Article Text

Abstract
Oncolytic viruses (OVs) are a new class of cancer therapeutics. This review was undertaken to provide insight into the current landscape of OV clinical trials. A PubMed search identified 119 papers from 2000 to 2020 with 97 studies reporting data on 3233 patients. The viruses used, presence of genetic modifications and/or transgene expression, cancer types targeted, inclusion of combination strategies and safety profile were reported. In addition, information on viral bioshedding across the studies, including which tissues or body fluids were evaluated and how virus was detected (eg, PCR, plaque assay or both), is also reported. Finally, the number of studies evaluating antiviral and antitumor humoral and cellular immune responses were noted. We found that adenovirus (n=30) is the most common OV in clinical trials with approximately two-thirds (n=63) using modified or recombinant viral backbones and granulocyte-macrophage colony-stimulating factor (n=24) was the most common transgene. The most common tumors targeted were melanoma (n=1000) and gastrointestinal (GI; n=577) cancers with most using monotherapy OVs given by intratumoral (n=1482) or intravenous (n=1347) delivery. The most common combination included chemotherapy (n=36). Overall, OV treatment-related adverse events were low-grade constitutional and local injection site reactions. Viral shedding was frequently measured although many studies restricted this to blood and tumor tissue and used PCR only. While most studies did report antiviral antibody titers (n=63), only a minority of studies reported viral-specific T cell responses (n=10). Tumor immunity was reported in 48 studies and largely relied on general measures of immune activation (eg, tumor biopsy immunohistochemistry (n=25) and serum cytokine measurement (n=19)) with few evaluating tumor-specific immune responses (n=7). Objective responses were reported in 292 (9%) patients and disease control was achieved in 681 (21.1%) patients, although standard reporting criteria were only used in 53% of the trials. Completed clinical trials not reported in the peer-reviewed literature were not included in this review potentially underestimating the impact of OV treatment. These data provide insight into the current profile of OV clinical trials reporting and identifies potential gaps where further studies are needed to better define the role of OVs, alone and in combination, for patients with cancer.
- oncolytic virotherapy
- immunotherapy
- clinical trials as topic
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Oncolytic viruses (OVs) are a new class of cancer agents that promote tumor regression through preferential replication in tumor cells, induction of immunogenic cell death and stimulation of host antitumor immunity.1 To date, three OVs have been approved globally for the treatment of advanced cancers. The first in 2004 was an RNA virus derived from the native ECHO-7 strain of a picornavirus, called Rigvir, and achieved approval for melanoma treatment in Latvia.2 Then, in 2005, China approved a genetically modified adenovirus, H101, for the treatment of nasopharyngeal carcinoma in combination with cytotoxic chemotherapy.3 In 2015, the U.S. Food and Drug Administration approved Talimogene laherparepvec (T-VEC), an attenuated herpes simplex virus, type 1 (HSV-1) encoding granulocyte-macrophage colony-stimulating factor (GM-CSF) for the local treatment of unresectable cutaneous, subcutaneous and nodal lesions in patients with recurrent melanoma after initial surgery.4 T-VEC was evaluated in a prospective, multi-institutional randomized clinical trial and has subsequently been approved in Europe, Australia and Israel. More recently, clinical trials have provided support for improved therapeutic responses when OVs are given in combination with immune checkpoint blockade.5 6 Despite the clinical trial results supporting the potential therapeutic benefit of OVs, there are many aspects of OV clinical development, including the viral species, genetic modifications, transgene expression, route and schedule of administration, type and stage of patients with cancer, optimal combination agents and predictive biomarkers of response that remain to be elucidated.
Preclinical studies have supported a large number of both DNA and RNA viruses as potential candidates for OV drug development.1 Indeed there is no standard method for OV selection with some viruses exhibiting natural tropism and predilection for preferential replication in tumors cells and others demonstrating improved replication in tumor cells following genetic modification.7 Since some viral genes are considered non-essential, in some viruses genetic deletions can help attenuate pathogenicity of viral infection and may promote tumor cell replication. In addition, larger viruses are able to express eukaryotic genes and, especially when non-essential viral genes have been deleted, OVs can be engineered to deliver additional gene expression to help promote anticancer activity. There has been considerable preclinical studies supporting expression of a variety of genes that help promote cytotoxic killing of tumor cells, induction of immune responses, inhibition of tumor neoangiogenesis, enhancing radiosensitization and other strategies.8–10 Other considerations in OV development includes selection of how to deliver the virus to the patient with cancer and, while initial studies used direct intratumoral (IT) injections, this may be logistically challenging for visceral and central nervous system (CNS) tumors. Other strategies have included intravenous administration which is logistically simple and allows targeting of multiple metastatic lesions but may be complicated by rapid dilution in the circulation, neutralization by antiviral antibodies and other serum proteins, and ultimately limited biodistribution to tumors.11 Other factors, such as which combination agents and how to sequence them with OVs, how to best select appropriate patients and lesions for OV therapy, and the need for alternative endpoint assessment criteria for IT therapy are highly controversial and require further clinical study.12
While preclinical models are useful for initial proof of concept for other forms of cancer therapy, this may be more problematic for OVs as many murine cells and tumor models are not permissive for viruses that can infect human tumors.13 Further, an intact immune response is necessary to fully assess the therapeutic potential for most OV approaches. Thus, clinical trials may be important for providing critical correlative data that can support presumed mechanisms of action for OV therapy and are ideally suited for evaluation of biomarkers, including viral shedding, antiviral immune responses and confirmation of antitumor immunity. Given the multiple mechanisms of action associated with OV delivery and the emerging safety profile of OV treatment, OVs are ideally suited for combination strategies and these also require clinical validation through well designed and statistically sound clinical trials.
There have been many thorough reviews on the potential therapeutic activity of OVs as a class as well as for specific agents alone and in combination.1 There are, however, few reports of collective data from OV clinical trials. This may be helpful to gain insight into the current status of OV clinical investigation as well as better understanding of how to standardize correlative studies to support OV safety, transmission and efficacy. In addition, an assessment of current clinical publications on OV clinical trials may identify gaps in the clinical literature that need to be addressed in future clinical trials. These data may guide better study designs to optimize the therapeutic potential for OV treatment in patients with cancer.
Methods
Literature review
An organized literature review was conducted by accessing PubMed database on June 20, 2020 and searched for keywords “oncolytic virus” and “oncolytic viruses”. The search was also filtered for clinical trials and randomized clinical trials. Using these search terms, 119 manuscripts were identified and selected for initial review. Of these, 97 were found to contain original reports of clinical trial data using an OV. The major reason for articles to be rejected from further review were reports of preclinical data, report of a clinical protocol only without data, review manuscript, subset analyses of clinical trials that reported clinical data in another publication, and imaging studies without clinical data. One report was in Chinese and the paper could not be accessed.
The 97 manuscripts reporting clinical trial results were selected for further review and each study was evaluated for multiple variables, which were recorded by investigators. Parameters assessed included the phase of the clinical trial, number of patients treated, the type of virus used, the nature of the viral backbone (ie, native virus, modified or recombinant virus, or activation of latent intracellular virus), expression of transgenes, the type of cancer treated, the use of single agent or combination regimens, including which agents were used in combination studies. Clinical trials were also evaluated for common OV treatment-related adverse events and all treatment-emergent grade III and IV adverse events. Viral shedding was also assessed by recording whether studies evaluated and reported viral bioshedding, which body tissues and/or fluids were assessed for virus and what assays were used for virus detection (eg, PCR (PCT), plaque assay or both) and whether virus was detected. In addition, studies were evaluated for reporting antiviral and antitumor immune responses. Humoral antiviral responses were further characterized as neutralizing or non-neutralizing antibody titers and whether cellular-mediated antiviral immune responses were assessed. Tumor immunity was also determined by the type of immune assays used. Finally, studies were evaluated for response rates and clinical efficacy. A complete list of the clinical reports included can be found in online supplemental table 1.
