cantilever beam with the cells during imaging. Before topographical imaging, the cells were fixed before induction, and 1 hour and 2 hour postinduction. These timepoints were determined based on confocal laser scanning microscopy images. At these timepoints, the cells showed interesting morphological changes: normal (control) morphology before induction, rounding of ferroptotic cells after 1 hour and plasma membrane permeabilization as indicated by PI exposure after 2 hours. First, the dishes were washed three times with phosphate-buffered saline (PBS) and then a 4% paraformaldehyde solution (PFA, Alfa Aesar, 043368.9M) was added for 10 min. Finally, the paraformaldehyde solution was replaced with PBS, and between measurements, the cells were stored at 4°C. Representative height images of the cells at the aforementioned timepoints were obtained.
Quantification of ATP release
Fibrosarcoma MCA205 cells were seeded and treated with RSL3 as described above. Mitoxantrone (MTX, 2 µM, Sigma Aldrich) was used as a positive control. After treatment, the cells were incubated for 0.5, 1, 6 and 24 hours in medium with 2% FBS. At each timepoint, supernatants were collected and centrifuged at 15 000 rpm at 4°C for 3 min. They were either stored at –80°C or used immediately for ATP measurement. ATP was quantified using CellTiter-Glo Luminescent Cell Viability Assay kit (Promega, G7571), as described by the manufacturer. Luminescence was measured on a Tecan Spark 20M multimode microplate reader.
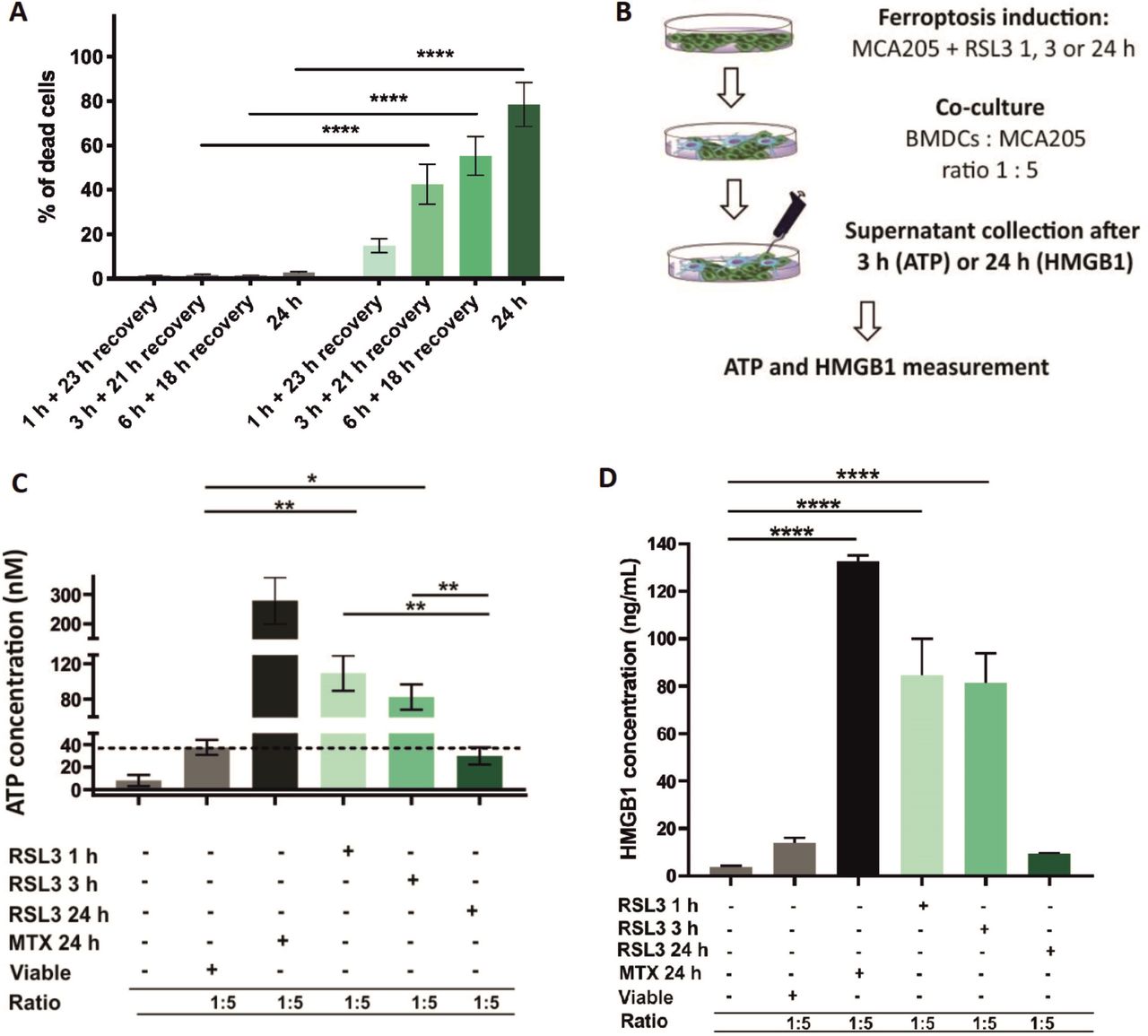
ATP release was also analyzed in the supernatants after 3 hours and 24 hours of cocultivation of BMDCs with MCA205 cells (ratio 1:5). For this analysis, MCA205 cells were pre-treated for 1, 3, or 24 hours with 2.5 µM of RSL3 or for 24 hours with 2 µM MTX. Controls were left untreated. BMDCs and MCA205 cocultivation and ATP measurement were performed as described above.
Quantification of HMGB1 release
MCA205 cells were induced with 2.5 µM RSL3 for 1, 3 or 24 hours or with 2 µM MTX (Sigma Aldrich, used as a positive control). After treatment, the supernatants were collected, cleared by centrifugation and frozen at −20°C for later HMGB1 quantification with an ELISA kit (IBL-Hamburg). All assays were performed in accordance with the respective manufacturers’ instructions and HMGB1 was quantified using Tecan Spark 20M multimode microplate reader. The data were analyzed with a four-parameter logistic curve fit.
HMGB1 release was also analyzed in the supernatants after 1, 3 and 24 hours of coculture of BMDCs with MCA205 cells (ratio 1:5). For this analysis, MCA205 cells were pretreated for 1, 3 or 24 hours with 2.5 µM of RSL3 or for 24 hours with 2 µM MTX. Controls were left untreated. BMDCs and MCA205 coculture and HMGB1 measurement were performed as described.
Generation of mouse BMDCs
BMDCs were harvested from the femurs and tibias of C57BL/6 J mice (age 7–10 weeks) and differentiated for 9 days using RPMI 1640 medium supplemented with 5% heat-inactivated FBS, 20 ng/mL mouse granulocyte macrophage colony stimulating factor (VIB Protein Core, IRC-UGent, Ghent, Belgium), 1% L-glutamine, 1 mM sodium pyruvate, 50 µM β-mercaptoethanol, 100 U/mL penicillin and 100 µg/L streptomycin. The culture medium was refreshed on days 3 and 6.
Phagocytosis assay
Target fibrosarcoma MCA205 cells were either left untreated (control, live cells) or induced to die by stimulation with 2.5 µM of RSL3 for 1 hour or 24 hours. The cells were collected, washed and labeled with 20 ng of pHrodo iFL Green STP Ester (Thermo Fisher, P36012) per 106 cells in PBS for 30 min. They were then washed and cocultured with BMDCs at ratio of 1:1 or 1:5 for 2 hours. Afterward, the cocultured cells were harvested, incubated with a mouse Fc block (eBioscience, 16016185), immunostained with phycoerythrin (Pe)-Cy7-anti-CD11c (BD PharMingen, 561022) and analyzed by a BD FACSCanto flow cytometer. The FlowJo software (V.10.0.8) was used for the flow cytometry data analysis. True engulfment of pHrodo-labeled dead cells by BMDCs was calculated based on a gating strategy that allows analysis of only single cells. They were defined as CD11c+pHrodo+ double-positive cells. Importantly, before setting up the phagocytosis assay, the percentage of the target MCA205 cell death was always measured by flow cytometry as described in the section on cell death assay.
