Article Text

Abstract
Background Cancer immunotherapy has evolved from interferon-alpha (IFNα) and interleukin-2 in the 1980s to CTLA-4 and PD-1/PD-L1 checkpoint inhibitors (CPIs), the latter highlighting the importance of enhancing T-cell functions. While the search for novel immunomodulatory pathways continues, combination therapies augmenting multiple pathways can also increase efficacy. The association of autoimmune-related adverse events with clinical efficacy following CPI treatment has been inferred and suggests that breaking tolerance thresholds associated with autoimmunity may affect host immune responses for effective cancer immunotherapy.
Results Here, we show that loss of autoimmune associated PTPN22, a key desensitization node for multiple signaling pathways, including IFNα receptor (IFNAR) and T-cell receptor, can augment tumor responses. Implantation of syngeneic tumors in Ptpn22-/- mice led to expansion and activation of peripheral and intratumoral T cells and, in turn, spontaneous tumor regression as well as enhanced responses in combination with anti-PD-L1 treatment. Using genetically modified mice expressing a catalytically inactive PTPN22 or the autoimmunity-associated human single-nucleotide polymorphism variant, augmentation of antitumor immunity was dependent on PTPN22 phosphatase activity and partially on its adaptor functions. Further, antitumor responses were dependent on both CD4+ and CD8+T cells and, in part, IFNAR function. Finally, we demonstrate that the autoimmune susceptibility Ptpn22(C1858T) variant is associated with lower risk of developing non-melanoma skin cancers, improved overall survival and increased risk for development of hyperthyroidism or hypothyroidism following atezolizumab (anti-PD-L1) treatment.
Conclusions Together, these data suggest that inhibition of PTPN22 phosphatase activity may provide an effective therapeutic option for cancer immunotherapy and that exploring genetic variants that shift immune tolerance thresholds may serve as a paradigm for finding new cancer immunotherapy targets.
- immunotherapy
- T-lymphocytes
- autoimmunity
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
PTPN22 (also known as LYP in humans and PEP in mice) is a PTPase preferentially expressed in immune cells.1–3 We and others have previously demonstrated that PTPN22 plays an inhibitory role in T-cell receptor (TCR) activation of antigen-experienced T cells and in interferon-alpha receptor (IFNAR) signaling.4–7 PTPN22 also plays inhibitory and activating roles in a number of additional innate and adaptive immune pathways including B-cell antigen receptor (BCR) signaling and repertoire selection, dectin-1 signaling in dendritic cells, toll-like receptor-mediated, NOD2-mediated and NLRP3-mediated functions in macrophages, regulatory T (Treg) cell development, adhesion and function, neutrophil adherence and IgE receptor signaling in mast cells.8–19
A single-nucleotide polymorphism (SNP) transition in human Ptpn22(C1858T) (rs2476601) resulting in an arginine to tryptophan amino acid substitution at position 620 (R620W) is associated with the development of a variety of autoimmune disorders including rheumatoid arthritis, type 1 diabetes mellitus and systemic lupus erythematosus among others.20 21 The coding variant resides within the first proline-rich (P1) motif of PTPN22 and results in decreased binding to c-Src kinase (CSK) family of protein tyrosine kinases.7 20 22 23 Phosphoproteome analysis of T cells expressing the mouse PTPN22(R619W) orthologue variant demonstrate enhanced phosphorylation of TCR-activated signaling components including phospholipase Cγ1, IκBα and MAPKs, but also phosphorylation of a broader set of substrates than Ptpn22-/- T cells.7 In addition, subcellular localization of human PTPN22(620W) differs from PTPN22(620R) to suggest a potential ‘switch of function’ for the at-risk variant.24 25 Human T and B cells expressing the Ptpn22(C1858T) variant have altered T-cell and B-cell functions and develop higher frequencies of autoreactive BCR repertoires.8 9 12 26 While aged Ptpn22-/- mice on a C57BL/6 genetic background demonstrate expanded numbers of antigen-experienced T cells and spontaneous formation of germinal centers, a histological finding often found in autoimmune prone strains of mice, they do not demonstrate any overt autoimmune features unless subjected to a secondary factor such as administration of IFNα, crossing to autoimmune-susceptible mice carrying an activating CD45 wedge mutation (E613R) or bred onto a mixed 129 genetic background.4 5 7 27
The development of certain immune-related adverse events (irAEs) is correlated with increased responses and prolonged survival to checkpoint inhibitors (CPIs).28 29 While immune tolerance is crucial for preventing autoimmunity, lowering the threshold required for T cell activation could enhance tumor control. Here, we evaluated whether absence of the autoimmune associated protein tyrosine phosphatase PTPN22 can confer increased tumor immunity.
Methods
Mice
Ptpn22619W/619W and Ptpn22227S/227S knock-in mice were generated on a C57BL/6 background using CRISPR technology. The sgRNA sequences, designed by Benchling, were AGACTCGGGTGTCCGTTCA for PTPN22(619W) and ACCTGCAGTGAATGCATAT for PTPN22(227S). 5’ and 3’ homology arms were 75 and 61 bps for 619W, and 75 and 42 bps for 227S, respectively. Ptpn22 -/- mice on a BALB/cJ background were generated by deleting exon 1 using GAGTTACCTATCTCCGACC and TGTAGGGATAACACGACCA as 5’ and 3’ gRNAs, respectively. Genotypes were confirmed by sequencing and PCR of genomic DNA and RNA, respectively. For tumor studies, female mice aged ~2 to 3 months were used.
Animal study oversight
All animal studies were reviewed and approved by Genentech’s Institutional Animal Care and Use Committee. Mice whose tumors exceeded acceptable size limits (2000 mm3) or became ulcerated were euthanized and removed from the study.