Supplemental material
Definitions
Although definitions often differed across clinical trials, for purposes of reporting we adopted standardized reporting criteria. Whenever possible, we report viruses at the family classification. Patients treated were defined as patients who were enrolled in the clinical study and received at least one administration of an OV. If patients were consented but did not receive treatment they were not included in the assessment. Cancer histology was recorded in the paper and considered “solid tumor, not otherwise specified” when further histological classification was not provided, and the trial criteria enrolled “solid tumors”. A similar definition was applied for hematological malignancies. In some cases, the tumor type was not reported and then they were categorized as “not specified”.
For safety reporting we defined treatment-related adverse events as any adverse event considered definitely, probably or possibly related to OV treatment. Whenever possible, if other agents were given this was reported. Treatment-emergent adverse events were defined as any adverse event that occurred after the first treatment was given. The Common Toxicity Criteria for Adverse Events (CTCAE, v2.0–4.0) was used to define adverse events.
Statistical analyses
This was a retrospective review of published clinical trials. All statistical analyses were descriptive in nature and summarized with median and IQRs used for continuous variables and frequencies with percentages used for dichotomous variables. In some cases, categories were grouped for ease of presentation (eg, anal, esophageal, gastric, pancreatic and colorectal cancer were listed as “gastrointestinal cancers”).
Results
Oncolytic viruses in clinical investigation
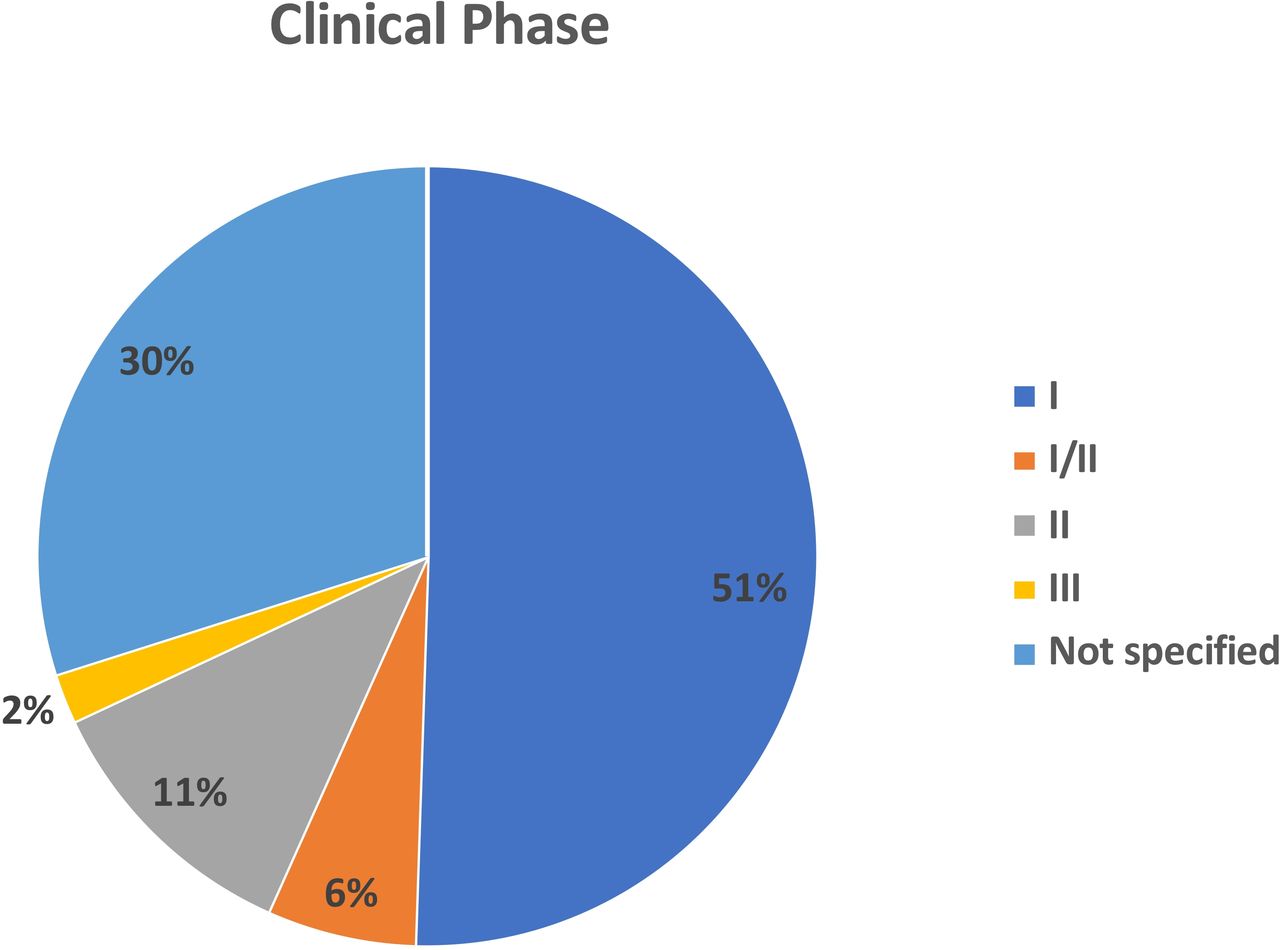
We identified 97 independent clinical trials reporting OV studies from 2000 to 2020 that included treatment of 3233 patients with cancer (table 1). The majority of the clinical trials were phase I (n=49; 50.5%) There were an additional 6 (6.2%) studies that were reported as phase I/II. There were 11 (11.3%) phase II clinical trials and only 2 (2.1%) phase III clinical trials. Another 29 (29.9%) clinical studies were not clearly specified but were largely early phase or first-in-man trials suggesting that most of the current literature focuses on early phase clinical trials (figure 1).