Analysis of the expression of BMDCs surface markers and IL-6 production
Cell death was induced in fibrosarcoma MCA205 cells by addition of MTX for 24 hours or RSL3 for 1 hour, 3 hours or 24 hours as described in the section on cell death assay. The dead MCA205 cells were collected, washed in RPMI culture medium and cocultured with 25×104 BMDCs at a ratio of 1:5. The cells were cocultured for 24 hours at 37°C in six-well plates in 2 mL of RPMI culture medium. The BMDCs that were left untreated or stimulated with 20 ng/mL lipopolysaccharide (LPS; Sigma Aldrich, L2630) were used as a control. After 24 hours of coculture, all the cells were harvested, spun down (200 × g for 5 min at 4°C) and washed once in Dulbecco’s PBS (DPBS; Gibco). Maturation of BMDCs was analyzed by immunostaining with Pe-Cy7-anti-CD11c (BD PharMingen, 561022), FITC-anti-MHC class II (eBioscience, 11532282), eFluor 450-anti-CD86 (eBioscience, 48086280), eFluor 450-anti-CD80 (eBioscience, 48080182) and mouse Fc block (eBioscience, 16016185). Mature BMDCs cells were identified as a percentage of CD11c+MHCII+, CD11c+CD80+ and CD11c+CD86+ cells. The BD FACS Canto II flow cytometer was used for sample acquisition and the data were analyzed with FlowJo software (V.10.0.8). Before the maturation assay, the percentage of death of the target MCA205 cells was always measured by flow cytometry as described in the section on cell death assay. After coculturing BMDCs with the ferroptotic MCA205 cancer cells, the supernatants were collected and IL-6 was measured by ELISA (BioLegend).
RNA isolation and BMDCs enrichment after coculture with glioma GL261 cells
For the RNA sequencing analysis, there were three experimental groups: BMDCs alone (a negative control), BMDCs cocultured for 6 hours with GL261 cells pretreated with either MTX (a positive control, 2 µM, 24 hours) or RSL3 (2.5 µM, 24 hours). BMDCs isolated from four mice were used for each experimental group and were cocultured with GL261 cells in a ratio of 1:5. For the BMDCs cocultures with GL261 cells treated with RSL3 or MTX additional technical replicates have been included. After 6 hours of coculture, all the cells were harvested and washed once with DPBS. For the first step of BMDCs enrichment, Dead Cell Removal Kit (MACS Miltenyi Biotec) was used to remove the dead cells from the coculture. After that, the final eluate was loaded onto MS columns (MACS Miltenyi Biotec) and treated with the CD11c MicroBeads UltraPure kit (MACS Miltenyi Biotec) to enrich for CD11c+ BMDCs. The enrichment quality was measured by flow cytometry on a BD FACS Canto II flow cytometer. After enrichment/purification of CD11c+ cells, total RNA was extracted using RNeasy Mini Kit (QIAGEN). The quantity and integrity of the isolated RNA were analyzed with a NanoDrop 8000 spectrophotometer (Thermo Fisher) and a Fragment Analyser 5200 system (Agilent), respectively. The purity of CD11c+ BMDCs after each step was tested on the BD FACS Canto II flow cytometer as described above.
RNAseq analysis
RNAseq library preparation was performed using the TruSeq Stranded mRNA kit (Illumina) according to the manufacturer protocol, followed by PE100 cycles sequencing on one lane of a NovaSeq 6000 S1 run (Illumina). Gene expression values were determined using Kallisto. Limma-Voom was used for differential expression analysis, with Benjamini-Hochberg-based multiple testing correction.
Mice
Immune-competent C57BL/6 J mice and immune-compromised Rag-2-/- mice on C57BL/6 background (7–9 weeks old) were housed in specific pathogen-free conditions. The experiments were performed according to the guidelines of the local Ethics Committee of Ghent University.
In vivo prophylactic tumor vaccination
Death of MCA205 cells was induced in vitro by treatment with RSL3 for 24 hours or 3 hours, as described above. Next, the cells were harvested, washed once in DPBS and resuspended at a density 5×105 cells per 200 µL of DBPS. Mice were injected subcutaneously with 5×105 ferroptotically dying MCA205 cells with or without the addition of 4 mg/kg oxiATP (adenosine 5′-diphosphate, periodate oxidized sodium salt, Sigma Aldrich) per mouse38 39 or with PBS. As a negative control, mice were vaccinated with 5×105 MCA205 cells subjected to freezing and thawing to cause accidental necrosis, which has been shown to be nonimmunogenic.22 40 On day 7 after vaccination, the mice were challenged subcutaneously on the opposite flank with 1×105 viable MCA205 cells. In some experiments, mice were vaccinated with a half dose of ferroptotically dying MCA205 cells (2.5×105 cells) and on day 7 challenged on the opposite flank with 1×105 viable MCA205 cells. Tumor growth at the challenge site was monitored for up to 21 days after the challenge by using a caliper.
Statistical analysis
Statistics was calculated in GraphPad Prism (V.8.0.1). Cell death was analyzed by two-way Analysis of Variance (ANOVA) followed by Dunnett’s multiple comparisons test. The release of ATP in the coculture after 3 hours and 24 hours was analyzed by two-way ANOVA. The results of the phagocytosis assay and the BMDCs activation and maturation assay were analyzed by two-way ANOVA with Tukey’s multiple comparisons test. Kaplan-Meier survival curves showing the timeline of tumor development were analyzed by log-rank Mantel-Cox test. Linear correlation between markers of phenotypic BMDCs maturation and cell death (Annexin V+ Sytox+ cells) was calculated by linear regression. We have chosen a 95% CI and all data showed p<0.001.
Results
GPX4 inhibition induces ferroptosis in cancer cells
First, we examined the possibility of inducing ferroptotic cell death by inhibiting GPX4 in murine fibrosarcoma MCA205 cells. Cell death was evident at an early timepoint (3 hours of stimulation with 2.5 µM of RSL3) and maximal after 24 hours of induction, when almost all the cells were dead (figure 1A). Phase-contrast and confocal fluorescent microscopy (figure 1B, online supplemental movie S1) revealed cell rounding, swelling and plasma membrane permeabilization, typical hallmarks of necrotic cell death. AFM further showed the appearance of uniform circular protrusions on the surface of ferroptotic cancer MCA205 cells ranging from 1 to 5 µm after 1 hour of stimulation with RSL3 (figure 1C). These morphological irregularities during ferroptosis are in agreement with the previously described AFM features of ferroptotic cancer cells41 as well as with confocal microscopic findings.42
Supplementary video

Ferroptotic cell death induction in cancer MCA205 cells. (A) Ferroptotic cell death analysis by staining with Sytox Green (% of dead cells). Cell death was significantly reduced by ferroptosis inhibitors (DFO, Fer-1 and α-toc) whereas inhibitors of apoptosis (zVAD-fmk) and necroptosis (Nec-1s) were ineffective. The following concentrations of inhibitors were used: 25 µM zVAD-fmk, 20 µM Nec-1s, 1 µM FER-1, 10 µM DFO and 100 µM α-toc. The values are the means±SEM and represent four independent experiments. Statistical significance was calculated by two-way ANOVA followed by Dunnett’s multiple comparisons test: ****p<0.0001. (B) Morphological analysis of ferroptotic MCA205 cells by fluorescence microscopy showing swelling and rounding of MCA205 cells undergoing ferroptosis. MCA205 cells were stained with propidium iodide (red) and Hoechst33342 (blue). The images were derived from online supplemental movies S1. (C) Characterization of ferroptotic MCA205 cells by AFM revealing the formation of membrane protrusions (white arrows) on the surface of ferroptotic MCA205 cells. Each panel is divided into two images: left: 3D AFM image; right: whole cell 2D AFM image. (A–C) MCA205 cells were stimulated with 2.5 µM RSL3. DFO, deferoxamine; Fer-1, ferrostatin-1; RSL3, RAS-selectivelethal 3; α-toc, α-tocopherol.
To identify the type of cell death induced by RSL3 in MCA205 cells, specific cell death inhibitors were used to block apoptosis (zVAD-fmk, a pan-caspase blocker), necroptosis (Nec-1s, a RIPK-1 inhibitor) or ferroptosis (DFO), an iron chelator, ferrostatin-1, an inhibitor of reactive oxygen species and lipid peroxidation and α-toc, a type of vitamin E).36 43 Importantly, the cell death induced by RSL3 was significantly blocked by the ferroptosis inhibitors ferrostatin-1, DFO and α-toc but not by the pan-caspase inhibitor zVAD-fmk or the RIPK-1 inhibitor Nec-1s (figure 1A). These results indicate that inhibition of GPX4 in fibrosarcoma MCA205 cells leads to induction of purely ferroptotic cancer cell death.
