Article Text

Abstract
Background Bintrafusp alfa is a first-in-class bifunctional fusion protein composed of the extracellular domain of transforming growth factor (TGF)-βRII (a TGF-β ‘trap’) fused to a human IgG1 mAb blocking programmed cell death ligand 1. This is the largest analysis of patients with advanced, pretreated human papillomavirus (HPV)-associated malignancies treated with bintrafusp alfa.
Methods In these phase 1 (NCT02517398) and phase 2 trials (NCT03427411), 59 patients with advanced, pretreated, checkpoint inhibitor-naive HPV-associated cancers received bintrafusp alfa intravenously every 2 weeks until progressive disease, unacceptable toxicity, or withdrawal. Primary endpoint was best overall response per Response Evaluation Criteria in Solid Tumors (RECIST) V.1.1; other endpoints included safety.
Results As of April 17, 2019 (phase 1), and October 4, 2019 (phase 2), the confirmed objective response rate per RECIST V.1.1 in the checkpoint inhibitor-naive, full-analysis population was 30.5% (95% CI, 19.2% to 43.9%; five complete responses); eight patients had stable disease (disease control rate, 44.1% (95% CI, 31.2% to 57.6%)). In addition, three patients experienced a delayed partial response after initial disease progression, for a total clinical response rate of 35.6% (95% CI, 23.6% to 49.1%). An additional patient with vulvar cancer had an unconfirmed response. Forty-nine patients (83.1%) experienced treatment-related adverse events, which were grade 3/4 in 16 patients (27.1%). No treatment-related deaths occurred.
Conclusion Bintrafusp alfa showed clinical activity and manageable safety and is a promising treatment in HPV-associated cancers. These findings support further investigation of bintrafusp alfa in patients with advanced, pretreated HPV-associated cancers.
- programmed cell death 1 receptor
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Human papillomavirus (HPV) causes almost all cervical cancers and a large proportion of anogenital and oropharyngeal cancers.1 Worldwide, approximately 630,000 new cases of HPV-associated malignancies are reported annually. Advanced HPV-associated cancers are often incurable and poorly palliated by traditional chemotherapies.2–5
Host immunity impacts HPV infection and progression to cancer,6 and several immune-related pathways are linked to HPV-associated cancers.6 7 Transforming growth factor β (TGF-β), a pleiotropic cytokine that suppresses tumor growth and inhibits tumor-promoting inflammation in the premalignant state, is associated with tumor growth, evasion of immune surveillance, invasion, and metastasis in the advanced cancer state.8 Genome-wide association studies showed that the TGF-β pathway is associated with cervical cancer and HPV-positive squamous cell carcinoma of the head and neck (SCCHN), and TGF-β receptor I is significantly overexpressed in these cancers compared with benign tissue.6 Another study found a positive correlation between HPV infection and TGF-β levels in saliva and serum of patients with oral squamous cell carcinoma (SCC).9 E6 and E7 oncoproteins induce activation of the TGF-β promotor in cervical cancer cell lines,10 and RNAseq analysis of HPV-positive oropharyngeal SCC showed that patients with poor survival were enriched for a TGF-β gene signature and had elevated levels of HPV-E6 protein expression.11 A recent study found that patients with HPV-positive SCCHN with a specific polymorphism in TGFB1 had significantly better overall and disease-specific survival compared with patients with the common genotype and that a similar benefit was not seen in patients with HPV-negative SCCHN cancers.12 Hence, dysregulation of the TGF-β pathway may play a critical role in HPV-mediated carcinogenesis, and this pathway may be a potential therapeutic target.
Results from two phase 1b, three phase 2 (including one basket trial), and one randomized phase 3 study showed objective response rates of 12%–24% for single-agent programmed cell death 1 (PD-1) inhibitors (nivolumab and pembrolizumab) in HPV-associated anal, cervical, and head and neck cancers.13–18 Studies in murine SCC models showed that anti-PD-1 therapy rarely led to complete regression, but adding anti-TGF-β synergistically enhanced antitumor responses.19 The synergy was partly driven by anti-TGF-β-mediated suppression of anti-PD-1 resistance and by attenuating epithelial-mesenchymal transition and stimulating immunosurveillance.