Cell lines
MC38 murine colon adenocarcinoma cells were obtained from Rienk Offringa. The CT26 mouse colon carcinoma cell line and E.G7-ovalbumin (OVA) mouse lymphoma cell line (EL4) expressing chicken OVA were obtained from ATCC (CRL-2638 and CRL-2113, respectively). The Hepa1-6 Sigma X1 (Hepa1-6.x1) cell line was generated from the Hepa1-6 mouse hepatoma cell line (Sigma, SKU92110305) by passaging in vivo tumor fragments grown in C57BL/6 mice (Charles River-Hollister). Tumor tissue from the second passage was minced with a pair of scalpels in a 10 cm Petri dish containing 20 mL RPMI 1640, 10% fetal bovine serum (FBS; HyClone, Waltham, Massachusetts, USA). Tissue fragments were transferred into a T75 tissue culture flask and cultured in an incubator at 37°C, 5% CO2 for 2 days to allow for cellular attachment. The medium was then removed and replaced with fresh growth medium. Contaminating fibroblasts were removed by differential trypsinization. Once a monolayer culture was established and passed a few times in vitro, cultures were passaged by trypsinization at appropriate intervals and split ratios.
All cell lines were tested for Mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza) and were cultured in RPMI 1640 medium plus 1% L-glutamine with 10% FBS (HyClone). Cells in log-phase growth were centrifuged, washed once with Hank’s balanced salt solution (HBSS), counted and resuspended in 50% HBSS and 50% Matrigel (BD Biosciences, San Jose, California, USA for injection into mice.
Antibodies
The following antibodies were used for flow cytometry: CD45 (clone 30-F11), TCRβ (clone H57-597), Thy1.2 (clone 30-H12), CD4 (clone RM4-5), ICOS (clone C398.4A), CD103 (clone 2E7) and T-bet (clone 4B10) from BioLegend; CD8α (clone 53–6.7) from BD Biosciences; CXCR3 (clone CXCR3-173), Foxp3 (clone FJK-16s), CD69 (clone H1.2F3), CD62L (clone Mel14) and CD44 (clones IM7) from eBioscience; anti-human granzyme B (GZMB) (cross-reactive with mouse) (clone MHGB05) from Life Technologies. LIVE/DEADTM Fixable Dead Cell Stain from Life Technologies was used to gate on live cells. Anti-PTPN22 antisera (P2) raised against a GST-fusion protein encoding PTPN22(aa 670–740) have been previously described.4 A rabbit monoclonal anti-PTPN22 antibody (Clone 3D5) was generated using an acylated N-terminal and amidated C-terminal peptide PERTLESFFLADEDC. Specificity was confirmed by Western blot analysis of wildtype (WT) and Ptpn22-/- splenocytes.
Syngeneic tumor studies
Tumor cells were harvested in log-phase growth and resuspended in HBSS containing Matrigel (BD Biosciences) at a 1:1 ratio. Cells were then implanted subcutaneously in the right unilateral thoracic area at 105 (MC38 and CT26), 3×106 (eg,7-OVA) or 107 (Hepa1−6.×1) cells in 100 µL of HBSS+Matrigel. For tumor growth studies, all animals were monitored and tumors were measured at least twice weekly. For efficacy studies, tumors were monitored until they became established and reached a mean tumor volume of ~190 mm3 (typically 7–18 days after cell inoculation depending on the tumor model). Mice with tumors in the range of 130–250 mm3 were then randomized into treatment groups. Treatment was initiated the next day with either isotype control antibody (anti-gp120 mIgG1) or anti-PD-L1 mIgG1 (clone 6E11) at an initial dose of 10 mg/kg intravenously, followed by 5 mg/kg intraperitoneally (IP) thereafter twice a week for 1 week for pharmacodynamic studies or 3 weeks for efficacy studies. All antibodies were diluted in 20 mM histidine acetate, 240 mM sucrose and 0.02% polysorbate 20, pH 5.5. Tumor volumes were measured in two dimensions (length and width) using Ultra Cal-intravenously calipers (Fred V Fowler, Newton, Massachusetts, USA) and volume was calculated using the formula: tumor size (mm3) = (length x width2) x 0.5. Mouse body weights were measured using an Adventura Pro AV812 scale (Ohaus, Pine Brook, New Jersey, USA).
To deplete circulating CD4+ or CD8+ T cells, mice were IP injected three times a week for 3 weeks with either 10 mg/kg rat antimouse CD8 IgG2b antibody (ATCC-2.43) or 25 mg/kg rat antimouse CD4 IgG2b antibody (GK1.5). Rat IgG2b anti-gp120 was used as the isotype matched control antibody. To block IFNAR, 10 mg/kg mouse anti-IFNAR IgG1 antibody (BP0241, BioXcell, Lebanon, New Hampshire, USA) was dosed intravenously for the first dose, followed by IP injection three times a week for 3 weeks. In tumor growth studies, depletion was initiated on the day of tumor inoculation.
Tissue processing
To generate single cell suspensions, tumors were cut into 2–4 mm pieces, digested for 30 min using the murine Tumor Dissociation Kit from Miltenyi (Miltenyi Biotec, Auburn, California, USA) following the manufacturer’s instructions (Cat. 130-096-730) and filtered through a 70 μm nylon filter (Corning, Corning, New York, USA). Tumor homogenates were then washed twice with RPMI 1640 media and resuspended in staining buffer (PBS, 0.5% FCS, 5 mM EDTA).
Spleens and draining lymph nodes (dLN) were minced on a 70 μm nylon filter (Corning). The flow-through was collected and centrifuged at 1500 rpm at 2°C–8°C. The supernatant was then aspirated and the cell pellets incubated with 5 mL (spleen) or 3 mL (dLNs) of ACK Lysis Buffer for 5 min at room temperature and followed by two washes with cold RPMI 1640 media. Cell pellets were then resuspended in staining buffer.