Late ferroptotic cancer cells are phagocytosed but do not induce maturation of BMDCs
After 24 hours of stimulation with RSL3, 90% of MCA205 cells were double positive for Annexin-V/Sytox Blue staining (figure 2A) and were thus in the late stage of ferroptosis. Therefore, we tested the immunogenic properties of late ferroptotic cancer cells in vitro. By coculturing primary BMDCs with untreated ferroptotic cells (viable MCA205 cells) or cells treated for 24 hours with RSL3 (late ferroptotic MCA205 cells), we observed that the late ferroptotic cells were effectively engulfed by BMDCs but the viable cells were not (figure 2B,C). Increasing the ratio of BMDCs to late ferroptotic MCA205 cells from 1:1 to 1:5 increased the rate of engulfment proportionately (figure 2B,C).

Phagocytosis assay showing effective uptake of early ferroptotic cancer cells by BMDCs. (A) Ferroptotic cell death measured by flow cytometry of the MCA205 cells used in the phagocytosis assay shown in figure 2B,C. Quantification was done by Annexin-V (AnV) and Sytox Blue (Sytox) staining. The values are the means±SEM and represent four independent experiments. (B) Ferroptotic MCA205 cancer cells are efficiently engulfed by BMDCs. Of note, ferroptotic MCA205 cells are efficiently engulfed after 1 hour stimulation with RSL3 even in the absence of Annexin-V/Sytox Blue staining (figure 2A). MCA205 cells were stimulated with RSL3 for 1 hour or 24 hours. Data represent mean values±SEM from four independent experiments. Statistical significance was calculated by two-way ANOVA followed by Tukey’s multiple comparisons test: **p<0.01, ***p<0.001, ****p<0.0001. (C) The flow cytometry dot plots show the uptake of pHrodo-labeled ferroptotic cells by BMDCs (CD11c+pHrodo+ double positive cells). The rate of phagocytosis is proportionally increased by increasing the ratio of BMDCs: MCA205 (1:1 to 1:5). BMDCs, bone-marrow derived dendritic cells; RSL3, RAS-selectivelethal 3.
In order to obtain a better insight into the functional status of BMDCs, we evaluated the immunogenic characteristics of late ferroptotic cells in vitro (ie, after 24 hours of RSL3 stimulation, figure 3A–E). For this, we compared BMDCs exposed to late ferroptotic cells to BMDCs exposed to viable cancer cells. For a positive control, we induced immunogenic apoptosis by treating MCA205 cells with MTX,10 22 and LPS was used as another positive control. Surprisingly, late ferroptotic cells did not induce a phenotypic maturation of BMDCs, as indicated by the absence of surface upregulation of the costimulatory molecules CD11c+CD86+, CD11c+CD80+ and CD11c+MHCII+ (figure 3A,B) and levels of IL-6 (figure 3D) when compared with BMDCs cocultured with viable cells. The nonimmunogenicity of late ferroptotic cancer cells was confirmed in vitro on another cancer cell line, murine glioma GL261 cells, which die ferroptotically after RSL3 stimulation (online supplemental figure S1A,B). RNAseq analysis revealed that unlike GL261 cells treated with MTX, a well-known chemotherapeutic that induces ICD,10 22 coculture of late ferroptotic glioma cells with BMDCs did not induce an immunogenic gene expression profile in the BMDCs (online supplemental figure S2).
Supplemental material
Supplemental material
Supplemental material
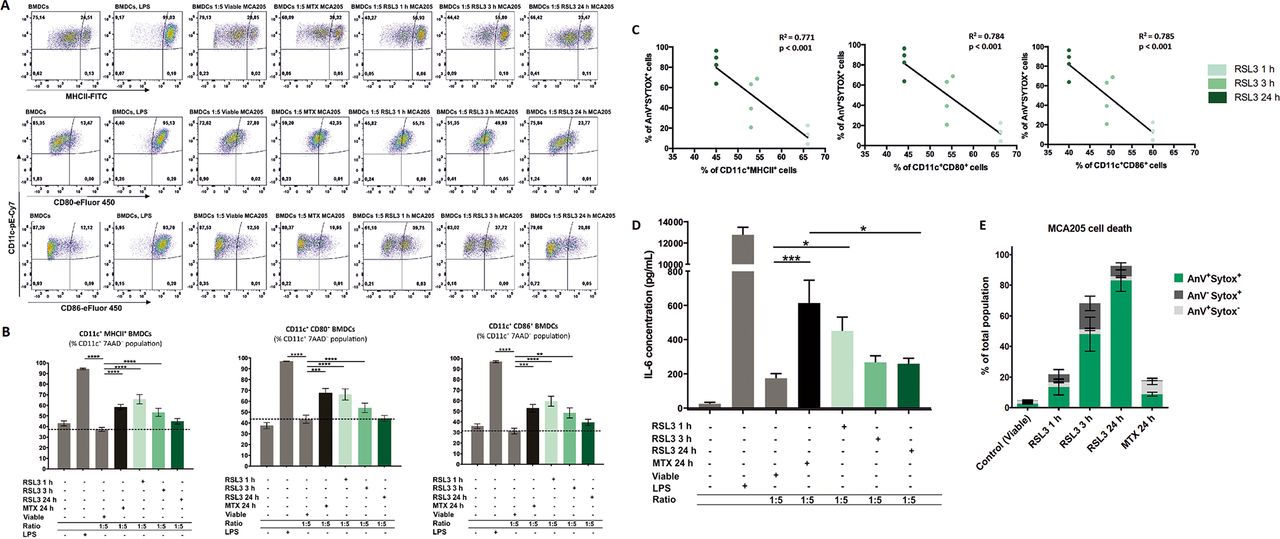
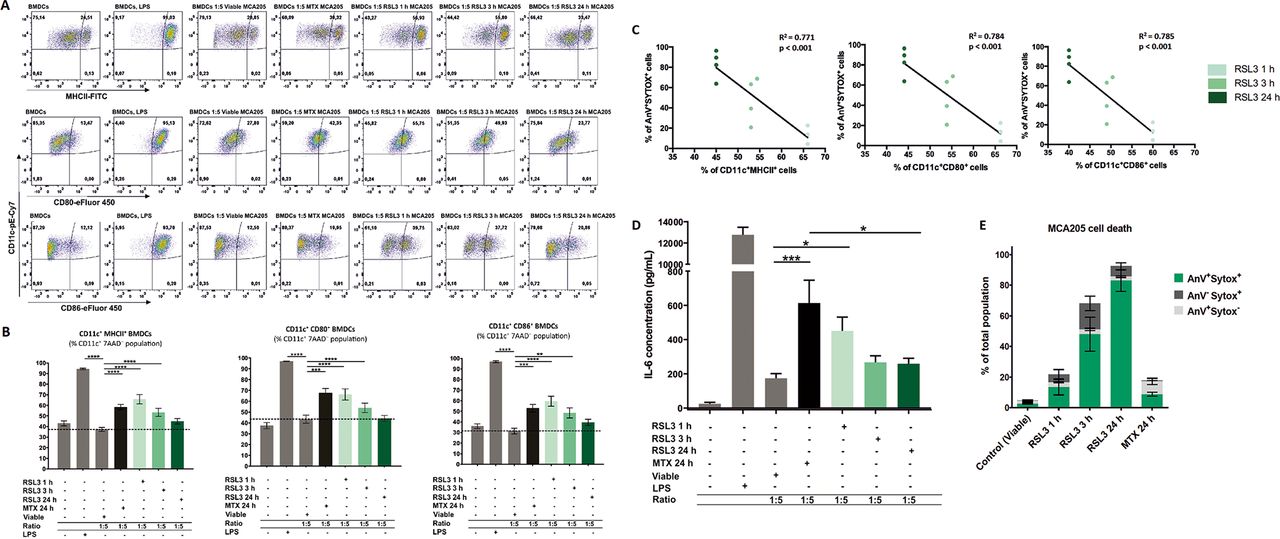
Ferroptotic cells induce maturation of bone-marrowderived dendritic cells (BMDCs) in a manner depending on the cell-death stage. (A) Representative flow cytometry dot plots demonstrating the gating and percentage of CD11c+MHCII+, CD11c+CD80+ and CD11c+CD86+ BMDCs cocultured in a 1:5 ratio with ferroptotic (RAS-selectivelethal 3 (RSL3) 1 hour, 3 hours, or 24 hours), untreated (viable), and mitoxantrone-treated (MTX) MCA205 cells. As a positive control BMDCs stimulated with lipopolysaccharide (LPS) were used. (B) Ferroptotic MCA205 cancer cells induce maturation of BMDCs in a manner that depends on the cell-death stage: early ferroptotic cells (after 1 hour or 3 hours of RSL3 stimulation) are more potent inducers of BMDCs maturation than late ferroptotic cells (after 24 hours of RSL3 stimulation). Notably, early ferroptotic cells (ie, after 1 hour of RSL3 stimulation) were as immunogenic as cancer cells treated with mitoxantrone (2 µM MTX, 24 hours), a positive control. BMDCs were cocultured with ferroptotic MCA205 cells in a 1:5 ratio. Dashed lines specify percentages of the CD11c+MHCII+, CD11c+CD80+ and CD11c+CD86+ populations, respectively, for BMDCs cocultured with viable MCA205 cells (control). BMDCs stimulated with LPS served as an additional positive control. Percentages of CD11c+MHCII+, CD11c+CD80+ and CD11c+CD86+ cells shown as mean values±SEM of four independent experiments for RSL3 1 hour, and seven independent experiments for other conditions. Two-way ANOVA was used to calculate the statistical significance: **p<0.01, ***p<0.001, ****p<0.0001. (C) Linear correlation of AnV+Sytox+ ferroptotic cells (figure 3E) and CD11c+MHCII+, CD11c+CD80+ or CD11c+CD86+ cells, respectively. (D) Absolute concentrations of IL-6 in cocultures of BMDCs with the respective target MCA205 cells at two different ratios (1:1, 1:5). LPS-treated BMDCs and coculture of BMDCs with MTX-treated MCA205 cells were used as a positive control. The values are the means±SEM of eight (for BMDCs alone, BMDCs cocultured with viable MCA205 cells, BMDCs cocultured with RSL3-treated MCA205 cells for 24 hours groups), seven (for BMDCs co-cultured with MTX-treated MCA205 cells, BMDCs co-cultured with RSL3-treated MCA205 cells for 3 hour groups) and six (for BMDCs treated with LPS, BMDCs cocultured with RSL-treated MCA205 cells for 1 hour groups) independent measurements performed in duplicates. Statistical significance was calculated by a Mann-Whitney nonparametric t-test: *p<0.05, ***p<0.001. (E) Ferroptotic cell death measured by flow cytometry of the MCA205 cells used in the maturation assay shown in figure 3A–C.