Bintrafusp alfa (M7824) is a first-in-class bifunctional fusion protein composed of the extracellular domain of the human TGF-β receptor II (TGF-βRII or TGF-β ‘trap’) fused via a flexible linker to the C-terminus of each heavy chain of an IgG1 antibody blocking programmed cell death ligand 1 (anti-PD-L1). In preclinical studies, compared with TGF-β sequestration or anti-PD-L1 antibody alone, bintrafusp alfa extended survival, conferred long-term protective immunity, decreased regulatory T-cell function,20 substantially increased CD8+ T cell and natural killer cell infiltration, and decreased myeloid-derived suppressor cell infiltration within tumors.21–23 In a phase 1 clinical trial, bintrafusp alfa efficiently sequestered plasma TGF-β1, TGF-β2, and TGF-β3 and bound to and saturated peripheral PD-L1.24 Treatment was well tolerated and clinically active, producing durable responses in several solid tumor types. Here, we report pooled safety and efficacy data from the subset of patients with checkpoint inhibitor-naive HPV-associated cancers from the phase 1 (study 001) and 2 (study 012) trials of bintrafusp alfa.
Methods
Study design and subjects
This is a post hoc analysis of an ongoing global, phase 1, open-label trial of bintrafusp alfa in patients with heavily pretreated advanced solid tumors and a phase 2 single-center trial of patients with advanced HPV-associated cancers. All patients with HPV-associated cancers from study 001 were from prospectively defined cohorts (cervical, SCCHN) or from the prospectively defined dose-escalation cohort, and the HPV population of study 012 was also prospectively planned. The primary results of the dose-escalation part (which included three patients from this analysis; online supplemental table S1) have been previously reported.24 Full inclusion and exclusion criteria for both studies are listed in the online supplemental file.
Supplemental material
The studies were conducted in accordance with all applicable regulatory requirements, and the protocols were approved by the institutional review boards of the participating institutions. International standards of Good Clinical Practice and the Declaration of Helsinki were followed. Each patient provided written informed consent before study enrollment. A full list of investigators and sites is listed in online supplemental table S2.
Procedures
Clinical procedure and assessments
Patients received bintrafusp alfa via 1-hour intravenous infusion every 2 weeks at doses of 0.3–30 mg/kg in the dose-escalation part of the phase 1 trial or at the recommended phase 2 dose of 1200 mg in the expansion part and the phase 2 trial.
The planned treatment duration was 1 year (for the phase 1 trial) or until progressive disease, unacceptable toxicity, or study withdrawal. Longer treatment and treatment past progression were permitted if clinically justified.
The primary objective of this post hoc analysis is to evaluate the efficacy of bintrafusp alfa monotherapy in checkpoint inhibitor-naive HPV-associated cancers. An exploratory analysis in checkpoint inhibitor-refractory HPV-associated cancers is also reported. Patients in both studies underwent tumor assessment scans every 6 weeks for the first 12 months and then every 12 weeks unless clinical symptoms warranted earlier imaging. Radiographic response was assessed by the investigator using Response Evaluation Criteria in Solid Tumors V. 1.1 (RECIST V.1.1) and reviewed by an independent radiologist at the investigational site for the dose-escalation part of the phase 1 study and the phase 2 study. A central facility reviewed radiographic responses for patients in the expansion part of the phase 1 study. Responses were confirmed by repeat assessment after a minimum of 4 weeks. Total clinical response rate was defined as the number of patients with best overall response (BOR) of complete response (CR) or partial response (PR) per RECIST V.1.1, or who experienced delayed response following initial pseudoprogression. The duration of response was defined as the time from initial response to the time of disease progression or death. Safety was evaluated according to the Common Terminology Criteria for Adverse Events versions 4.03 and 5 in the phase 1 and 2 studies, respectively.
HPV status
For determination of HPV-positive disease, prior documentation of tumor sample HPV status was accepted. For patients without documentation in the dose-escalation cohort of the phase 1 or phase 2 study, HPV status was determined by PCR, when fresh or archived tissue was available, using the cobas 4800 HPV Test (Roche Molecular Systems) or BD Onclarity HPV Assay (Becton Dickinson). In the expansion cohort of the phase 1 study, HPV status was determined by RNA sequencing using formalin-fixed paraffin-embedded (FFPE) tissue samples according to standard protocols, with HPV content in each sample assessed as the fraction of reads mapping to any papillomavirus genome.