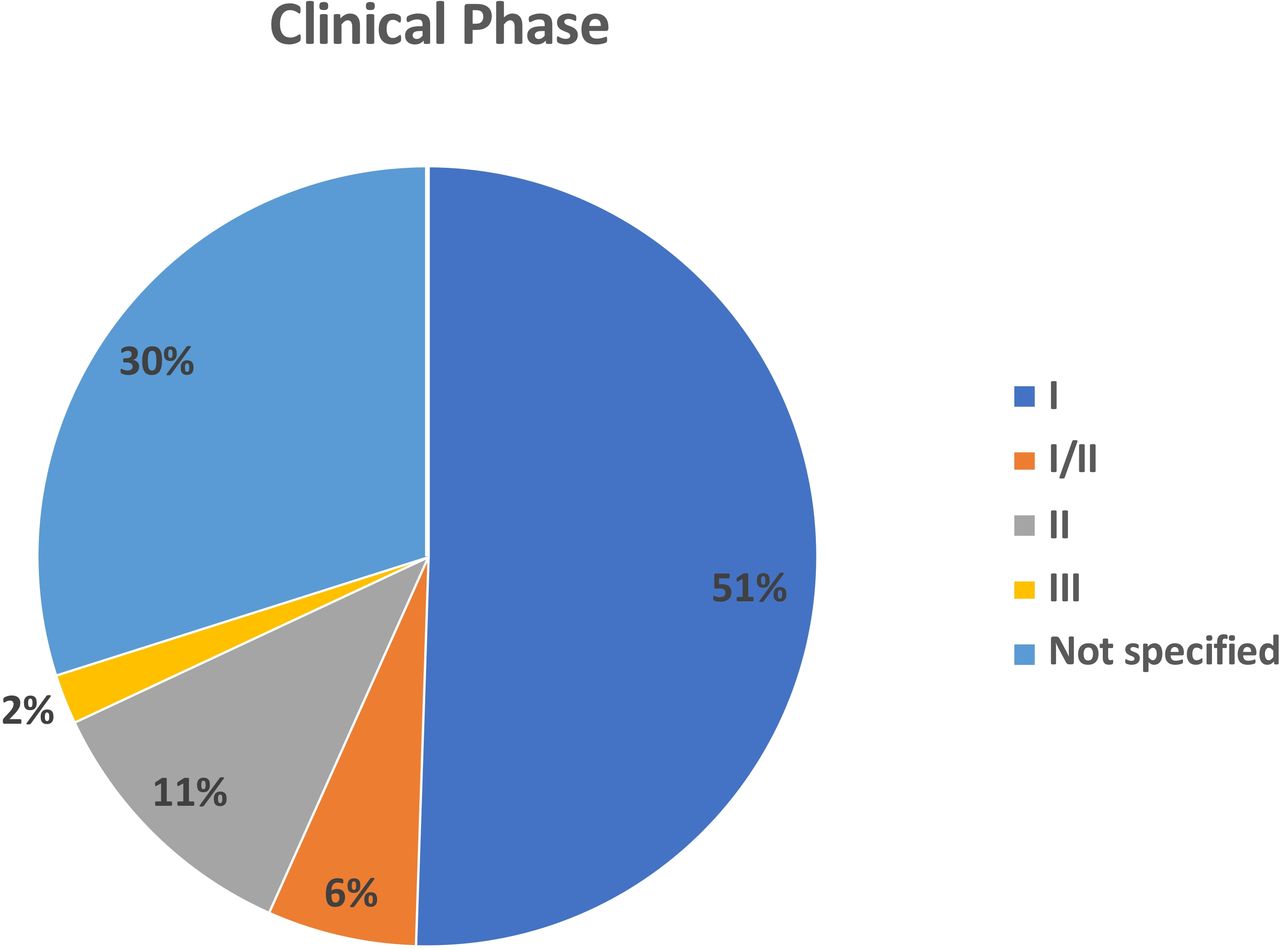
Pie chart showing the distribution of oncolytic viruses by clinical stage. The majority of studies were phase I (n=49; 51%) or not specified (n-=29; 30%). There were 6 (6%) phase I/II trials, 11 (11%) phase 2 and only two phase 3 clinical trials.
Patient characteristics in oncolytic virus clinical trials
There are many DNA and RNA viruses that may be used as OVs and the majority of clinical studies used DNA viruses (figure 2A). The most common viruses were adenovirus (n=30; 30.9%) followed by HSV-1 (n=23; 23.7%), reovirus (n=19; 19.6%) and poxviruses (n=12; 12.4%). There were an additional five studies (5.2%) using Newcastle Disease virus, three studies (3.1%) using measles virus and two studies (2.1%) using Seneca Valley virus and the hemagluttinating virus of Japan Envelope (HVJ-E) virus. Other viruses that were reported in single clinical trials included gamma-herpes virus, parvovirus, and retrovirus. Although prime boost with two different viruses have been proposed, no studies used more than one OV in the reported clinical trials.
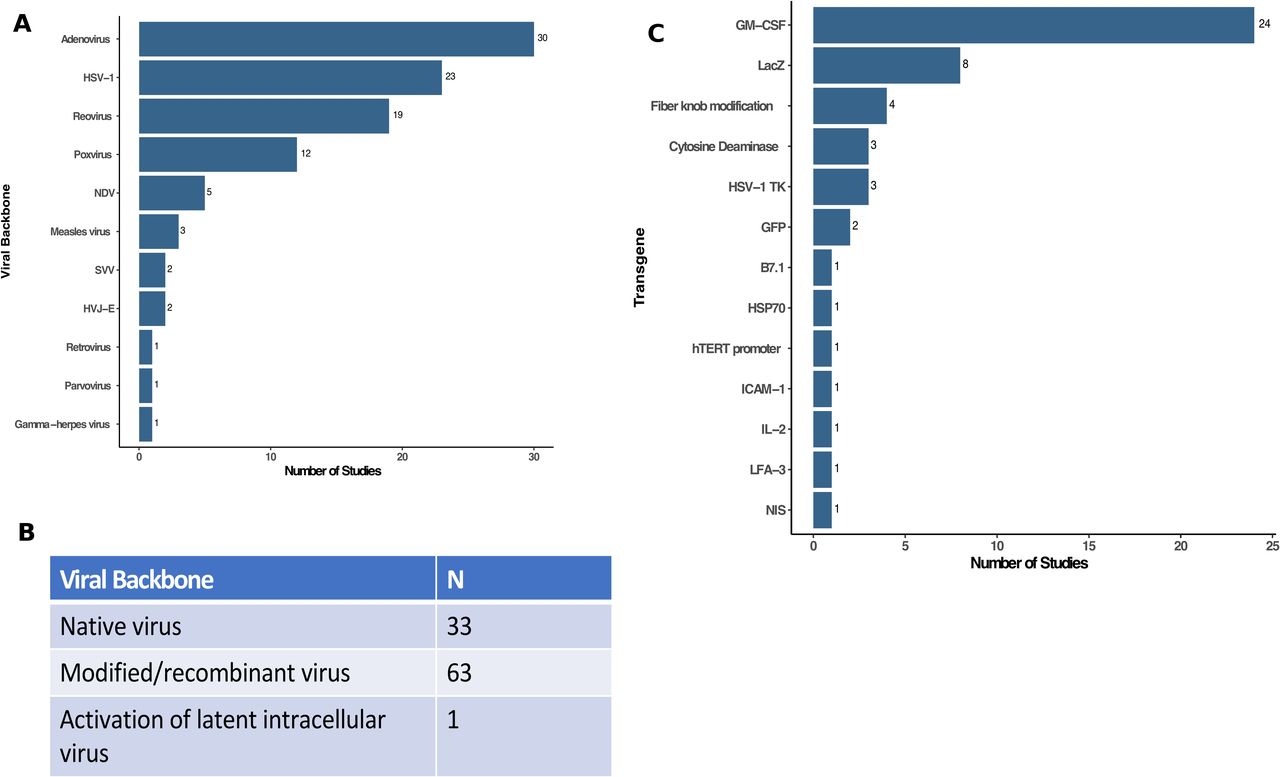
Characterization of viruses used in oncolytic virus clinical trials. (A) The type (family) of viruses reported in clinical trials were dominated by adenovirus (n=30), HSV-1 (n=23), reovirus (n=19) and poxviruses (n=12) with several other viruses as shown. (B) Table showing the viral backbone used in clinical oncolytic virus studies (eg, native virus, modified viruses, including recombinant or attenuated viruses, or activation of latent intracellular viruses). (C) Transgenes used as payloads in oncolytic viruses. Of the 97 independent clinical trials, 57 used oncolytic viruses without transgenes while 40 had recombinant transgene(s) expressed with GM-CSF (n=24) and LacZ (n=8) being the most common. GM-CSF, granulocyte-macrophage colony-stimulating factor; HSV-1, herpes simplex virus, type 1; HSP70, heat shock protein 70; hTERT, human telomerase reverse transcriptase; HVJ-E, hemagglutinating virus of Japan—Envelope; IL-2, interleukin 2; ICAM-1, intercellular adhesion molecule 1; LFA-3, lymphocyte function associated antigen 3 gene; NDV, Newcastle Disease virus; NIS, sodium iodide symporter; SVV, Seneca Valley virus.