Together, these in vitro results indicate that late ferroptotic cancer cells that were treated with a GPX4 inhibitor for 24 hours are potent inducers of BMDC-mediated phagocytic clearance. However, our findings indicate that two different types of cancer cells (fibrosarcoma MCA205 cells and glioma GL261 cells) undergoing late ferroptosis cannot induce an immunogenic signature in BMDCs.
Early ferroptotic cancer cells induce maturation of BMDCs
It is conceivable that the immunogenicity of dying cancer cells may vary significantly depending on the stage of cell death. Therefore, to assess the dependency of the immunogenic properties of ferroptotic cancer cells on the cell-death stage, we cocultured BMDCs with cancer cells treated for 1 hour with RSL3, which leads to so-called early ferroptotic cell death. After 1 hour of RSL3 treatment, the cancer cells showed early signs of cell death. Less than 6% of the cancer cells were double positive for Annexin-V/Sytox Blue staining (figure 2A), indicating that they were still in an early stage of ferroptosis. By coculturing BMDCs with untreated, viable MCA205 cells or with MCA205 cells treated for 1 hour with RSL3 (early ferroptotic MCA205 cells), we observed that the early ferroptotic cells were efficiently engulfed by BMDCs but the viable cells were not (figure 2B,C). Increasing the ratio of BMDCs to early ferroptotic MCA205 cells from 1:1 to 1:5 increased the rate of engulfment proportionately (figure 2B,C). Importantly, only 15% of the cell population were Annexin-V+/Sytox Blue−, indicating that clearance of these early ferroptotic cells probably occurs via mechanisms other than PS exposure.44–46 Furthermore, when we cocultured these early ferroptotic cancer cells with BMDCs for 24 hours, we observed that early ferroptotic cancer cells induced CD80, CD86 and MHCII expression in CD11c+ BMDCs (figure 3A,B,E). Importantly, the substantial surface expression of these molecules was very similar to that induced by MTX, a positive control, when BMDCs were cocultured with early ferroptotic cells (figure 3A,B). Of note, coculture with the same amount of cancer cells undergoing late ferroptosis did not alter the maturation status of the BMDCs (figure 3A,B). Moreover, ferroptotic MCA205 cells after 3 hours of RSL3 treatment were also able to induce intermediate levels of CD80, CD86 and MHCII expression in CD11c+ BMDCs (figure 3A,B). There is, indeed, an inverse correlation between the stage of ferroptotic cell death in MCA205 cells and maturation status of the BMDCs (figure 3C).
To gain a deeper understanding of the functional status of BMDCs, we measured the production of the proinflammatory cytokine, IL-6. We compared BMDCs exposed to early ferroptotic MCA205 cells with those exposed to live MCA205 cancer cells. We found that BMDCs exposed to early ferroptotic cancer cells displayed a distinctive and ratio-dependent pattern of functional activation characterized by IL-6 production (figure 3D).
Early but not late ferroptotic cancer cells induce antitumor immunogenicity in mice that results in an adaptive immune response
To test the ability of early ferroptotic cancer cells to activate the adaptive immune system, we took advantage of the well-accepted prophylactic tumor vaccination model in immunocompetent C57BL/6 J mice (figure 4A).7 40 47 We immunized C57BL/6 J mice with early or late ferroptotic MCA205 cells (figure 4B, online supplemental figure S3B) and injected the negative control mice with PBS or with accidental necrotic MCA205 cells (F/T).40 47 The immunized mice were then challenged with viable MCA205 tumor cells. Protection against tumor growth at the challenge site was interpreted as a sign of successful priming of the adaptive immune system. Interestingly, mice immunized with late ferroptotic MCA205 cells (24 hours of RSL3 induction) remained significantly less tumour-free at the challenge site (figure 4B), indicating that late ferroptotic cells are not immunogenic in vivo, confirming our initial in vitro observations (figures 2B,C and 3A–E and online supplemental figure 2C–H). In contrast, mice immunized with early ferroptotic MCA205 cells showed attenuation of the appearance of tumors at the challenge site (figure 4B), confirming the strong immunogenic potential of early ferroptotic cells seen in vitro (figure 3A–D). Notably, the mice vaccinated with late ferroptotic cells developed tumors very similar to mice vaccinated with F/T cells, which are known to be nonimmunogenic.22 40 48 It is noteworthy that early ferroptotic cancer cells were significantly more immunogenic than accidental necrotic cells. This is in line with the immunogenicity of late ferroptotic cancer cells, but it was completely lost when the mice were vaccinated with half the number of cells, whereas the early ferroptotic cancer cells remained immunogenic (figure 4, online supplemental figure S3A,B). These results indicate that the immunogenicity of ferroptosis depends on the cell death stage: only early but not late ferroptotic cancer cells induced protection in the murine vaccination experiments.