Laboratory correlates
Immune responses to HPV were analyzed in patients from the dose-escalation cohort of the phase 1 study and the phase 2 study as previously described.25 Briefly, HPV-specific T-cell responses were assessed in cryopreserved peripheral blood mononuclear cells (PBMCs) isolated before and 2 weeks after one and/or three cycles of bintrafusp alfa, by intracellular cytokine staining following in vitro stimulation with a mixture of overlapping 15-mer peptide pools encoding HPV-16 E6 and E7. Peptide pools encoding human leukocyte antigen and CEFT (a mixture of peptides of cytomegalovirus, Epstein-Barr virus, influenza, and tetanus toxin) served as negative and positive controls, respectively. The absolute number of viable CD4+ or CD8+ T lymphocytes producing cytokine or positive for the degranulation marker CD107a at the end of expansion was calculated per 1×106 cells plated at the start of the stimulation assay. This calculation takes into account not only the percentage but also the total number of viable antigen-specific CD4+ and CD8+ T cells expanded in the stimulation assay.
Finally, PD-L1 expression was detected by immunohistochemistry staining of FFPE tumor tissue using an anti-PD-L1 antibody clone 73-10 (Dako PD-L1 IHC 73-10 pharmDx; Dako, Carpinteria, California, USA). PD-L1 expression was measured on tumor cells and on cells of the tumor microenvironment (TME). Data herein are reported based on the percentage of tumor cells expressing PD-L1. A threshold of 1% was used to characterize tumors as either PD-L1 positive (≥1%) or negative (<1%).
Outcomes
The primary endpoint of the dose-escalation part of the phase 1 trial was safety. The primary endpoint of the expansion part of the phase 1 trial and the phase 2 trial was the BOR according to RECIST V.1.1, and the secondary endpoint was safety. Progression-free survival (PFS), overall survival (OS), duration of response, and the relationship of immune responses to clinical responses were exploratory endpoints for the phase 1 trial and secondary endpoints for the phase 2 trial (except for immune response).
Statistical analysis
The sample size for the dose-escalation component of the trial followed a 3+3 design for dose-finding studies. Enrollment into multiple expansion cohorts was opened after the recommended phase 2 dose of bintrafusp alfa had been established (1200 mg intravenously every 2 weeks). All patients with HPV-associated cancers who received bintrafusp alfa were included in the safety and full analysis sets described here. Safety and tolerability were analyzed using descriptive statistics. The durations of PFS, response, and OS were analyzed using the Kaplan-Meier method.
Results
From January 26, 2016, to August 21, 2017, 17 patients with advanced HPV-associated cancer (cervical (n=10), anal (n=4), p16+ SCCHN (n=3)) were enrolled in the dose-escalation cohort, and 26 patients with advanced cervical cancer (n=15) or HPV-positive SCCHN (n=11) were enrolled into the expansion part of the phase 1 study. Overall, 14 patients with SCCHN and confirmed HPV-positive status from the phase 1 study are included in this analysis. The results for the overall SCCHN cohort are reported in a separate manuscript.26 HPV status for all patients was determined post hoc and not required for enrollment. Thirty-six patients were enrolled in the phase 2 study from February 27, 2018, to July 16, 2019, including 20 patients with checkpoint inhibitor-refractory disease.
Fifty-nine patients, including 43 from the phase 1 trial and 16 from the phase 2 trial, with checkpoint inhibitor-naive disease were included in this post hoc analysis. At the phase 1 analysis cutoff of April 17, 2019, and phase 2 analysis cutoff of October 4, 2019, the median duration of bintrafusp alfa treatment among all patients in this post hoc analysis was 3.9 months (range, 0.5–29.9 months) and 3.0 months (range, 0.5–7.8 months), respectively. Treatment was ongoing in 7 of 59 checkpoint inhibitor-naive patients (11.9%). The primary reasons for treatment discontinuation were disease progression (n=35), adverse events (n=8), non-treatment-related death (n=1), withdrew consent (n=5), investigator decision (n=1), lack of clinical benefit/patient decision (n=1), and completion of treatment (n=1).
Baseline demographic data and disease characteristics are summarized in table 1. Fifty-two patients (88.1%) had confirmed HPV-positive tumors, three patients (5.1%, all with cervical cancer) had HPV-negative disease (by RNA sequencing), and HPV status was missing or not available for four patients (6.8%, all with cervical cancer). Although the phase 2 study primarily enrolled female patients from a single center in the USA (National Cancer Institute) and enrolled patients with many more different tumor types than in the phase 1 study, age and Eastern Cooperative Oncology Group performance status were similar in both studies.