Figure 2B shows the status of the viruses employed in the OV clinical trials. Native virus was used in about one-third of the clinical trials (n=33) whereas two-thirds of the studies used genetically modified viruses (n=63). The modifications were largely deletion of viral non-essential genes to promote selective tumor cell replication and attenuate viral pathogenicity. In 40 clinical trials, the genetic modifications also include expression of one or more transgenes employing 51 independent recombinant genes. The genes used are shown in figure 2C and GM-CSF was the most common transgene (n=24; 24.7%), which is designed to promote and mature local dendritic cells to help stimulate host immune responses. The next most commonly expressed transgene was LacZ, which encodes the bacterial β—galactosidase, and is used for selection of recombinant virus and can be used as a marker to identify OVs after host infection. There were six viruses encoding prodrug enzyme genes, such as cytosine deaminase (n=3) and the HSV-1 thymidine kinase (n=3), which result in enhanced tumor cell death when patients are treated with chemotherapy prodrugs. Other transgenes included immune enhancing genes, such as interleukin-2 (IL-2; n=1), B7.1 costimulatory molecule (n=1), lymphocyte function associated antigen 3 gene (n=1), and intercellular adhesion molecule 1 (n=1). In addition, one study each used transgenes encoding the heat shock protein 70, human telomerase reverse transcriptase promoter and sodium iodide symporter, which allows for radiolocalization and sensitizes cells to radiation therapy. Finally, four studies used adenoviruses expressing a modified type 5 fiber knob designed to enhance viral cell entry.14
Types of cancer targeted in OV clinical trials
OV clinical trials are targeting a large number of patients with cancer (see table 1). Overall, the most common tumors evaluated were melanoma and GI cancers. For melanoma, there were 30 clinical studies accruing the largest number of patients (n=1000). This includes the phase III T-VEC clinical trial that enrolled 436 patients with melanoma. There were a total of 76 clinical trials targeting patients with GI cancer and enrolled 577 patients. Figure 3A shows the cancer types targeted in specific OV clinical trials and other common cancers targeted included head and neck cancer, (n=15 studies) breast and gynecological cancers (n=31 studies), genitourinary cancers (n=26 studies), and sarcomas (n=16 studies).

Types of cancer being targeted in oncolytic virus (OV) clinical trials. The histological type of cancer being treated by OVs in clinical trials is shown by (A) number of clinical studies and by (B) patients enrolled. Melanoma and GI cancers were the most common. CSCC, cutaneous squamous cell carcinoma; GI, gastrointestinal; N.S., not specified; NOS, not otherwise specified.
Figure 3B shows the number of patients enrolled into OV clinical trials and, while melanoma was most common, this was followed by GI cancers (n=577; 17.8%), genitourinary cancer (n=207; 6.4%), non-small cell lung cancer (n=197; 6.1%), gynecological cancers (n=185; 5.7%), brain tumors (n=154; 4.8%), breast cancer (n=136; 4.2%), and head and neck cancer (106; 3.3%). There were 494 (15.3%) patients enrolled with solid tumors that were not otherwise defined. An additional 71 (2.2%) patients with a variety of hematological malignancies were treated with OVs in the clinical trials reported. Sixty-two patients (1.9%) across 10 clinical trials were f pediatric tumors.
Other drugs being used in combination with OVs
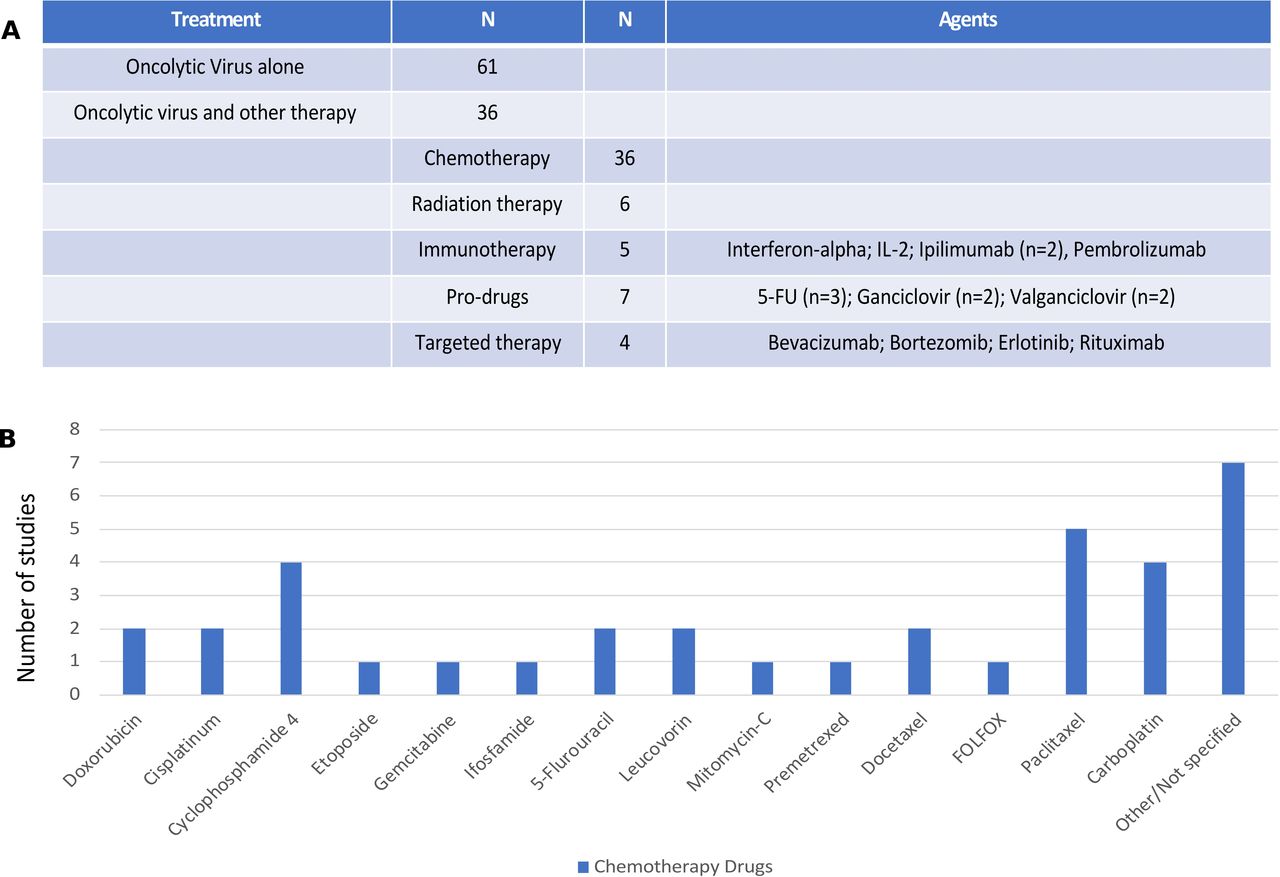
Overall, of the 97 clinical trials reviewed, 61 (62.9%) clinical trials were conducted with OV monotherapy while 36 (37.1%) reported OV was given in combination with at least one other treatment or anticancer drug. Of the combinations (see figure 4A), the most common other drugs were cytotoxic chemotherapy agents (n=36; 37.1%) and chemotherapy prodrugs (n=7; 7.2%). Other modalities used in OV combination studies included radiation therapy (n=6; 6.2%), immunotherapy (n=5; 5.2%) and targeted therapy (n=4; 4.1%).