Blood samples were incubated with 1 mL/50 µL blood of ACK Lysis Buffer for 5 min at room temperature and followed by two washes with cold RPMI1640. The cell pellet was then resuspended in staining buffer.
Flow cytometry
For surface staining, cells were first incubated with LIVE/DEADTM Fixable Aqua Dead Cell Stain Kit (Thermo Fischer Scientific, Waltham, Massachusetts, USA) and with TruStain FcX antimouse CD16/32 (BioLegend, San Diego, California, USA) for 5 min in PBS. Cells were then washed with staining buffer, resuspended and surface-stained for 25 min at 4°C. For intracellular staining, cells were fixed and permeabilized using the eBioscienceTM Intracellular Fixation and Permeabilization Buffer Set (eBioscience, San Diego, California, USA) following the manufacturer’s instructions. FACS analyzes were performed using a Symphony flow cytometer (BD Biosciences).
For tetramer staining tumor lysates were stained with BV421-conjugated peptide–MHCI tetramers for 30 min at 4°C followed by staining with cell surface markers. Tetramers specific for the following sequences were pooled to a final concentration of 20 µg/mL: Reps1: AQLANDVVL, Adpgk: ASMTNMELM, Irgq: AALLNSAVL, N4bp2l2: ATINFRRL, Aatf: MAPIDHTTM, Cpne1: SSPYSLHYL, Med12: SSVLFEYM, Dpagt1: SIIVFNLL, Spire1: SAIRSYQV. Sequences for these peptides have previously been reported.30
Western blot analysis
Whole-cell lysates were generated with RIPA lysis buffer (Thermo Fisher Scientific) with protease and phosphatase inhibitors, DTT (1 mM), NaF (10 mM) and PMSF (1 mM). Cell lysates were loaded on a NuPAGE 4%–12% Bis-Tris gel (Life Technologies) and transferred using an iBlot Gel transfer device (Invitrogen). Membranes were blocked with 5% milk and 0.2% Tween in PBS. All antibodies were diluted in blocking buffer and incubated overnight at 4°C. HRP-conjugated goat anti-rabbit secondary antibody (Cell Signaling Technology, Danvers, Massachusetts, USA), ECL reagent (Bio-Rad, Hercules, California, USA) and Azure 600 (Azure Biosystems, Dublin, California, USA) for band visualization were used in Western blot analysis.
Analysis of rs2476601 skin cancer association in the UK Biobank
Prior to any analysis, we used individuals in the British white ancestry cohort. We removed heterozygosity outliers, individuals where inferred gender did not match reported gender, individuals with evidence of sex chromosome aneuploidy, individuals excluded from kinship inference, and individuals with excess relatives. We constructed our cases on the basis of a prefix match to ICD10 code C44 ‘other and unspecified malignant neoplasm of skin’ in phenotype field 40006. All other remaining individuals served as controls. We associated the presence of the rs2476601 risk variant with case control by use of logistic regression (glm in R v3.6.1). The analysis was limited to patients of European ancestry (EUR >0.7 as estimated by admixture v1.23). The computed OR was adjusted for 10 genotype eigenvectors (as computed by UK Biobank) and gender.
Analysis of atezolizumab-treated patients that are homozygous and heterozygous carriers of the autoimmune risk allele at rs2476601
The association analysis between the genotype status of individuals at the risk allele of rs2476601 and time to hyperthyroidism or hypothyroidism irAEs and overall survival was performed using a mixed-effects Cox model (coxme package in R v3.6.1). The model used a binary indicator for homozygous or heterozygous risk status of the individual that also included a random effects term to account for the differing effect size in each of the atezolizumab trial arms. The mixed-effects Cox model was also stratified by trial arm to account for differing baseline risk of hyperthyroidism and hypothyroidism irAEs and risk of death in each of the trial arms. In the multivariable analysis of time to hypothyroidism irAEs, we included five genotype eigenvectors, to account for any remaining population stratification, baseline thyroid-stimulating hormone (TSH) levels, and gender as covariates. TSH levels were quantile normalized to the quantiles of a standard normal distribution. The multivariable analysis of overall survival included the following covariates: genotype eigenvectors, presence of liver metastases at baseline, an indicator variable set to one if BECOG status was greater than 0 and zero otherwise, PD-L1 positivity on immune cells, and the free T3 over free T4 ratio measured at baseline.
Statistical analysis
Flow cytometry data are presented as means±SD. Comparisons between groups or treatments was generated using non-parametric, Mann-Whitney tests. Prism v6.0 (GraphPad) was used to process the statistical analyzes.