Supplemental material
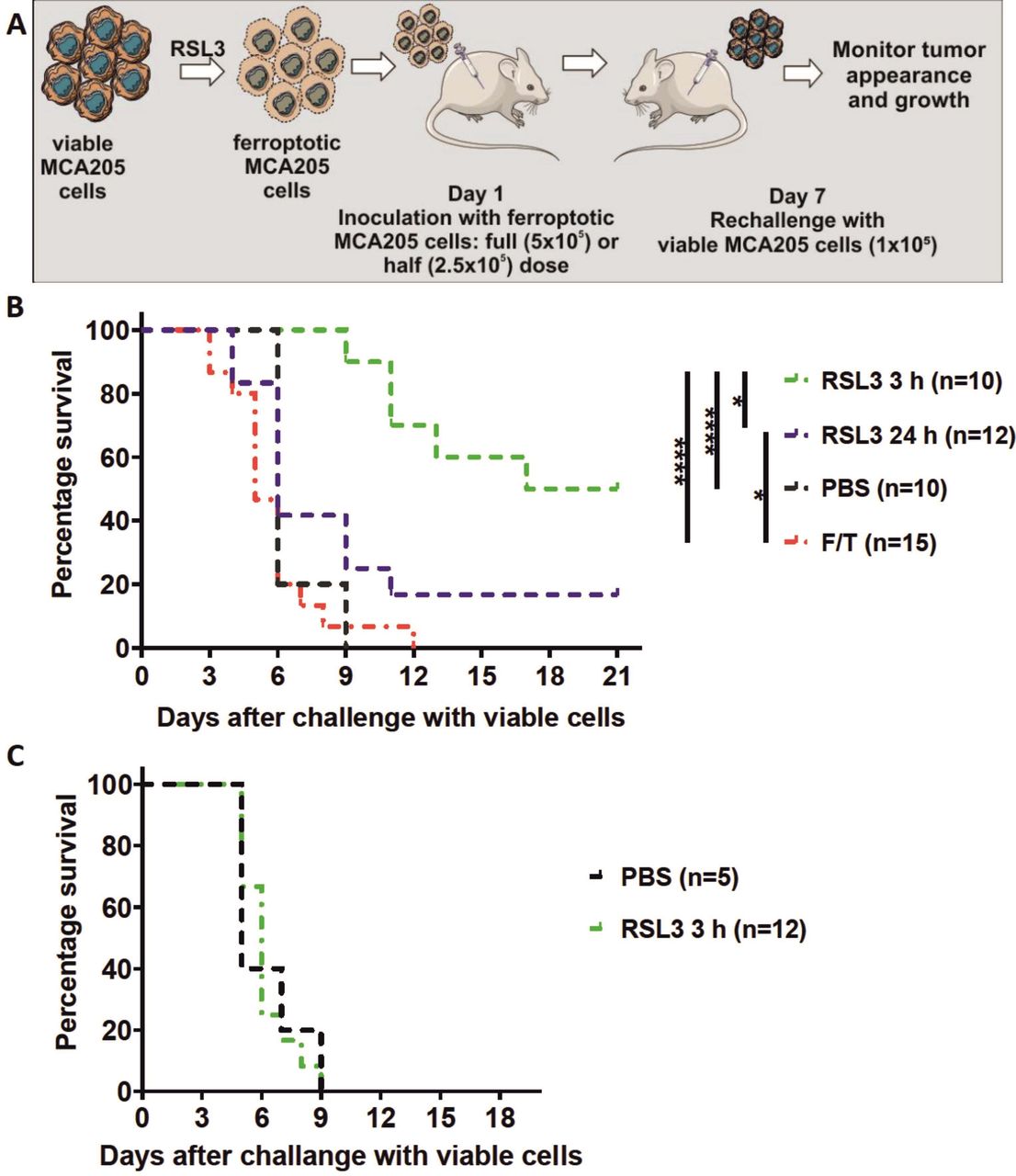
Early ferroptotic cells are immunogenic in vivo. (A) The in vivo prophylactic tumor vaccination model. The MCA205 cells used for immunization were stimulated with RSL3 for 3 hours or 24 hours and resuspended in PBS before injection. (B) Kaplan-Meier curve of the progression of tumor development over time. C57BL/6 J mice were vaccinated with 5 × 105 MCA205 cells treated with RSL3. Then 1 week late the mice were challenged with viable MCA205 cells. MCA205 cells induced by RSL3 for 3 hours triggered an anti-tumor immune response in mice immunized with 5 × 105 cells. Negative control mice were injected with PBS or with MCA205 cells undergoing accidental necrosis (F/T, freeze and thaw group). (C) Kaplan–Meier curve of the progression of tumor development over time. Rag-2-/- mice were vaccinated with 5 × 105 MCA205 cells treated with RSL3 for 3 hours. after 1 week, the mice were challenged with viable MCA205 cells. No antitumor immune response was triggered in mice Rag-2−/− immunized with ferroptotic MCA205 cells. The statistical differences were calculated by a log-rank (Mantel-Cox) test. Survival curves comparison: *p<0.05, ****p<0.0001. PBS, phosphate-buffered saline; RSL3, RAS-selectivelethal 3.
To examine the role of the adaptive immune system, we determined whether vaccination of immune compromised mice with early ferroptotic cancer cells would lead to protective immunity against challenge with viable cancer cells. For this, Rag-2-deficient mice were inoculated with early ferroptotic cancer cells or with PBS and 1 week later challenged with viable MCA205 cancer cells (figure 4A). Interestingly, the tumor-protective effect of early ferroptotic cancer cells was absent in Rag-2−/− mice and was comparable to the mice vaccinated with PBS (figure 4C). These data suggest that the adaptive immune system is required for induction of tumor protection by early ferroptotic cancer cells.
Immunogenicity of early ferroptosis is associated with time-dependent ATP and HMGB1 release
One of the main characteristics of the immunogenicity of cancer cells undergoing ICD is the spatiotemporally coordinated emission of DAMPs (eg, ATP and HMGB1), which is beneficial in anticancer therapy owing to the interaction of these DAMPs with the immune system.8 39 49–52 As ATP is one of the most critical DAMPs, we first analyzed the kinetics of the ATP release induced by RSL3 in MCA205 cells. We noticed that ATP release temporally followed the cell death process (figure 5A,B). After 3 hours of RSL3 treatment, ferroptotic cancer cells started releasing ATP until 6 hours but by 24 hours, all released ATP was depleted (figure 5B). Of note, the pattern of HMGB1 release, another well-known DAMP, from ferroptotic cancer cells, was different from that of ATP release. The maximal level of HMGB1 release was reached only after 24 hours of stimulation with RSL3 (figure 5C,D), which corresponds to the late ferroptotic stage. The levels of HMGB1 in the supernatant from the early ferroptotic cells were not statistically different from the level in viable cancer cells (figure 5C).

Release of HMGB1 and ATP from ferroptotic cancer cells. (A) Cell death progression in cancer MCA205 cells on stimulation with RSL3 measured by Sytox Green fluorescence. Pool of three independent experiments. (B) Time-dependent ATP release from ferroptotic cancer MCA205 cells stimulated with RSL3. Note that the peak of ATP release corresponds to 6 hours of RSL3 stimulation but for 24 hours of stimulation ATP release is minimal. The values are the means±SEM of four independent measurements performed in triplicates. Two-way ANOVA was used to calculate the statistical significance: ***p<0.001, ****p<0.0001. (C) Release of HMBG1 from ferroptotic MCA205 cancer cells stimulated with RSL3. The peak of HMGB1 release was at 24 hours of ferroptosis induction with RSL3. This is in reverse to the release of ATP: release of HMGB1 was minimal for early ferroptotic time-points (1 and 3 hours). The values are the means±SEM of four independent measurements performed in duplicates. One-way ANOVA with Tukey’s multiple comparison test was used to calculate the statistical significance: *p<0.05, ****p<0.0001. (D) Ferroptotic cell death measured by flow cytometry of the MCA205 cells used for HMGB1 release. The values are the means±SEM of four independent measurements performed in duplicates. HMGB1,high-mobility group box 1; MTX, mitoxantrone; RSL3, RAS-selective lethal 3.
Next, we performed the cell death recovery experiment to better mimic in vitro coculture and prophylactic vaccination experiments in which the dead cells are detached and washed with PBS before their use in these experiments. Even after removing the RSL3 stimulus, the cells continued to undergo ferroptosis (figure 6A). To further confirm this hypothesis, we measured ATP and HMGB1 concentrations during coculture of early and late ferroptotic cancer cells with BMDCs (figure 6B), which most accurately reflects the actual situation occurring in vitro and in vivo. In cocultures of BMDCs with early ferroptotic cells (1 hour or 3 hours with RSL3), ATP concentration was 3.5 and 2.8 times higher, respectively, than in cocultures with late ferroptotic cells (24 hours with RSL3) (figure 6C). ATP levels of the coculture of BMDCs and cancer cells treated for 24 hours (late ferroptotic cells) were similar to those in BMDCs cocultures with viable cells (figure 6C). Surprisingly, we observed that the level of HMGB1 was at its maximum in the coculture of BMDCs with early ferroptotic cancer cells (1 hour or 3 hours with RSL3, figure 6D). The HMGB1 level was not statistically different in the cocultures of BMDCs with the late ferroptotic cells (24 hours with RSL3) from that in the control groups (ie, BMDCs alone or BMDCs cocultured with viable cancer cells) (figure 6D). All these data suggest a potential role of ATP and HMGB1 in the immunogenicity of early ferroptotic cancer cells.