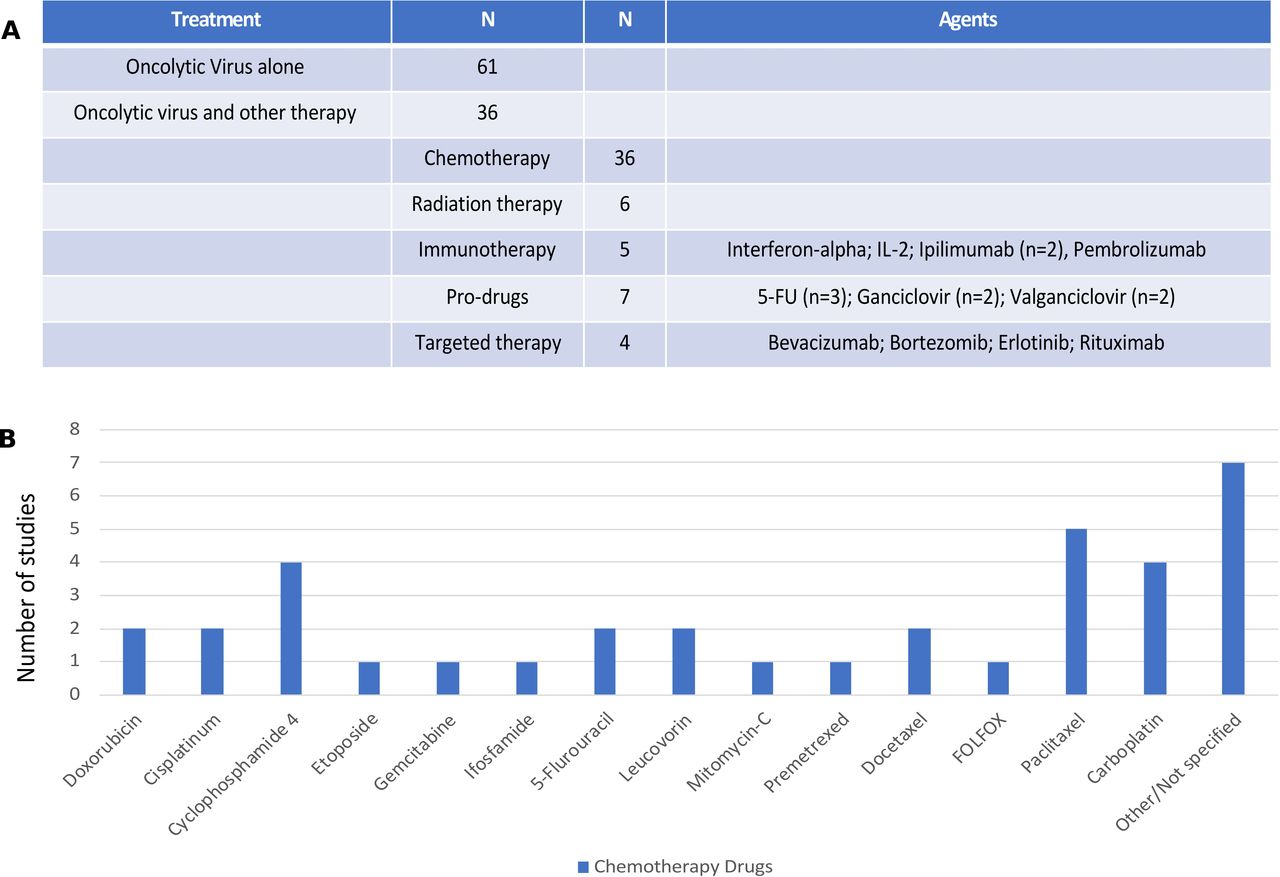
Combination agents used with oncolytic viruses (OVs) in clinical trials. (A) The number of clinical studies using monotherapy OVs (n=61) or combination trials (n=36) with the breakdown by types of other drugs or regimens combined with OVs. The specific agents are listed for immunotherapy, prodrugs and targeted therapy; (B) chemotherapy agents used in combination OV clinical trials. IL-2, interleukin 2; 5-FU, 5-fluorouracil.
The types of chemotherapy agents used are shown in figure 4B with the most common not reported and largely from studies that allowed investigator choice or standard chemotherapy to be given with OV treatment and the type of chemotherapy was not explicitly reported. Where specific agents were prespecified within the clinical protocol, the most common agents used were paclitaxel (n=5) and carboplatin (n=4), often used together. In addition, four studies used cyclophosphamide, which was given as preconditioning chemotherapy to help promote antitumor immune responses. There were seven studies that combined OV treatment with a prodrug, including three studies with 5-fluorocytosine, a precursor to 5-fluorouracil, two studies with ganciclovir and two studies with valganciclovir.
There were few clinical studies reporting on the combination of OV and immunotherapy, but all studies used immune checkpoint blockade or cytokines. Two trials used ipilimumab and one study used pembrolizumab. There was one study each using interferon-alpha and IL-2. There were four studies that reported on OV and targeted therapy with one each evaluating combinations with erlotinib, rituximab, bortezomib and bevacizumab.
Routes of OV administration
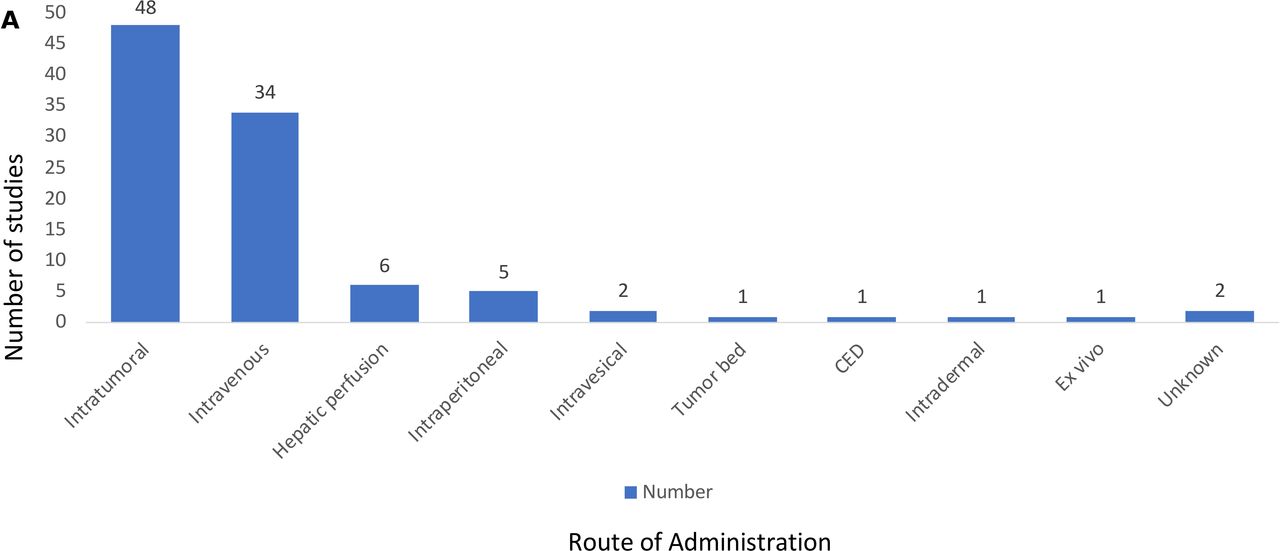
The delivery of OVs has been a controversial area and so we sought to determine which routes of administration were used in the reported OV clinical trials (figure 5). The most common route was IT delivery used in 48 of the clinical trials (49.5%) followed by intravenous delivery used in 34 of the clinical trials (35%). Other routes of delivery included hepatic artery infusion in six studies (6.2%), intraperitoneal delivery in five studies (5.1%) and additional delivery modalities included intravesical delivery (n=2), direct injection of a resected tumor bed (n=1), convection-enhanced delivery to brain tumor bed (n=1), intradermal injection (n=1), ex vivo infection of tumor cells (n=1), and two studies did not report how the OV was given. There were no reported clinical trials using stem cell or nanovesicle delivery, although these have been described in preclinical studies.15 16

Routes of administration for oncolytic viruses in clinical trials. Method of oncolytic virus delivery in clinical trials; most were by intratumoral (n=48) or intravenous (n=34) routes of administration with 18% using alternative delivery routes. CED, convection-enhanced delivery.