Results
Loss of PTPN22 enhances anti-PD-L1 antitumor immunity
To analyze whether loss of Ptpn22 may enhance CPI activity, we engrafted the colon adenocarcinoma tumor cell line MC38 into WT or Ptpn22-/- mice, aged ~2 to 3 months. Tumor growth was not affected with loss of Ptpn22 (figure 1A). When tumors grew to ~190 mm3 in size, mice were randomized into isotype control or anti-PD-L1 mAb treatment groups. While 20% of WT mice achieved a complete response (CR) with anti-PD-L1 treatment, 45% of Ptpn22-/- mice achieved CRs with anti-PD-L1 treatment. Consistent with greater tumor shrinkage, there was a significant increase in absolute numbers of intratumoral CD8+ T cells, known to play a central role in cancer immunity as well as an associated increase in the CD8/Treg ratio (figure 1B), as well as increased numbers of tumor antigen specific CD8+ T cells as measured by pooled tetramer staining (figure 1C). An increase in CD8+ and CD4+ T cells was also observed in dLNs of anti-PD-L1 treated Ptpn22-/- mice (figure 1D and online supplemental figure S1A). In dLNs, there was also expansion of CD8+ central memory T cells (Tcm) as well as CD4+ and CD8+ effector T cells (Teff) of control treated Ptpn22-/- mice when compared with WT mice, which were further increased following anti-PD-L1 treatment (figure 1E and online supplemental figure S1B). This effect was also present in spleens of treated mice where an increase in CD8+ Tcm and CD8+ and CD4+ effector memory T cells (Tem) were observed in control treated mice as well as increased CD4+ and CD8+ Tem T cells following PD-L1 blockade (online supplemental figure S1C,D). Expansion of antigen experienced CD8+ and CD4+ T cells in Ptpn22-/- mice was accompanied by increased expression of the chemokine receptor CXCR3, highly expressed on effector cells and critical for T cell trafficking and function,31 as well as increased expression of the activation marker ICOS (figure 1F and online supplemental figure S1E). In addition, CD8+ T cells in blood of anti-PD-L1 treated Ptpn22-/- mice showed higher expression of CXCR3 as well as PD1, Ki-67 and GZMB confirming their higher activation and proliferation states (online supplemental figure S2A). A similar phenotype for PD1, Ki-67 and GZMB was also observed in blood CD4+ T cells of anti-PD-L1 treated Ptpn22-/- mice (online supplemental figure S2B). Consistent with the expansion of effector/memory T cells in aged (>4 mo) Ptpn22-/- mice,4 these MC38 tumor studies in younger mice suggest that Ptpn22 deficiency by itself has an effect on antigen experienced cells in dLNs and spleen, but little impact on intratumoral T cell numbers. With PD-L1 blockade, however, these effects were significantly augmented in secondary lymphoid organs leading to a pronounced increase in intratumoral CD8+ T cells. This effect on peripheral T cells is particularly important given recent findings that intratumoral T cells, especially in responsive patients are replenished by non-exhausted T cells from outside the tumor and that CPIs (eg, anti-PD-L1) are likely playing an important role in expanding cells in secondary lymphoid organs which then infiltrate the tumor.32
Supplemental material
Supplemental material

Ptpn22 deficiency enhances anti-PD-L1 mediated antitumor responses. WT or Ptpn22-/- mice were inoculated with MC38 tumor cells. On tumor establishment (~190 mm3) mice were treated with anti-PD-L1 or isotype control antibody. (A) Tumor volume of treated mice are presented on a log2 scale. Complete regression (CR) was assigned to mice whose tumor volume were <32 mm3. n=10–11 mice/group. Nine days after isotype control or anti-PD-L1 treatment, tissues were collected for immune analysis. Shown in (B) are the number of tumor infiltrating CD8+ T cells and the CD8/Treg ratio for each group and (C) the number of tumor infiltrating tetramer+ CD8+ T cells. (D) Number of CD8+ T cells in draining lymph nodes (dLNs), (E) Frequency of CD8+ T cell subsets in dLNs and (F) expression of CXCR3 and ICOS on dLN CD8+ and CD4+ T cells. n=4–8 mice/group. (G) Tumor volume and frequency of CRS in CT26 tumor bearing mice presented on a log2 scale. n=9–10 mice/group. Results from (A) and (G) representative of two independent experiments (except for C). (B–F) Data are mean±SD. P values determined by using non-parametric, Mann-Whitney U tests. *P<0.05, **p<0.01, ***p<0.001. Treg, regulatory T; WT, wildtype.
The frequencies of macrophages, monocytes and dendritic cells were not affected in Ptpn22-/- in MC38 tumor-bearing mice (online supplemental figure 3A). However, there was a reduction in the frequency of CD40 and CD86 expressing macrophages (online supplemental figure 3B). While no changes in the M1-like, M1/M2-intermediate and M2-like subsets was observed based on CD206 and MHCII expression, there is indication that the functionality of macrophages could be affected and will require additional investigation. Further, no changes in dendritic cell phenotypes were observed (online supplemental figure 3C).
Supplemental material
Given the enhanced expansion and activation of peripheral CD8+ T cells following anti-PD-L1 treatment in MC38 tumor bearing Ptpn22-/- mice, we tested whether enhanced antitumor immunity was also observed in other tumor models and mouse genetic backgrounds. As BALB/cJ mice are more prone to autoimmune and lymphoproliferative diseases,33 we assessed the effects of Ptpn22 deficiency on a BALB/cJ genetic background. We observed that in CT26 tumor-bearing mice, characterized by infiltrating, yet poorly activated CD8+ T cells,34 that anti-PD-L1 treatment significantly increased tumor control leading to 50% CRs in Ptpn22-/- mice when compared with no CRs in WT mice (figure 1G). These results suggest that Ptpn22-/- deficiency can augment antitumor responses in different tumor immune contextures.