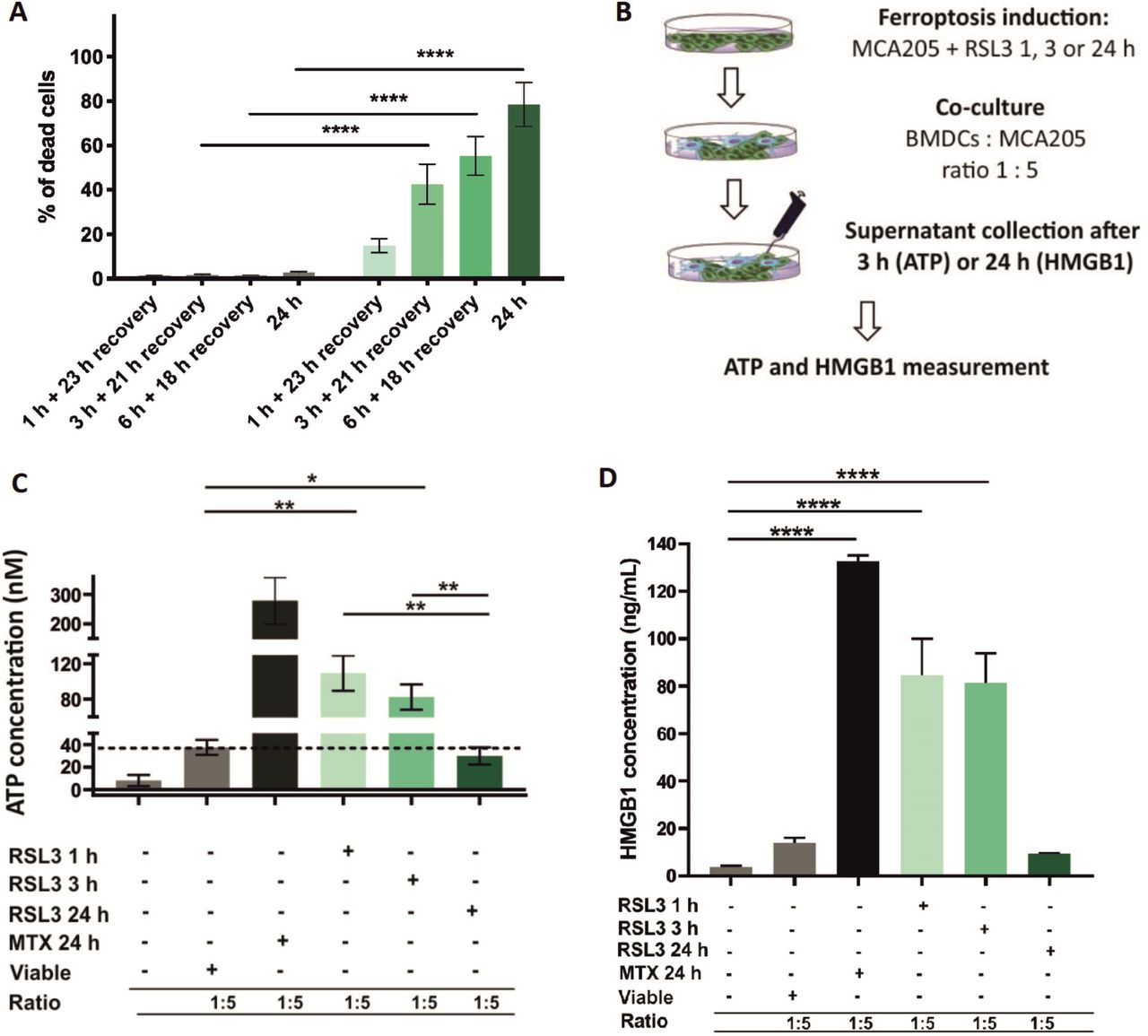
Early but not late ferroptotic cancer cells induce immunogenic cell death in vitro. (A) Cell-death recovery was measured by Sytox Green fluorescence in cancer MCA205 cells on stimulation with RSL3. After 1 hour, 3 hours and 6 hours of RSL3 stimulation, the cells were washed in PBS and reseeded in fresh culture medium (without RSL3) and cultured further for 23 hours, 21 hours or 18 hours, respectively, to give a total culture time. (B) Schematic representation of the measurement of ATP and HMGB1 release in the coculture of BMDCs with ferroptotic MCA205 cells shown in figure 6C,D. Viable or dying cancer MCA205 cells (induced with either RSL3 or MTX as positive controls) were harvested and cocultured with BMDCs for 3 hours (for ATP) or 24 hours (for HMGB1). Next, the supernatants were collected and analyzed for ATP using CellTiter-Glo Luminescent Cell Viability Assay (Promega) and for HMGB1 with ELISA kit (IBL-Hamburg). (C) The concentration of ATP in the coculture of BMDCs with ferroptotic MCA205 cells after 3 hours of coincubation. The values are the means±SEM of four independent measurements performed in triplicates. Two-way ANOVA was used to calculate the statistical significance (*p<0.05, **p<0.01). (D) The concentration of HMGB1 in the coculture of BMDCs with ferroptotic MCA205 cells after 24 hours. The values are the means±SEM of three independent measurements performed in duplicates. One-way ANOVA with Tukey’s multiple comparison test was used to calculate the statistical significance: ****p<0.0001. For positive control, BMDCs were cocultured with MCA205 cells killed by MTX (2 µM, 24 hours). Activation and maturation profiles of BMDCs from these experiments are shown in figure 3A–C. BMDCs,bone-marrow derived dendritic cells; HMGB1, high-mobility group box 1; MTX, mitoxantrone; PBS, phosphate-buffered saline; RSL3, RAS-selectivelethal 3.
In the light of the in vitro and in vivo data, we hypothesize that ATP along with HMGB1 might be crucial for the immunogenicity of early ferroptosis. Indeed, it has been shown that ATP release by cells undergoing immunogenic apoptosis is one of the major hallmarks of ICD.3 8 50 To verify this hypothesis in vivo, we used the strategy of modulating the function of extracellular ATP by blocking purinergic receptors with the 2′,3′-dialdehyde derivative of ATP ('oxidized ATP', oxiATP), which inhibits the P2X7 subclass of purinergic receptors.9 38 50 Blockade of purinergic receptors in early ferroptotic MCA205 cells (3 hours of RSL3 induction) significantly abolished their ability to protect the host against a lethal rechallenge with viable MCA205 cells (online supplemental figure S3C). Taken together, all these results underscore the cardinal contribution of ATP as one of the potential DAMPs in the immune response elicited by early ferroptotic cancer cells and underline the immunogenic potential of early ferroptotic cells.