Baseline patient characteristics
Between the two studies, 5 patients (8.5%) with checkpoint inhibitor-naive disease had a confirmed CR and 13 patients (22%) had a confirmed PR, as determined by investigator-assessed RECIST V.1.1 (table 2, figure 1A, online supplemental figure S1). The confirmed objective response rate was 30.5% (95% CI, 19.2 to 43.9) in the full analysis set. Patients with confirmed CRs had cervical (n=2), anal (n=1), vaginal (n=1), and rectal SCC (n=1) cancers; the confirmed PRs occurred in four patients with SCCHN, eight with cervical cancer (including one patient with neuroendocrine cervical cancer), and one with anal cancer (figure 1B, online supplemental figure S2). Treatment responses occurred irrespective of PD-L1 expression in the phase 1 study (online supplemental figure S3). The response durations ranged from 2.8+ to 30.4 months (median, 19.1 months (95% CI, 9.6 to 27.4)); as of the data cut-off, 5 responses have lasted >18 months, and 11 responses (including one delayed response) were ongoing.
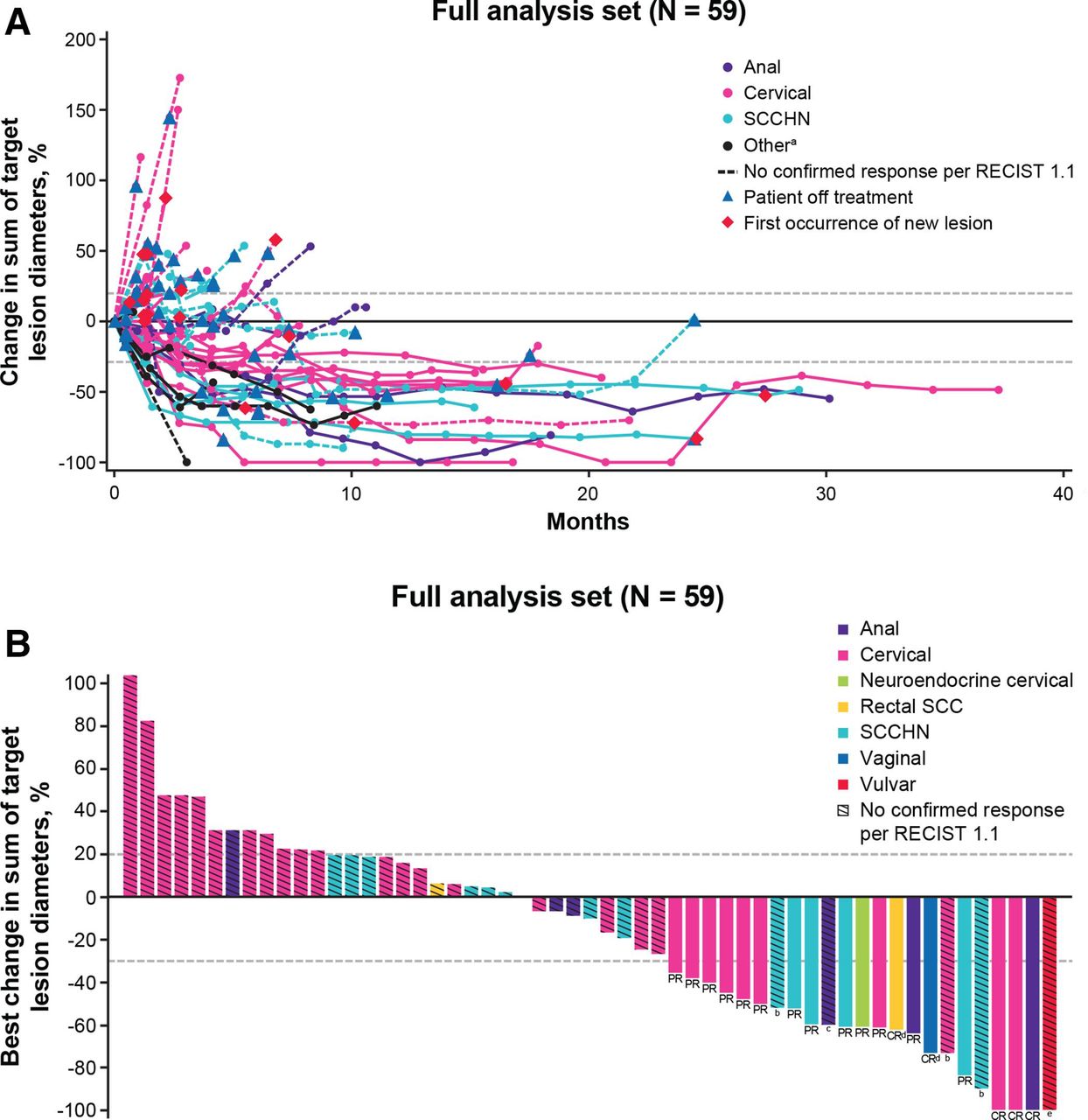
Clinical responses to bintrafusp alfa. (A) Percentage change in tumor diameters over time per investigator-assessed RECIST V.1.1 in the full analysis set. (B) Best percentage change in target lesions from baseline by cancer type as assessed by the investigators in the full analysis set. Data from three patients are missing due to lack of post-baseline tumor assessments. aIncludes two patients with rectal SCC tumors and one patient (each) with neuroendocrine cervical, vaginal, and vulvar tumors from study 012. Additional details can be found in online supplemental figure S1B). bDelayed PR. Due to confirmed progressive disease (PD) before onset of response, these patients did not meet response criteria by RECIST V.1.1. cPatient had a PR of target lesions but had progression of a non-target lesion requiring radiotherapy to the isolated non-target lesion (best response of PD by RECIST V.1.1). dPatients had disease limited to lymph nodes, which shrank to <1 cm in the short axis and did not completely disappear (best response of CR by RECIST V.1.1). ePatient had a CR but died of an unrelated medical illness (osteoporotic hip fracture with resulting sequela) prior to confirmation of response by investigator. CR, complete response; PR, partial response; SCC, squamous cell carcinoma; SCCHN, squamous cell carcinoma of the head and neck.