Spontaneous antitumor activity with loss of Ptpn22
To further expand on the possibility that Ptpn22 deficiency can confer intrinsic tumor control, we used the Hepa1-6.x1 hepatocellular carcinoma tumor model (on a C57BL/6 background) which is highly infiltrated by CD8+ T cells.34 We hypothesized that in this tumor model, increased activation of infiltrating T cells, rather than increased numbers, could tilt the balance in favor of tumor control. Indeed, we observed 50% spontaneous remissions (SRs) in Ptpn22-/- mice compared with no SRs in WT mice (figure 2A). This was associated with increases in CD8+ Tcm cells in dLNs and blood as well as increases in CD4+ Tem T cells in blood, but not dLNs (figure 2B and online supplemental figure S4A,B). These dLN and blood CD8+ and CD4+ T cells also had increased expression of CXCR3 and ICOS similar to the observations in MC38 tumor bearing Ptpn22-/- mice (figure 2B and online supplemental figure S4C). As this tumor model is already highly infiltrated with CD8+ T cells, we did not observe any increase in intratumoral CD4+ or CD8+ T cells (figure 2C). In fact, a decrease in intratumoral CD4+, CD8+ and Tregs was observed in Ptpn22-/- mice. However, the tumor infiltrating CD8+ T cells from Ptpn22-/- mice expressed higher levels of the cytolytic marker GZMB, activation markers ICOS and CD69, adhesion marker CD103 and transcription factor T-bet, markers all associated with activated effector cells (figure 2D). Expression of GZMB and ICOS were also enhanced in intratumoral CD4+ T cells from Ptpn22-/- mice. SRs were not limited to Hepa1-6.x1 tumors. In E.G7-OVA tumors (C57BL/6 background), characterized by low infiltration of CD8+ T cells despite expression of a highly immunogenic antigen,34 we also observed SRs in 30% of Ptpn22-/- mice when compared with no SRs in WT mice (online supplemental figure S5). Tumor infiltrating CD8+ T cells from Hepa1-6.x1 tumors of Ptpn22-/- mice also showed increased frequency of cells co-expressing PD1, LAG3 and TIM3 (figure 2E). We have previously shown that PD1+LAG3+TIM3+CD8+ T cells represent highly activated cells which are still functional (not yet exhausted) and correlate with response to anti-PD-L1 treatment.34 These PD1+LAG3+TIM3+ cells also expressed the highest levels of GZMB (figure 2F) suggesting that Hepa1-6.x1 tumors from Ptpn22-/- mice have a higher fraction of activated cytotoxic cells.
Supplemental material
Supplemental material
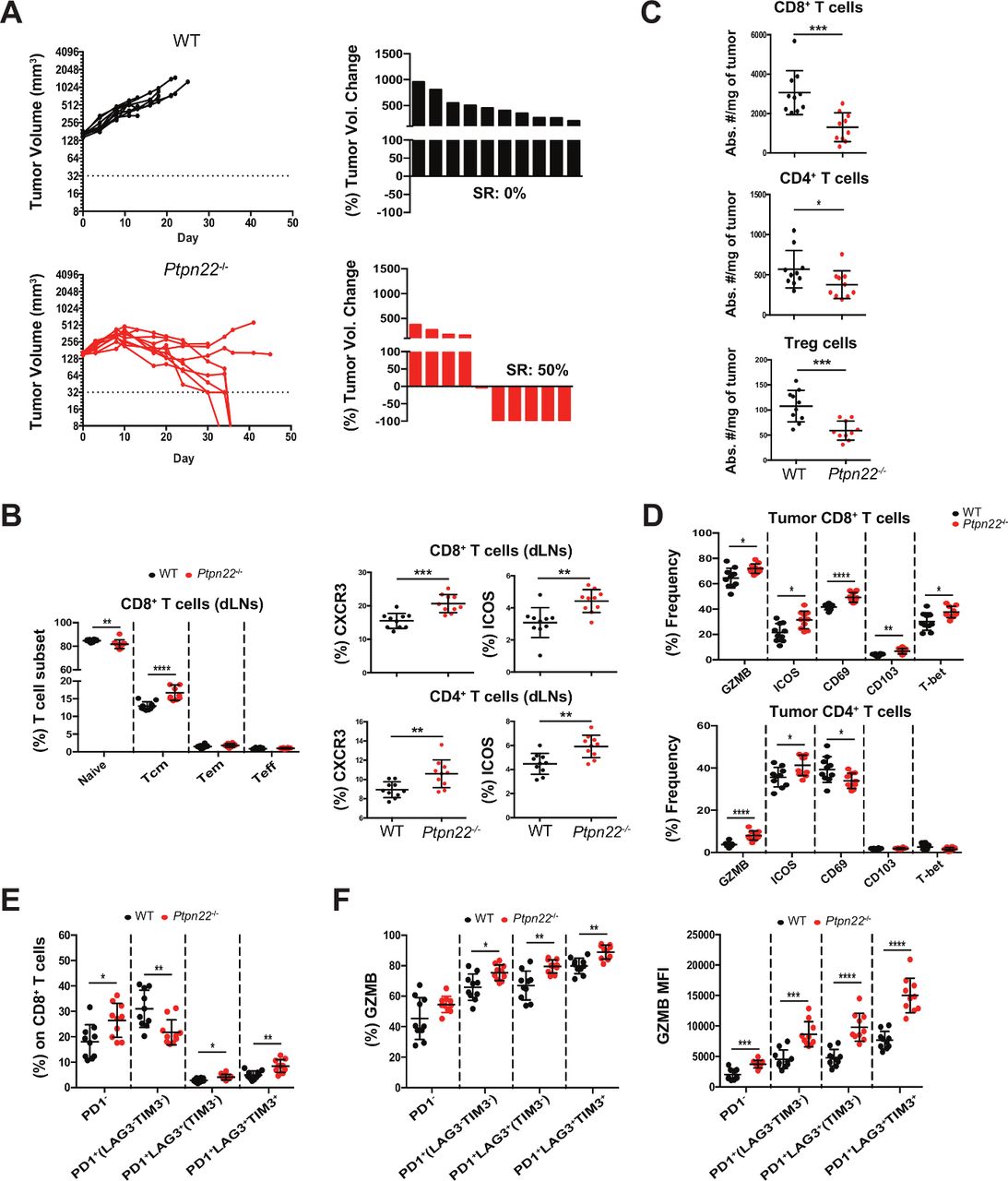
Ptpn22 deficiency results in spontaneous rejection of Hepa1-6.x1 tumors. WT or Ptpn22-/- mice were inoculated with Hepa1-6.x1 subcutaneously in the left flank. (A) Tumor volume of untreated mice and waterfall plots depict the frequency of spontaneous regressors (SRs). n=10 mice/group. Seven days after tumors were established (~190 mm3), tissues were collected for further analysis. Shown in (B) is frequency of CD8+ T cell subsets in dLNs and percent CXCR3+ and ICOS+ expressing CD8+ and CD4+ T cells, (C) number of intratumoral CD8+, CD4+ and Treg cells in WT and Ptpn22-/- mice, (D) frequency of GZMB, ICOS, CD69, CD103 and T-bet on intratumoral CD8+ and CD4+ T cells. (F) Frequency of PD1/LAG3/TIM3 subsets on intratumoral CD8+ T cells. (G) Frequency and MFI of GZMB expression on PD1/LAG3/TIM3 CD8+ T cell subsets. n=10 mice/group. Results from (A) are representative of more than four independent experiments. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. dLNs, draining lymph nodes; GZMB, granzyme B; Treg, regulatory T; WT, wildtype.