Discussion
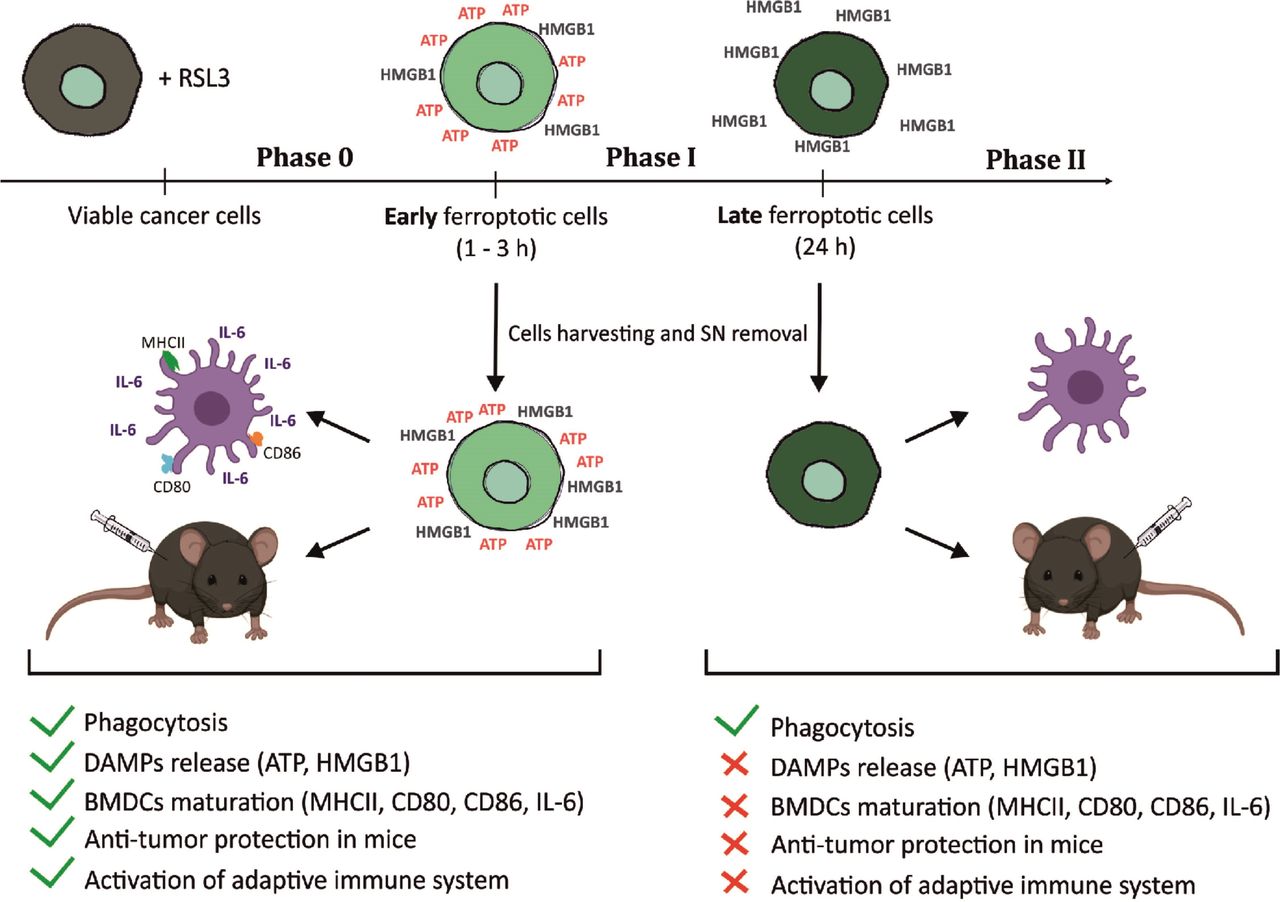
ICD is a finely regulated process through which dying cells can stimulate the adaptive immune system. Apoptotic and necroptotic ICD modalities have been extensively described and characterized22 53 54 but no conclusive evidence is available about the immunogenicity of ferroptosis.34 Here, we demonstrate for the first time that ferroptosis induces ICD both in vitro and in vivo (figure 7). We show that the ferroptotic stage of dying cells is crucial in determining their immunostimulatory capacity and that several DAMPs, including ATP and HMGB1, might be necessary for promoting ferroptotic ICD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Graphical abstract—timespan of immunogenicity of ferroptotic cancer cell death. After receiving ferroptotic stimuli, cancer cells start to undergo cell death (Phase 0) and release DAMPs (eg, ATP and HMGB1). After 3 hours of ferroptosis induction, cancer cells are in phase I, which is the early ferroptotic cell death stage, where the highest levels of ATP and HMGB1 are reached. At this time point, early ferroptotic cancer cells are engulfed by BMDCs and promote their activation and maturation, as evidenced by analyses of their phenotypic markers (MHCII, CD80, and CD86) and the pro-inflammatory cytokine IL-6. In addition, if early ferroptotic cells in Phase I are injected into mice, they lead to protective immunity in the mouse tumor prophylactic vaccination model if the adaptive immune system is intact. In the late ferroptotic phase II, cancer cells are no longer immunogenic. Because all the supernatants (SN) from the late ferroptotic cancer cells (treated with RSL3 for 24 hours) are removed and no DAMPs (ie, HMGB1, ATP) are present anymore, which are required for their immunogenicity. Notably, although late ferroptotic cells were engulfed by BMDCs, they failed to induce BMDC phenotypic activation and maturation in vitro or to induce an effective antitumor immune response. It is crucial that phase I ferroptotic cell death occurs in the presence of antigen-presenting cells and should preferably proceed in vivo. During that stage (ie, phase I), most of ATP and HMGB1 and possibly other DAMPs are released or reaching the maximal levels, thereby creating the best adjuvanticity effect of early ferroptotic cancer cells. These data show that the stage of cell death is a key aspect of the immunogenicity of ferroptotic cancer cells and demonstrate that ATP and HMGB1 released after ferroptosis induction may act as immunogenic signals. BMDCs,bone-marrow derived dendritic cells; DAMPs, damage-associated molecularpatterns; HMGB1, high-mobility group box 1.
The first discovery we made is that only early dying ferroptotic cells can induce ICD efficiently in vitro and in vivo. We noticed that, in vitro, early ferroptotic cells were engulfed by BMDCs, but to a lesser extent than late ferroptotic cells. When we cocultured BMDCs with ferroptotic cells, we noted an inverse relationship between how long tumor cells were treated with RSL3 before coculture and the extent of BMDCs maturation (figure 3C). MCA205 cells pretreated for 1 hour increased the expression of all BMDCs maturation markers and IL-6 levels to the same or even greater extent than the bone fide ICD inducer MTX, which was used as a positive control (figure 3A–D). Tumor cells treated for 3 hours were somewhat less effective than cells treated for 1 hour with RSL3, while no BMDCs phenotypic maturation was observed when late ferroptotic MCA205 cells were used (24 hours of RSL3 treatment).
Our results show that early dying cells, which are characterized by an intact membrane, are more immunogenic than late-dying cells, which are spilling out all their intracellular material. Studies on immunogenic apoptosis and necroptosis have disclosed that the immune stimulation is a tightly regulated mechanism, with each stimulus, i.e., DAMP, activated at a precise moment and that the activation of all DAMPs in a specific sequence executes effective activation of the immune system.2 7 Depending on the stimulus, a generic chemotherapeutic agent exerts a cytotoxic effect that immediately determines whether cell death will be immunogenic, silent or tolerogenic. Indeed, every cell death program enacts the exposure of different ‘eat me’ and ‘don’t eat me’ signals, which, combined with different receptors that either promote or inhibit engulfment and, thus regulate uptake of the cells.55 For instance, PS externalization on the outer cell surface is one of the best-known crucial events in the recognition and elimination of dying cells. In apoptotic cells, it represents a point of no return and flags apoptotic cells to alert professional antigen presenting cells (APCs), such as BMDCs, to clear them.56–58 Similarly, in necroptotic cells, PS promotes their clearance. PS-positive necroptotic cells can be revived and start clonal growth.26 59–61 Interestingly, we found that early ferroptotic cancer cells were efficiently cleared by BMDCs but remained largely negative for Annexin-V staining (figure 2A,B), a marker often used to detect PS on the cell surface of dying cells.62 63 Thus, it is conceivable that other ‘eat me’ signals besides PS could be involved in the clearance of early ferroptotic cells. For instance, during immunogenic apoptosis, translocation of CRT to the outer cellular membrane (Ecto-CRT) is usually the first ‘eat me’ signal emitted, happening even before PS exposure.10 64 Ecto-CRT on a membrane on which no PS has been exposed has been proven to provide the switch to a cognate immune response by APCs such as macrophages and dendritic cells.10 64–67 Ecto-CRT could be the missing piece that would explain why early ferroptotic cells can be significantly engulfed by BMDCs, while viable cells are not (figure 2B,C), even if they share the lack of exposed PS (figure 2A).
Notably, it has been shown that the stage of apoptotic cell death is not important for its immunogenicity. Indeed, no correlation was found between the immunogenicity of early dying (Annexin-V+/PI–) and late-dying (Annexin-V+/PI+) apoptotic cells. This means that both early10 68 69 and late14 70 apoptotic cells trigger ICD both in vitro and in vivo. This is at odds with our finding that immunogenicity of ferroptosis depends on the cell-death stage.