Summary of tumor response and survival data
In addition, three patients with checkpoint inhibitor-naive disease (cervical (n=1) and SCCHN (n=2)) had delayed PRs after initial disease progression that lasted 14.6, 6.1, and 15.9+ months, respectively (figure 1A, online supplemental figure S1), resulting in a total clinical response rate of 35.6% in the full analysis set (table 2). Additionally, one patient with checkpoint inhibitor-naive disease (vulvar cancer) had an unconfirmed CR but died of an unrelated medical illness (osteoporotic hip fracture with resulting sequela) prior to confirmation of response. The total clinical response rates were ≥30% for most HPV-associated tumor types, including cervical cancer (10/33 (30%)), anal cancer (2/6 (33%)), and SCCHN (6/15 (40%)). In addition, confirmed responses were seen in 3 of 5 (60%) rare tumor types, including vaginal (1/1 (100%)), rectal SCC (1/2 (50%)), and neuroendocrine cervical (1/1 (100%)) cancer. More than half (31/59 (53%)) of all patients had reduction in tumor diameters with bintrafusp alfa treatment, and 23 patients (39%) had tumor diameter reductions of >30% (figure 1B, online supplemental figure S2). The disease control rate according to RECIST V.1.1 was 44.1% in the full analysis set (table 2).
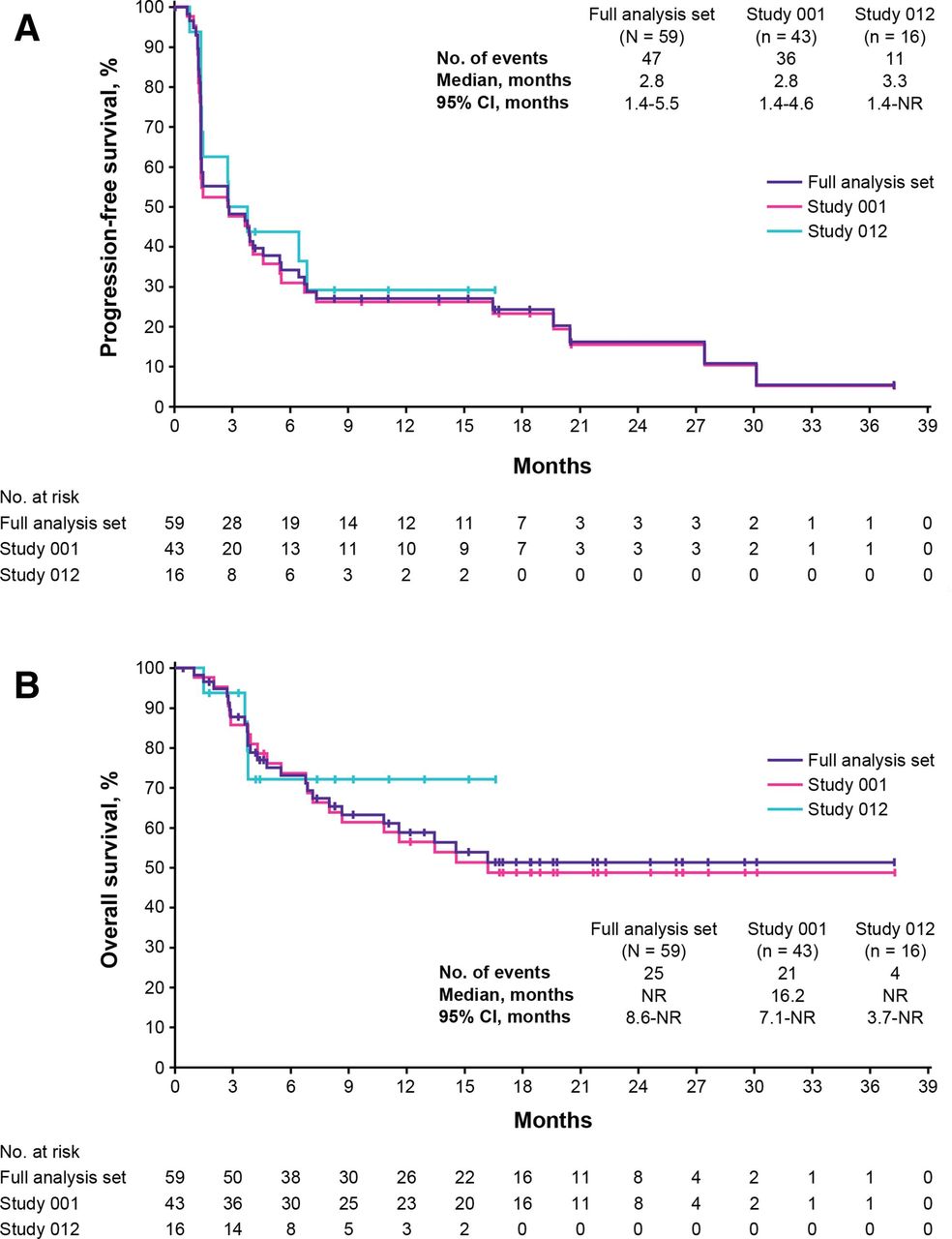
The median PFS was 2.8 months in the full analysis set (95% CI, 1.4 to 5.5 months; table 2, figure 2A). The median OS was not reached (95% CI, 8.6 months to not reached) in the full analysis set (table 2, figure 2B). Kaplan-Meier estimated proportions of patients with PFS and OS at different time points from baseline are shown in table 2. Of 59 patients in the full analysis set, 34 (58%) were alive at the cutoff, after a median follow-up of 9.2 months.

{kind=link}
{kind=link}
Kaplan-Meier analyses of PFS and OS. PFS according to investigator-assessed RECIST V.1.1 (A) and OS (B) in the full analysis set (purple), study 001 (pink), and study 012 (cyan). Marks on the curve indicate patients who were censored. OS, overall survival; PFS, progression-free survival.
Twenty patients refractory to immune checkpoint inhibitors were also enrolled and were not part of the full analysis set. The confirmed objective response rate for this group was 10% (95% CI, 1.2% to 31.7%; 1 CR (anal) and 1 PR (SCCHN)), with both responses ongoing and with durations of 1.4+ and 3.7+ months. Neither patient had received checkpoint inhibitor therapy for several months prior to enrolling, suggesting that these responses were not due to prior checkpoint therapy. The median PFS and OS for patients with checkpoint inhibitor-refractory disease was 1.4 months (95% CI, 1.3 to 3.3 months) and 3.4 months (2.3 months to not reached), respectively.