Tumor control dependent on T cells and partly on IFNAR signaling
As PTPN22 plays inhibitory roles in TCR and IFNAR signaling, we further explored the contribution of CD4+ T cells, CD8+ T cells and IFNAR signaling to Hepa1−6.×1 tumor rejection. Ptpn22-/- mice were treated with depleting CD4 or CD8 antibodies at the time of tumor inoculation. Total depletion of both intratumoral and blood CD4+ and CD8+ T cells was achieved (online supplemental figure S6A). Antitumor immunity was significantly compromised with CD4+ or CD8+ T cell depletion (figure 3A–C). The requirement for CD4+ T cells for antitumor immunity in this model is interesting as recent reports have described the importance of CD4+ T cell help for mounting effective CTL responses and tumor immunity.35 36
Supplemental material

Spontaneous tumor rejection in Ptpn22 -/- mice is dependent on both CD4+ and CD8+ T cells and partly on IFNAR signaling. Ptpn22-/- mice were inoculated with Hepa1-6.x1 subcutaneously in the left flank. Mice were treated with (A) isotype (B) anti-CD4 or (C) anti-CD8 depleting antibodies or with (D) a blocking antibody against IFNAR. Tumor volume and waterfall plots are shown. Results pooled from three independent experiments. n=34–35 mice/group. IFNAR, interferon-alpha receptor; SR, spontaneous remissions.
Since PTPN22 also downregulates IFNAR signaling,5 we investigated whether IFNAR function contributed to the SRs observed in Ptpn22-/- mice. Hepa1−6.×1 tumor-bearing Ptpn22-/- mice were treated with an IFNAR blocking antibody and achieved >90% blockade on CD4+ and CD8+ T cells and >80% blockade on CD11b+ granulocytes (online supplemental figure S6B). Blocking IFNAR signaling had a partial effect and inhibited about half of the SRs observed in control treated mice (figure 3D). Together, these results demonstrate that PTPN22 contributes to the spontaneous antitumor activity in this model predominantly through both CD4+ and CD8+ T cells and, in part, through enhancement of IFNAR activity.
Requirement for PTPN22 catalytic activity, but not P1 proline interactions, for full augmentation of antitumor immunity
Since the ability of PTPN22 to modulate antitumor immunity can be mediated through its P1 proline rich domain’s interaction with CSK’s SH3 domain and/or through its PTPase activity, we generated mice encoding mouse PTPN22(619W), the orthologue of human PTPN22(620W) with compromised ability to interact with CSK, or PTPN22(227S), a catalytically inactive PTPN22 (figure 4A). Levels of PTPN22 expression in WT, Ptpn22619W/619W and Ptpn22227S/227S in total splenic cells as well as in purified CD4+ and CD8+ T cells were comparable (figure 4B,C). Similar to previous characterization of Ptpn22-/- mice and Ptpn22619W/619W mice,4 6 7 immune cell composition of Ptpn22619W/619W mice aged 6–8 weeks old was comparable in the number of naïve and effector/memory T cells in thymus, spleen and LNs when compared with WT mice (online supplemental figure S7A). Thymocyte, spleen and LN composition of Ptpn22227S/227S mice aged 6–8 weeks old were also similar to WT mice (online supplemental figure S7B). Analysis of older (>4 months) mice demonstrated increased numbers of splenic effector/memory T cells in Ptpn22619W/619W and Ptpn22227S/227S knock-in mice when compared with WT mice and phenocopies age-matched Ptpn22-/- mice (online supplemental figure S7C).
Supplemental material

Ptpn22227S/227S mice phenocopy the spontaneous regressions observed in Hepa1−6.×1 tumor-bearing Ptpn22-/- mice. (A) Schematic designs for Ptpn22-/- , Ptpn22619W/619W and Ptpn22227S/227S knock-in constructs. Point mutations were introduced by CRISPR using a 141 bp and 130 bp donor oligonucleotide sequences for R619W and C227S, respectively. PTPN22 protein expression of naïve total splenic T cells (B) or purified splenic CD4+ and CD8+ T cells (C) from each genotype was analyzed by Western blot analysis with an anti-PTPN22 (3D5) mAb or a polyclonal anti-PTPN22 (P2) antisera, respectively. (D) WT, Ptpn22-/- , Ptpn22619W/619W and Ptpn22227S/227S mice were inoculated with Hepa1-6.x1 subcutaneously in the left flank. tumor volume of untreated mice and waterfall plots depict the frequency of spontaneous regressors (SRs). n=12 mice/group. WT, wildtype.