When delivery was evaluated by numbers of patients (table 1), the most common approaches were again IT (n=1482; 45.8%) and intravenous (n=1147; 35.5%). Fifty-four patients received OVs by multiple routes in the same study, most commonly a combination of intravenous and IT. Another 550 (17%) of patients received OVs through other routes, as described above.
Safety profile of OVs in clinical development
OVs have been purported to have a tolerable safety profile and, thus, we sought to determine the type and incidence of adverse events being reported in OV clinical trials. Consistent with this notion, we found that the vast majority of common treatment-related adverse events attributed to OVs were low grade (CTCAE grade 1–2) constitutional symptoms and local injections site reactions. As shown in table 2, the most common OV-related adverse event reported across all 97 clinical trials was fever seen in 60 studies (56 grade 1–2 and 4 studies reporting grade 3–4). Other frequent low-grade constitutional symptoms included chills and rigors (n=40), nausea and vomiting (n=30), flu-like symptoms (n=27), fatigue (n=36), and pain (n=19). Local injection site pain was reported in 15 studies. Common grade 3 or greater OV-related adverse events included nausea/vomiting (n=8 studies), pain (n=7 studies), fever (n=4 studies), fatigue (n=4 studies) and flu-like symptoms (n=2 studies).
Most common treatment-related adverse events in oncolytic virus clinical trials
Overall, there were 98 grade 3 and 21 grade 4 treatment-emergent adverse events reported across the clinical studies (online supplemental tables 2 and 3). Many of these events were associated with disease progression or due to other drugs used in combination clinical trials. Given the large number of early phase clinical studies often enrolling late stage patients, the safety profile for OVs appears to be tolerable. As shown in (online supplemental tables 2 and 3), most adverse events were comparable across IT and intravenous routes of delivery with only one case of infusion reaction reported following intravenous delivery of monotherapy reovirus. While myelosuppression was common in the trials, high grade events were more frequently associated with cytotoxic chemotherapy administration. Immune-related adverse events (irAEs) were rare and while there were a few reported in the studies, high grade irAEs were uniformly associated with coadministration of immune checkpoint inhibitors.
Supplemental material
Viral bioshedding in OV clinical trials
Since OVs are typically replication competent viruses, transmission to close contacts and/or the environment is possible, and patients treated on OV clinical trials are often evaluated for evidence of viral shedding. Of note, none of the studies reported any transmission of OV to household contacts or healthcare providers. Of the 97 studies reported, viral bioshedding was evaluated in 71 trials (73.2%) with 21 (21.6%) not including an assessment of viral shedding and five additional studies did not specify whether shedding had been performed but none of these studies reported shedding data (figure 6A).

Summary of viral bioshedding assessment in oncolytic virus clinical trials. (A) The number of studies that collected information on viral shedding (n=71) while 21 studies did not assess viral shedding and five could not be determined. (B) Pie chart of the anatomic sites or fluid biospecimens collected for virus determination showing that blood/serum and urine were the most commonly tested sites followed by tumor tissue. (C) The frequency of positive viral detection and by method of detection (PCR, plaque assay or both).
The presence of virus in tissue is an important parameter to ensure biodelivery of virus to tumor sites and to understand which tissues and/or fluids may be reservoirs or sites of viral shedding. Figure 6B shows the tissues and fluids that were evaluated across the 97 OV clinical trials. The most common site evaluated for OV shedding was blood or serum in 56 (57.7%) of the studies. This was followed by urinary shedding in 36 (37.1%) studies and tumor biopsy specimens in 26 (26.8% studies). An additional 18 studies (18.6%) reported virus in salivary fluid or oral swabs and 12 studies (12.4%) reported virus in sputum samples. There were 26 studies where other fluids or tissues were collected, including cerebrospinal fluid, peritoneal washings, injection sites and so on. Five studies reported viral shedding but did not specify the fluids or tissues used.
In the 71 studies that evaluated viral bioshedding, all studies reported evidence of virus detection (figure 6C). There are different assays that can be used, and we found that the most common method for detection was PCR, which detects specific viral genome sequences, and was used to detect OVs in 58 (81.7%) of the studies. Plaque assay detects infectious viral particles, and this was done alone in one study, and as a complement to PCR assay in 12 (16.9%) of the reported studies.
Antiviral immunity in OV clinical trials
The immune response against the virus is an important correlative biomarker in OV clinical trials. The number of studies reporting humoral and cellular viral-specific immune responses is summarized in figure 7. Across the 97 studies evaluated, we found that measurement of antiviral antibody titers was conducted in 63 (64.9%) of the clinical trials. This included an assessment of neutralizing antibodies in 27 (27.8%) studies and the remainder evaluated non-neutralizing titers. Assessment of viral-specific T cell responses was less common and reported in only 10 (10.3%) clinical trials.
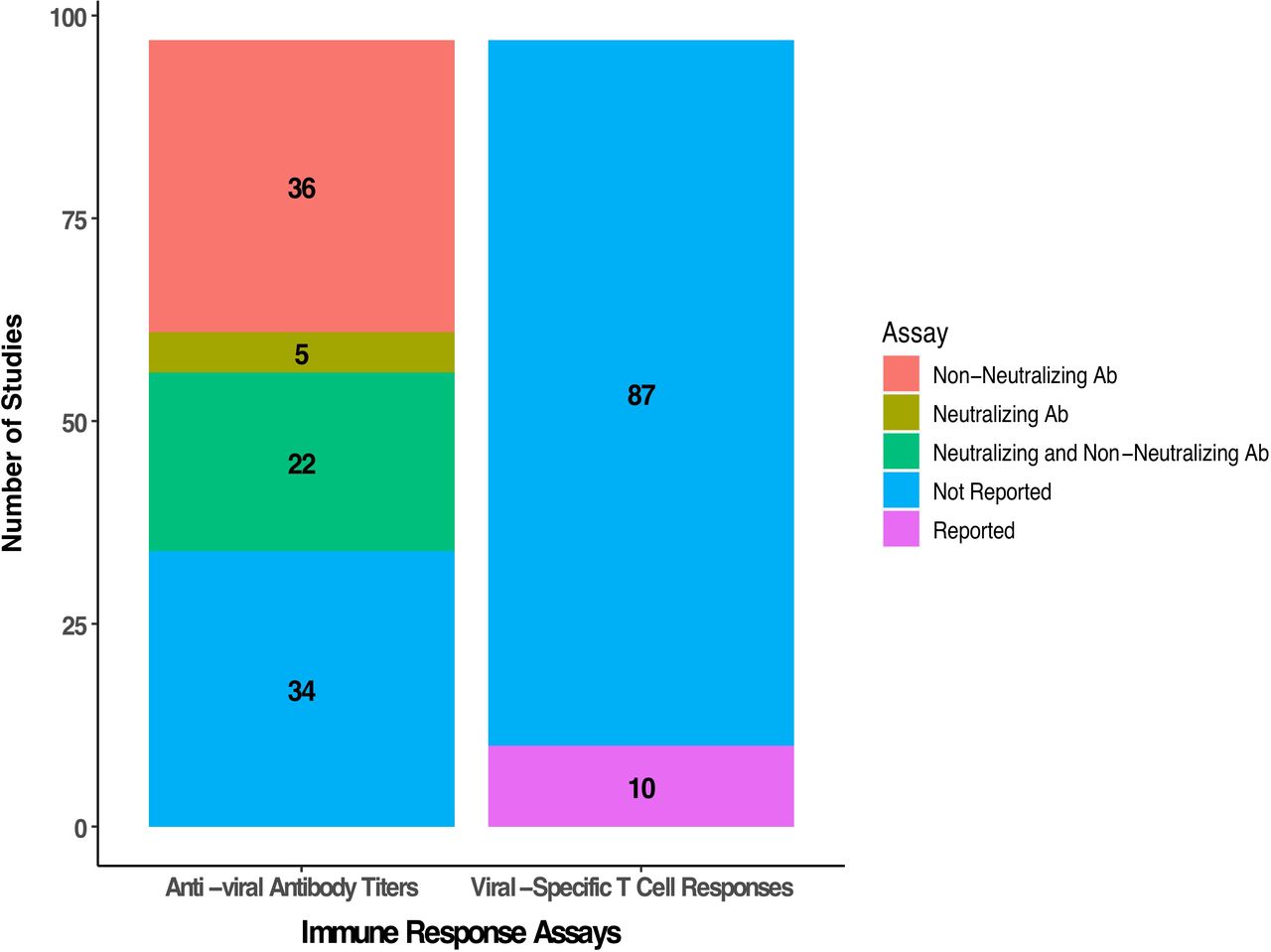
Summary of antiviral humoral and cellular immunity reported in oncolytic virus clinical studies. Detection of antiviral antibody titers was performed in 63 studies with 36 reporting non-neutralizing antibody titers, 5 reporting neutralizing titers and 22 reporting both neutralizing and non-neutralizing titers. Only 10 studies reported viral-specific T cell responses.