An evaluation of immune responses to HPV-16 in patients who had a BOR to bintrafusp alfa therapy of stable disease or better versus patients who had a BOR of progressive disease was performed in the dose-escalation cohort and phase 2 study patients after one and/or three cycles of bintrafusp alfa. Sufficient PBMCs to test for HPV-16-specific T-cell responses before and after treatment were available from 33 patients; 31 patients were evaluated before and after cycle 1, and 23 patients were evaluated before and after cycle 3 according to PBMC availability (online supplemental table S3). HPV-specific T cells were calculated as the absolute number of CD4+ or CD8+ T cells producing cytokine or positive for CD107a (lysosome-associated membrane protein 1, a functional marker of T-cell and natural killer cell activity) after expansion per 1×106 PBMCs plated at the start of the stimulation assay, which takes into account not only the percentage of positive lymphocytes, but also the number of total antigen-specific T cells that are expanded. In this analysis, after cycle 1, 9 of 14 patients (64.3%) with stable disease or better developed HPV-16-specific T cells versus 4 of 17 (23.5%) with progressive disease (p=0.03; online supplemental table S3). After cycle 3, 9 of 12 patients (75%) with stable disease or better developed HPV-16-specific T cells versus 6 of 11 (54.5%) with progressive disease (p=0.40; online supplemental table S3). Patients who had a BOR of stable disease or better had, on average, sixfold more HPV-specific T cells that produced cytokines or were positive for CD107a after cycle 1 (p=0.04) and cycle 3 (p<0.001) than patients who had a BOR of progressive disease (online supplemental figure S4). Trends in differences between responders and non-responders were also noted when HPV-specific T cells were quantified as a percentage of viable lymphocytes; using a twofold change as a cutoff, 11 of 14 patients (78.6%) with a BOR of stable disease or better, and 8 of 17 patients (47.1%) with progressive disease had an increase in HPV-specific T cells after one cycle of bintrafusp alfa.