To analyze the contributions of scaffolding and PTPase functions of PTPN22 to the SRs observed in Ptpn22-/- mice with the Hepa1−6.×1 hepatocarcinoma tumor model, we implanted tumor cells in WT, Ptpn22-/- , Ptpn22619W/619W or Ptpn22227S/227S mice. While SRs were observed in 0%–17% of WT mice as compared with 67% in Ptpn22-/- mice, 67% SRs were also observed in catalytically inactive Ptpn22227S/227S mice, suggesting that loss of PTPN22 catalytic activity was essential to drive anti-tumor immune responses (figure 4D). In contrast, only 25% SRs were observed in Ptpn22619W/619W mice compared with WT mice suggesting that PTPN22 scaffolding function with CSK and potentially other binding partners through its P1 motif only partly contributes to antitumor immunity in this model. Pooled from three independent studies, an average of 36%±9% SRs was observed with Ptpn22619W/619W mice (N=39) when compared with 14%±24% SRs with WT mice (N=35) and 69%±13% SRs with Ptpn22-/- mice (N=39). Our data suggest that loss of PTPN22 phosphatase activity is more important than interrupting P1-SH3 interactions to optimally enhance tumor control.
Protective effect of rs2476601 in development of non-melanoma skin cancers
The Ptpn22(C1858T) autoimmune susceptibility SNP encoding PTPN22(R620W) is also identified as rs2476601. Previous studies have shown that variants in this region are associated with non-melanoma skin cancer.37 The SNPs identified in these analyses include rs679677 which is in high linkage disequilibrium with rs2476601 (r2=0.97 in the European population of 1000 Genomes38). We further investigated the direction of effect of the autoimmune risk variant rs2476601 in a non-melanoma skin cancer cohort of 17 426 cases and 319 712 controls defined by cancer ICD10 code C44 in the UK Biobank.39 We found that carriers of the variant corresponding to the Ptpn22(1858T) autoimmune risk allele were protected from non-melanoma skin cancer in this cohort (OR 0.89, 95% CI 0.86 to 0.93, p=4.6×10-8). Consistent with this protective association, the Ptpn22(1858T) allele frequency was highest in populations with lighter skin pigmentation, a risk factor for non-melanoma skin cancer, (0.1007 non-Finnish Europeans and 0.1490 Finnish, gnomAD) as compared with populations with darker skin pigmentation (0.0154 in Africans and 0.0131 South Asians, gnomAD).40
Improved overall survival and development of hyperthyroidism/hypothyroidism in patients that carry the risk allele of rs2476601 following CPI treatment
Development of irAEs, which are thought to be autoimmune in origin, is associated with CPI treatment.28 We assessed whether the Ptpn22(C1858T) variant was associated with risk of thyroid irAEs in patients treated with atezolizumab (anti-PD-L1) as hyperthyroidism and hypothyroidism are both common and can, in general, be attributed to the CPI when used in combination with chemotherapies. We scanned an internal genetic database of 3468 patients of European ancestry in the safety-evaluable population of 10 randomized controlled trials in which 2075 patients were treated with atezolizumab as a monotherapy or in combination with standard of care chemotherapy (online supplemental table 1). In total, 14 and 343 atezolizumab patients were homozygous and heterozygous, respectively, for the autoimmune risk allele, reflecting a slightly lower allele frequency (0.087) than observed in European populations (0.1007) in gnomAD. Hyperthyroidism irAEs are often detected before hypothyroidism, but at lower rates than hypothyroidism in patients treated with cancer immunotherapy, including atezolizumab.41 We found that carrying a single risk allele was sufficient to increase individual risk of hyperthyroidism (figure 5A; univariable p=0.0072; HR=1.89; 95% CI 1.19 to 3.02). In contrast, patients homozygous for the risk allele of rs2476601 were more likely to develop atezolizumab-induced hypothyroidism than heterozygous carriers and non-carriers (figure 5B; univariable p=0.0029; HR=4.89; 95% CI 1.72 to 13.91). We confirmed this association was present in a multivariable model that accounted for genotype eigenvectors, baseline TSH levels, and gender (adjusted, p=0.002; HR=5.03; 95% CI 1.81 to 14.00). Endocrine irAEs, including hyperthyroidism and hypothyroidism, have been shown to be associated with longer patient survival during anti-PD-1 treatment.42 Consistent with these observations, atezolizumab-treated patients homozygous at rs2476601 also had longer overall survival when compared with all other patients (figure 5C; univariable, p=0.012; HR=0.45; 95% CI 0.13 to 0.78). We confirmed this association with survival was robust to inclusion of prognostic covariates including PD-L1 positivity in a multivariable model (see Methods section; p=0.018; HR=0.30; 95% CI 0.11 to 0.81). We observed no association with OS (p=0.55; 95% CI 0.35 to 7.04) and hypothyroidism irAEs (p=0.58; 95% CI 0.72 to 1.77) in the control arms of the trials we analyzed indicating the variant was not prognostic. We additionally found no evidence for an association between rs2476601 with other common irAEs including rash (p=0.83), pneumonitis (p=0.14), and hepatitis (p=0.69) in atezolizumab treated patients. Hence, the Ptpn22(C1858T) variant contributes to higher risk of thyroid irAEs and improved OS with anti-PD-L1 therapy.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PTPN22(R620W) is associated with increased risk of thyroid irAEs and longer overall survival in atezolizumab (anti-PD-L1) treated cancer patients. (A) Cumulative event plot for hyperthyroidism and (B) hypothyroidism irAEs in patients treated with atezolizumab as a monotherapy or in combination with chemotherapies across ten randomized controlled trials (online supplemental table 1). (C) Kaplan-Maier plot of overall survival of atezolizumab treated patients across the same clinical trials. Tick marks designate censoring events. irAEs, immune-related adverse events.