Antitumor immunity in OV clinical trials
OVs are expected to mediate antitumor activity, at least in part, through induction of host antitumor immunity. To determine if the clinical trials were evaluating patients for evidence of tumor-specific immune responses, studies of antitumor immunity were sought in the clinical trial reports and are summarized in table 3. Around half of the studies, 48 (49.5%) did report some evidence of antitumor immune response. This was usually limited to a general assessment with immunohistochemistry of tumor biopsy specimens (n=25; 52%) and serum cytokine measurement (n=19; 39.6%) being the most common assays employed. Another six studies (12.5%) evaluated the phenotype of peripheral blood immune cells by flow cytometry. There were seven clinical trials (14.5%) that used tumor antigen-specific enzyme-linked immunosorbent T cell assay (ELISPOT) assays to define tumor immunity. Three studies used other assays.
Summary of antitumor immunity analyzed in clinical oncolytic virus studies
Antitumor activity in OV clinical trials
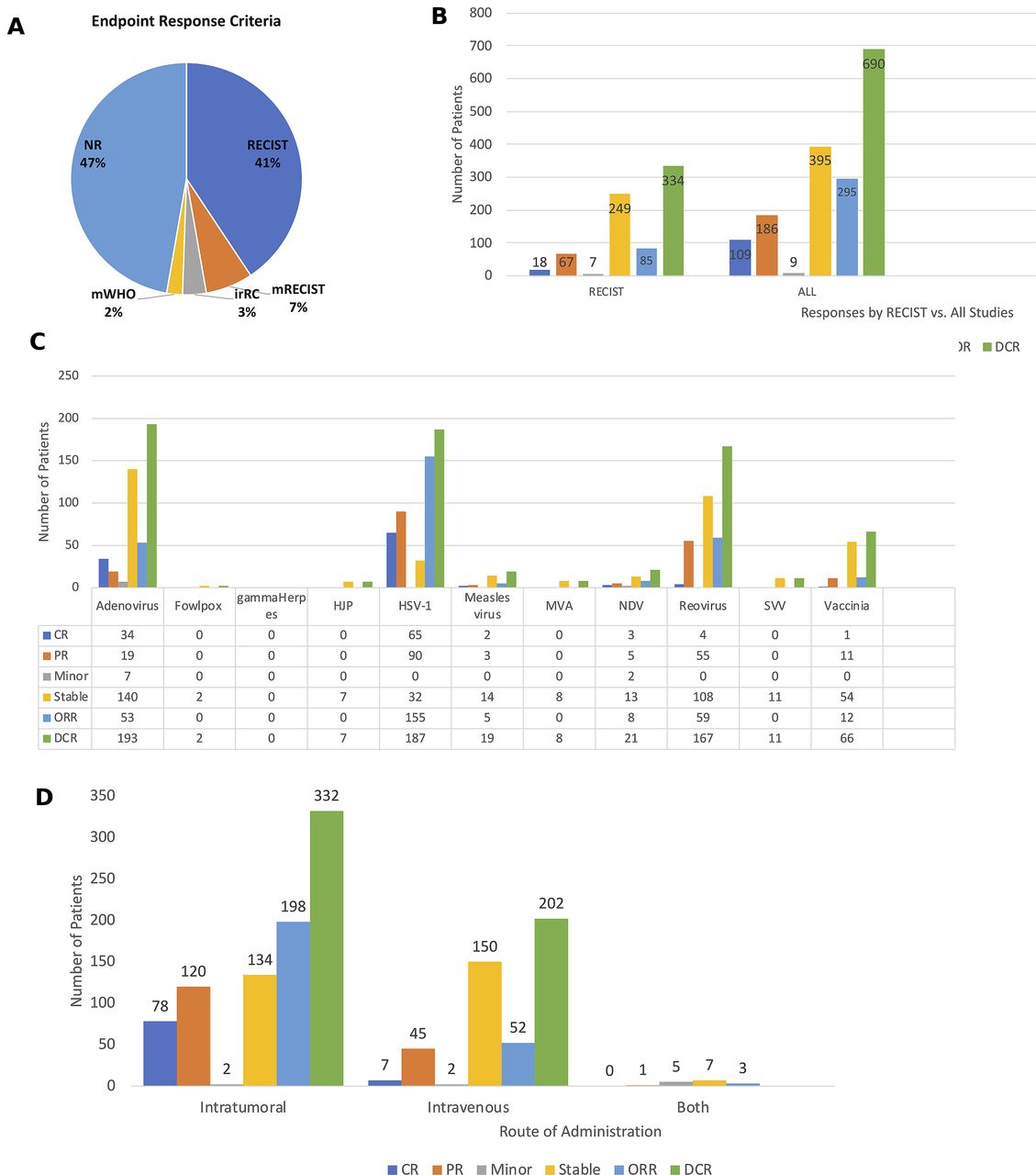
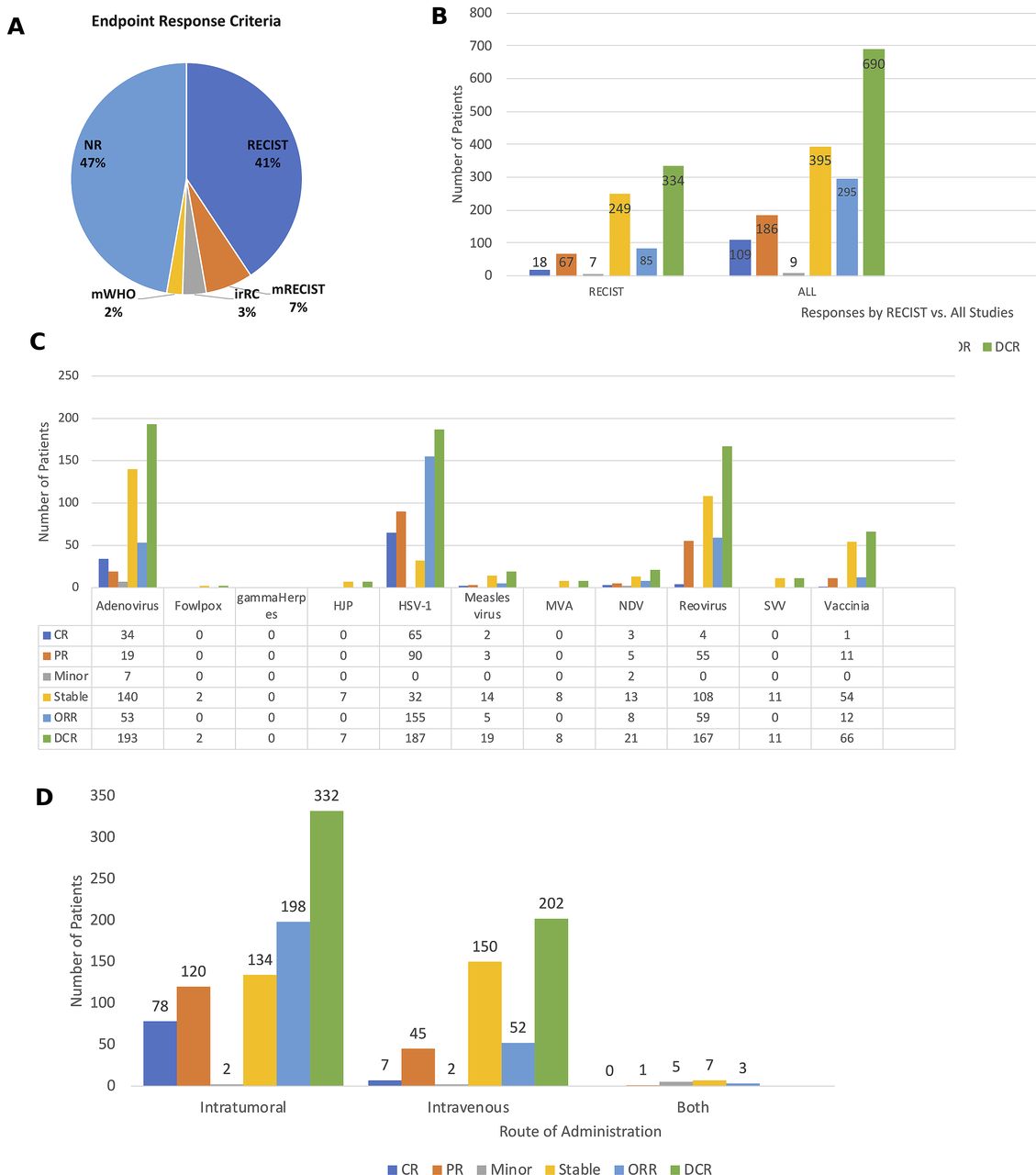
Although most of the clinical trials were early phase studies and not designed to detect therapeutic responses, the majority of the studies did report clinical responses. Importantly, 46 (51%) clinical trials used some form of the Response Evaluation Criteria in Solid Tumors (RECIST) criteria to report clinical responses, including 37 trials (41%) using standard RECIST, 6 studies (7%) using modified RECIST, and 3 studies (3%) using irRECIST (3%) criteria (figure 8A). Two clinical trials (2%) used modified WHO criteria, including the phase III OPTiM T-VEC study while the remaining 49 (47%) of the studies did not report using specific criteria. Of the 3233 patients treated across the studies evaluated, there was an overall objective response rate observed in 292 (9.0%) of the patients, and this included 109 (3.4%) patients with complete responses and 183 (5.7%) patients with partial responses. In addition, 389 (12.0%) patients had stable disease resulting in disease control in 681 (21.1%) of the patients. Nine (0.3%) of the patients were reported to have minor responses. Figure 8B shows the overall responses in those studies using RECIST criteria and for all clinical trials. The clinical responses associated with specific OV treatments are shown in figure 8C and responses were most pronounced in patients treated with HSV-1 with objective responses seen in 155 patients and disease control in 187 patients; adenovirus, which was associated with objective responses 53 patients and disease control in 193 patients; reovirus, which was associated with objective responses in 59 patients and disease control in 167 patients; and vaccinia virus, which was associated with objective responses in 12 patients and disease control in 66 patients. Finally, we explored whether the route of administration impacted responses and found that clinical responses were higher in patients treated by IT administration with an objective response seen in 198 patients and disease control in 332 patients compared with intravenous administration, which was associated with an objective response in 52 patients and disease control in 202 patients (figure 8D). A few trials allowed treatment with bot IT and intravenous delivery and disease control was seen in seven patients but no objective responses were reported.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Antitumor activity of oncolytic viruses (OVs) in clinical studies. (A) Pie chart showing the endpoint response criteria used to monitor clinical responses in the OV trials. (B) The number of patients with specific clinical responses in clinical trials using RECIST criteria (left panel) and in all studies (right panel). (C) Responses by type of OV used in the clinical study. (D) Responses by route of administration. Abbreviations: CR, complete response; DCR, disease control rate; irRC, immune-related RECIST criteria; Minor, minor response; mWHO, modified WHO criteria; NR, not reported; ORR, objective response rate; PR, partial response; RECIST, Response Endpoint Criteria in Solid Tumors; Stable, stable response.
Discussion