Treatment-related adverse events (TRAEs) occurred in 83.1% (49/59) patients (table 3). The most common TRAE was pruritus, which occurred in 15 patients (25.4%). Grade 3 TRAEs occurred in 16 patients (27.1%); the most common was anemia, which occurred in four patients (6.8%). A patient who had grade 3 gastroparesis developed asymptomatic grade 3 hypokalemia, which worsened to grade 4 (one grade 4 event (1.7%)) and led to permanent study treatment discontinuation. This was medically managed without corticosteroids, gradually improved, and resolved completely within 2 months. No treatment-related deaths occurred.
TRAEs occurring at any grade in ≥5% of patients or grade ≥3 in any patient and any AEs of special interest (AESIs) from the full analysis set
Seven patients (11.9%) discontinued bintrafusp alfa due to TRAEs (colitis, gastroparesis (described above), infusion-related reaction, non-infective cystitis, pneumonitis, acneiform dermatitis, and psoriasiform dermatitis). Treatment-related infusion-related reactions occurred in three patients (5.1%). Adverse events of special interest, including potential TGF-β-related skin lesions, which included Medical Dictionary for Regulatory Activities V.21.0 preferred terms of keratoacanthoma, SCC of skin, basal cell carcinoma, hyperkeratosis, actinic keratosis, lip SCC, and Bowen’s disease, occurred in 12 patients (27.9%) (table 3, online supplemental table S4). These skin lesions were well managed with observation or local therapy (cryotherapy or excision) and did not require any patient to discontinue treatment. Thirty-eight patients (64.4%) in the full analysis set experienced treatment-emergent bleeding; nine patients (15.3%) had grade 3 bleeding events, and no patients had grade 4 or 5 events.
Discussion
Safety and efficacy data are presented from a post hoc combined analysis of 59 patients with advanced pretreated, checkpoint inhibitor-naive HPV-associated cancers who were enrolled in global phase 1 and 2 studies of bintrafusp alfa. Responses to bintrafusp alfa occurred in patients with several different types of HPV-associated cancers (SCCHN, cervical (including neuroendocrine), anal, vaginal, vulvar, rectal SCC). These responses were durable, ranging from 2.8+ to 30.4 months. While responses were observed irrespective of PD-L1 expression, given that PD-L1 expression was determined from archival samples, the age of the sample or previous therapy may have influenced the results.
Historical data observed with PD-1 inhibitors pembrolizumab and nivolumab in patients with HPV-associated cancers demonstrated objective response rates of 12%–24% and median OS of 7.5–11.5 months.13–18 Based on safety and efficacy data from these phase 1 and phase 2 studies, bintrafusp alfa in patients with advanced HPV-associated malignancies compares favorably with historical data of these PD-1 inhibitors. Survival also seems to be longer, with a median OS not reached after 18 months of follow-up; however, data from these studies cannot be compared directly due to differences in study design, eligibility criteria, and patient characteristics. To increase the response rates in HPV-associated cancers and other solid tumors, checkpoint inhibitors are being evaluated in combination with other novel immunotherapies, and our findings may support TGF-β as a therapeutic target in HPV-associated cancers.6
The safety profile of bintrafusp alfa was consistent with historically observed safety profiles of bintrafusp alfa in other tumor types. The severity and type of immune-related adverse events observed with bintrafusp alfa were also comparable to those observed with PD-(L)1 inhibitors.13 16 27 Additional toxicities that were seen with bintrafusp alfa that have not been described with PD-(L)1 inhibitors included keratoacanthomas and low-grade mucosal bleeding (eg, epistaxis, gingival bleeding). Study limitations from this combined analysis include the post hoc nature of this analysis and absence of a comparator treatment arm. Patients were selected for this analysis based on HPV-associated disease. Therefore, this analysis does not provide any conclusions about whether HPV-positive status is an independent biomarker predictive of response in all HPV-associated cancers; however, in the SCCHN expansion cohort, response rates in those with HPV-positive disease (determined by viral RNA detected in tumor samples) were 33% (3 of 9 patients) compared with 5% (1 of 22) in those without evidence of HPV infection.26 Finally, the small numbers of patients with rare tumors (cervical neuroendocrine, anal, vaginal, vulvar, rectal SCC) limits conclusions for safety and efficacy in these tumors.