Discussion
PTPN22 serves as a rheostat in antigen receptor signaling to shape TCR and BCR repertoires and likelihood of autoimmunity in mice and humans.4 6 7 12 26 Consistent with this role, the rs2476601 variant in Ptpn22 increases lifetime risk for autoimmunity while conferring lower risk for non-melanoma skin cancer. In anti-PD-L1-treated cancer patients who are carriers, we observe that heterozygosity is sufficient to increase the risk of developing hyperthyroidism. Yet, homozygosity is required to confer a higher risk for developing hypothyroidism and longer overall survival following atezolizumab treatment. This may reflect differences in the sensitivity of these events to immune tolerance thresholds conferred by the rs2476601 variant. Taken together, these observations support the notion additional analysis of genetic variants that alter tolerance thresholds may provide a fertile source for new targets for cancer immunotherapy.
Targeting PTPN22 affords the opportunity to augment cancer immunotherapy through at least two clinically validated classes of therapies and pathways—IFNAR and TCR signaling. As PTPN22 plays important roles in other signaling pathways, additional mechanisms can further contribute to tumor immunity. As Ptpn22-/- mice under C57BL/6 and BALB/cJ genetic backgrounds are healthy and require other factors to manifest autoimmunity, PTPN22 inhibition may provide an improved safety profile as compared with other strategies that combine two or more targeted immunotherapies, which often is associated with significant adverse toxicities. While autoimmunity can be exacerbated in Ptpn22-/- mice with administration of IFNα5 or when bred to an activating CD45(E3613R) wedge mutation,27 whether PTPN22 inhibition combined with checkpoint inhibition will result in autoimmunity to limit therapeutic dosing will require human studies.
While we have previously shown that Ptpn22 deficiency can expand the number and responsiveness of memory and effector T cells in aged mice,4 this effect is present in young tumor bearing mice and further augmented following anti-PD-L1 treatment. Ptpn22 deficiency and PD-L1 blockade combined effectively to expand and activate peripheral T cells as well as increase CXCR3 expression and, in turn, translate into increased activation and numbers of tumor infiltrating CD8+ T cells in the MC38 adenocarcinoma tumor model. The resulting effects of Ptpn22 deficiency and checkpoint blockade led to a pronounced enhancement of tumor responsiveness in a variety of tumor models with different levels and activation status of infiltrating T cells. Given that anti-PD-L1 might work by both expanding non-exhausted peripheral T cells and invigorating the responsiveness of pre-exhausted cells, targeting PTPN22 could combine with CPI at multiple stages of the cancer immunity cycle. Even in the absence of checkpoint blockade, Ptpn22 deficiency led to spontaneous regressions in the Hepa1-6.x1 tumor model characterized by high levels of infiltrating CD8+ T cells. In this tumor immune contexture, absence of Ptpn22 expression also led to increased activation of tumor infiltrating T cells and a higher frequency of activated PD1+LAG3+TIM3+CD8+ T cells. While in various syngeneic tumor models co-expression of these receptors is observed in highly activated and functional cells,34 prolonged antigenic stimulation in the context of Ptpn22 deficiency could ultimately favor an exhausted-like phenotype.
Most human solid tumors can be classified to immune inflamed (~25%), excluded (~50%) or desert (~25%) phenotypes.43 While CPIs appear to provide greatest benefit to inflamed tumors, efficacy is suboptimal in immune excluded or desert phenotypes. TGFβ appears to play an important role in establishing the fibroblast and collagen-rich peritumoral stromal microenvironment to exclude immune cells44 45 and in preclinical models, inhibition of TGFβ alters the stromal microenvironment and enhances immune cell entry and antitumor immunity. As deletion of Ptpn22 in CD8+ T cells can overcome the suppressive effects of TGFβ,46 PTPN22 inhibition might, therefore, combine with CPIs to provide effective cancer immunotherapy in a broader set of tumors.
Conclusion
Augmentation of two clinically validated pathways, TCR and IFNAR signaling, through inhibition of PTPN22 may provide a novel approach to achieve effective cancer immunotherapy. As Ptpn22-/- mice on a C57BL/6 or BALB/c genetic backgrounds have normal livelihoods, PTPN22 inhibition may also afford a safer approach to target multiple pathways to augment antitumor immunity. The potential benefit of PTPN22 is further underscored by the observation that individuals carrying the Ptpn22(C1858T) variant associated with autoimmunity have lower risk of developing non-melanoma skin cancer and that individuals homozygous for the Ptpn22(C1858T) variant have improved overall survival when treated with atezolizumab (anti-PDL1). Finally, as breaking tolerance thresholds may be linked with improved antitumor responses, exploring host autoimmune associated genetic variants may provide additional targets for effective cancer immunotherapy.
Acknowledgments
The authors thank Eric Brown, Sanjeev Mariathasan, Mark McCarthy, Ira Mellman, David Shames, Andrey Shaw and Shannon Turley for critical discussions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AC and RC conceived and designed the study. ZK conceived and performed bioinformatic data analysis and generated figures. HX processed and prepared samples for flow cytometry and provided analysis. MM performed the in vivo mouse tumor studies and processed and analyzed the data. QG, QO, RR and CP generated and characterized the different mouse strains. QG also generated figures. JC performed flow cytometry analysis to determine the level of IFNAR blockade and analyzed data. RC analyzed flow cytometry data and in vivo mouse tumor studies and generated figures. AC, RC, ZK and QG contributed to discussions and writing of the manuscript. All authors reviewed and edited the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests Authors are employees of Genentech and receive salary and stocks from Roche.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Data are available on reasonable request. Qualified researchers may request the individual-level clinical trial data used in our study by following Roche data sharing policies outlined here: https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm. After completing this process, the data requester may contact the corresponding authors for the variant status at rs2476601.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.