Driven by these results and in order to choose the best stage of cell death for in vitro and in vivo vaccination, we simulated in vitro what happens to tumor cells after the ferroptotic stimulus is removed. In the in vitro experimental setup, we simulated the in vivo prophylactic vaccination and BMDCs coculture conditions by washing the dying cells in PBS before injection into mice. We observed that cells treated with RSL3 continue to die after reseeding in fresh medium without the inducer (figure 6A). We were looking for a relationship between the immunostimulatory effect and cell death. The amount of cell death after 1 hour of treatment with RSL3 was not enough for the cells to be used in the in vivo experiments. Therefore, we compared the in vivo effect of ferroptotic cells treated for 3 hours with those treated for 24 hours. The results confirmed the abovementioned trend: vaccination experiments showed significant activation of the adaptive immune system by injection of early ferroptotic cells (3 hour stimulation) but no effect of late ferroptotic cells (24 hours stimulation). This was confirmed by using immune-deficient (Rag-2−/−) mice, in which the immunogenicity of early ferroptotic cancer cells was absent (figure 4C). The role of the adaptive immune system in vaccination with apoptotic14 71 and necroptotic24 72 cells has been previously shown.
Even if we find an explanation for the immunogenicity of early ferroptotic cells, the lack of immunogenicity of the late death stage remains, in a way, counterintuitive and challenges the current concept of the high immunogenic potential of cells in the later stages of cell death. Indeed, it is commonly believed that the release of the complete intracellular content, including a large amount of DAMPs (ie, inflammatory stimuli), occurs mainly in the later stages of cell death when the plasma membrane ruptures, and so the later stages represent the best way to induce an antigen-specific adaptive immune response mediated by CD8+ T cells.73–75 However, our in vivo vaccination experiments showed that ferroptotic cells in the late stage of death, like accidental necrotic cells killed by freezing and thawing, were unable to protect mice from tumor development at the challenge site. This challenges the notion that for local inflammation and infiltration, the massive release of intracellular content during late cell-death stages is always proinflammatory and immunogenic. The nonimmunogenicity of cancer cells undergoing accidental necrosis has been described,14 22 73 and in this study, we show that ferroptotic cancer cells are immunogenic in the early stages of cell death.
As mentioned above, DAMPs release during ICD is not a continuum, and exposure or secretion of ‘eat me’ and ‘find me’ signals probably does not persist for the duration needed for cells to die. Recently, it has been shown that ferroptotic cells release HMGB1.76 77 In contrast, we show here that several DAMPs, including ATP and HMGB1, are released from ferroptotic cancer cells in a strict time-dependent manner. In other words, with sequential presentation of DAMPs, they can coexist at the same stage, and synergy of their signals will lead to development of an effective immune response.65 Our data indicate that the process of cell death from its very early stages would occur in situ (figure 7), which will enable the release of the DAMPs in a consequent manner during the cell death process, leading to the induction of effective immune responses. Thus, from a practical point of view, it could be best to perform vaccination with cells in the middle of the process of DAMPs release so that early, middle and late danger signals might occur in situ (figure 7).
Key DAMPs might be depleted in cancer cells in the latest stage of cell death. Furthermore, the experimental setting requires that dying cancer cells are washed before injection in mice or testing for BMDC studies, so soluble DAMPs already secreted in the medium will be lost. If those missing DAMPs are important in the activation of adaptive immunity, the result would be an immunologically silent cell death or even immune suppression. Therefore, the time dependency of ICD in terms of DAMPs activation and presentation to the immune system effectors, in contrast to the mere spilling of all signals, might explain the lack of protection against tumor development in mice when late ferroptotic cells are used as a vaccine.
Together with CRT, HMGB-1 and ATP represent two limiting features of the immunogenicity of apoptosis and necroptosis.11 22 50 78 It has been demonstrated that active secretion of ATP and passive release of HMGB1 are crucial for the generation of an adaptive immune response to apoptotic ICD,50 while necroptotic immunogenic cells can passively release this crucial DAMP.22 23 Here, we show that RSL3 promotes a perimortem, time-dependent ATP and HMGB1 release, most probably due to rupture of the cell membrane, which is maximal when cells are treated with RSL3 for up to 6 hours for ATP and up to 24 hours for HMGB1. The kinetics of ATP and HMGB1 release overlaps that of cell death (figure 5A–D), and thus the cells are most likely not releasing ATP and HMGB1 actively. Even if we did not observe premortem ATP secretion, the interesting point is that after 24 hours, ATP was completely degraded (figure 5B). This is in line with previous reports on ATP exhaustion after cell death stimulation for a long time.22 79 80 So, to assess whether the lack of ATP and HMGB1 explains the inability of late ferroptotic cells to activate the adaptive immune system, we measured their levels in the supernatants of BMDCs cocultured with ferroptotic MCA205 cells treated with RSL3 for different times. Importantly, the levels of ATP and HMGB1 were maximal when the cells were treated for a short time and therefore in the early stages of ferroptosis (figure 6C,D). Additionally, if we blocked the cognate ATP P2X7 purinergic receptors in vivo, 3-hour ferroptotic cells were not protective in mice, demonstrating their inability to trigger an adaptive immune response. All these data together suggest that passive release of ATP and possibly also HMGB1 are essential for the immunogenicity of ferroptosis. These results also explain why late stages of ferroptotic cancer cell death are not immunogenic. Moreover, it is important to emphasize that the nonimmunogenic nature of late ferroptotic cancer cells is not tumor-type specific. As RNA-seq analysis shows, the gene expression profile of BMDCs is not significantly affected by 24-hour RSL3-treated GL261 cells compared with MTX treatment, pointing to absence of significant immunogenic potential in late ferroptotic cancer cells (online supplemental figure S2).
In addition, it is important to stress that an adaptive immune response depends on the recognition of different tumor associated antigens (TAA). Thus, ICD should have a balanced combination of adjuvanticity (effects related to DAMPs) and antigenicity (attributed mainly to tumor antigens) to induce efficient antitumor immunity. In this regard, it was recently illustrated that the strong expression of the immunodominant TAA, AH1, was indispensable for the immunogenicity of apoptotic cancer cells, whereas necroptotic cancer cells could also engage other neoepitopes.72 This finding highlights the importance of the immunodominant TAA and demonstrates its antigenic contribution to ICD, which is required, in addition to DAMPs, for induction of efficient antitumor immunity. Taken together, the results suggest that early and late ferroptotic cancer cells could have different patterns of TAA and neoantigens that contribute to their antigenicity and thus their immunogenicity and antitumor effect. This is a challenging area for future research.
Taking these data together, we have shown that the exhaustion of crucial effector DAMPs could explain the cell-death stage dependency of ferroptotic cells. Late ferroptotic cells used either for vaccination experiments or for coculture with BMDCs have already lost some key DAMPs, such as ATP and HMGB1, and so late-stage ferroptotic cells do not present all the necessary elements needed to promote the stimulation of the immune system and cannot trigger a vaccination-like effect (figure 7). We conclude that the stage of cell death is a key aspect of the immunogenicity of ferroptotic cancer cells and their effect on antitumor immunity. Further studies are needed to comprehend its molecular mechanism of action fully and determine whether other DAMPs, including Ecto-CRT and IFNs, are critical in this process.
Acknowledgments
D.V.K. lab is supported by FWO-Flanders (Research Foundation Flanders, 1506218N, 1507118N, G051918N). A.G.S. and D.V.K. acknowledge the support of FWO-Flanders (G043219N) and Ghent University BOF (Special Research Fund; IOP 01/O3618). Optimization of RNAseq protocol and DAMPs identification are supported by a grant from Russian Science Foundation (RSF, project No.18-15-00279) to D.V.K. S.L. held a postdoctoral fellowship (12R3618N) and IE holds a PhD fellowship (11F7721N) from FWO-Flanders.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
IE and EC contributed equally.
Contributors IE, EC, LVdM, OK, VT: performed the experiments and analyzed the data; TM, CB, MV, SL, CF, FC, AS, OK, DK: supervised the experiments; HH: provided resources; IE, EC, LVdM, SL, OK, HH, FC and DK: participated in manuscript preparation; DK: conceived of and designed the project. All authors read, discussed and approved the final manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.