In conclusion, targeting TGF-β and PD-L1 bifunctionally with bintrafusp alfa is a promising therapeutic approach for patients with HPV-associated cancers. Bintrafusp alfa had a manageable safety profile and resulted in an objective response rate of 30.5% in patients with pretreated checkpoint inhibitor-naive HPV-associated cancers, with clinical activity observed in patients who were refractory to PD-(L)1 treatment. Bintrafusp alfa continues in a range of phase 2 studies, including studies of patients with HPV-associated malignancies.
Acknowledgments
We thank the patients involved in this study and their families. We express appreciation to the nurses, medical oncology fellows, and consultation services at the National Institutes of Health Clinical Center for their excellent patient care. We thank the study teams at Merck KGaA, Darmstadt, Germany, and EMD Serono Research & Development Institute, an affiliate of Merck KGaA, as well as Angie Schwab (Laboratory of Tumor Immunology and Biology, Center for Cancer Research, National Cancer Institute [NCI]) for technical assistance with immune assays. The authors also thank Christian Ihling, of Merck KGaA, for his substantial contribution to the immune phenotype analysis. This research was supported by the Intramural Research Program of the Center for Cancer Research, NCI, National Institutes of Health, and through a Cooperative Research and Development Agreement with EMD Serono Research & Development Institute and GSK. This study was also funded in part by Merck KGaA and is part of an alliance between Merck KGaA and GSK. Medical writing support was provided by Ravi Subramanian, PhD, of ClinicalThinking Inc, which was also funded by Merck KGaA and GSK in accordance with Good Publication Practice guidelines (http://www.ismpp.org/gpp3).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @DrGattiMays, @HsaterMD, @mbilusic, @gulleyj1
JS and MEG-M contributed equally.
Presented at Oral presentation at the 2018 American Society of Clinical Oncology annual meeting; Oral presentation at the 2019 American Association for Cancer Research annual meeting.
Contributors All authors contributed to the design, data collection, interpretation, analysis, and writing of the article and approved the final version.
Funding Merck KGaA, Darmstadt, Germany provided the study drug and worked with investigators on the trial design and plan, collection and analysis of data, and interpretation of results. Funding for a professional medical writer with access to the data was provided by Merck KGaA and GlaxoSmithKline.
Competing interests JStrauss, CSH, and JLG disclose inventorship on a National Institutes of Health patent related to the work. JSchlom and JLG disclose that the National Cancer Institute has a Cooperative Research and Development Agreement (CRADA) with EMD Serono Research & Development Institute; an affiliate of Merck KGaA, Darmstadt, Germany. BCC discloses research funding from Novartis, Bayer, AstraZeneca, MOGAM Institute, Dong-A ST, Champions Oncology, Janssen, Yuhan, Ono, Dizal Pharma, and Merck Sharp & Dohme; discloses consultancy with Novartis, AstraZeneca, Boehringer-Ingelheim, Roche, Bristol Myers Squibb, Ono, Yuhan, Pfizer, Eli Lilly, Janssen, Takeda, and Merck Sharp & Dohme; discloses stock ownership with TheraCanVac; and discloses royalty from Champions Oncology. CSH is an employee of Merck KGaA. LSO and PAR are employees of EMD Serono Research and Development Institute. CSH discloses CRADA with Kite Pharma.
Patient consent for publication Not required.
Ethics approval This study was conducted following international standards consistent with the International Council for Harmonisation E6 Guideline for Good Clinical Practice. The study protocol was approved by ethics committees at all participating institutions, and each patient provided written informed consent before study enrollment.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck’s Data Sharing Policy. All requests should be submitted in writing to Merck’s data sharing portal (https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html). When Merck has a co-research, co-development, or co-marketing or co-promotion agreement, or when the product has been out-licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck will endeavor to gain agreement to share data in response to requests.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.