OVs represent a new class of cancer therapeutics with considerable flexibility to induce tumor cell death in a manner that promotes both innate and tumor-specific adaptive immune responses. Indeed, preclinical studies have identified a large number of viral species with native permissiveness for tumor cell replication.1 17 In addition, viruses can be easily modified to improve antitumor activity by promoting preferential entry and replication in cancer cells and enhancing antitumor immunity through altering antiviral immune responses or expressing eukaryotic genes that increase direct cell killing or promote host immunity. The clinical implementation of OV therapy, despite preclinical support for their antitumor properties and tolerable safety profile, has been slow. Challenges include an incomplete understanding of the underlying mechanisms of tumor regression with specific OV agents, lack of biomarkers to better match effective viral species with permissive tumor types and patient features and limited standardization of immune correlates in clinical trials. In order to better understand the current clinical landscape of OV clinical development, we reviewed 97 published OV clinical trials and confirmed a wide range of approaches are under study spanning many different viruses, cancer populations, routes of delivery and combination treatment strategies. Most of the studies were early phase trials likely reflecting the novelty of this approach, but it is also possible that later stage, negative studies may not have been published creating a missed opportunity to better understand the complex responses of OV treatment in patients with cancer. Furthermore, we found that while most studies did evaluate antiviral humoral responses and made limited attempts to identify viral shedding, few studies focused on defining cellular immune responses against virus or tumor cells.
The most common viruses used in OV cancer clinical trials were adenovirus, HSV-1, reovirus and poxviruses. This likely reflects the better understanding of DNA viruses and the ability of large DNA viruses to allow deletion of non-essential viral genes to alter cell selective replication and reduce pathogenicity while being able to accept foreign transgenes for expression. We also found that approximately two-thirds (65%) of the OV studies conducted used some type of viral modification. While transgene expression was reported in 40 clinical trials encompassing 51 specific recombinant genes, the majority were GM-CSF (see figure 2), which is a cytokine thought to promote recruitment and maturation of dendritic cells, and ultimately to help generate adaptive immune responses by promoting cross presentation of tumor antigens.1 18 Other commonly used transgenes included LacZ, included for viral detection purposes, and enzymes designed to activate pro-drugs that aid in tumor cell killing. GM-CSF was used in T-VEC, and the common inclusion of GM-CSF likely represents its integration in the T-VEC product, but is was surprising that other cytokines and genes encoding immune stimulatory proteins were not commonly used in viruses entering the clinic. Although not as amenable to genetic manipulation, reovirus was one RNA virus that has been widely studied in the clinic. The selection of the optimal virus and potential transgenes should be based on further biological analyses of tumor cells, host factors and mechanisms that promote Th1 and CD8+ effector T cell immune responses. Studies have identified intracellular sensors, such as the cGAS-STING complex and Toll-like receptors that are critical for tumor cell induction of host innate immunity.19 Interestingly, these same sensors are used for detection of DNA and RNA viruses, and the status of these intracellular sensors in human cancers are not well defined.20 Further translational and biomarker studies are needed to better define the status of these antiviral machinery elements across human cancers and preclinical studies should continue to evaluate which viruses and genetic modifications are best utilized in tumor cells based on their genomic profile at the time of treatment. While most studies used exogenous OVs, we also identified two trials that used non-traditional approaches to OV treatment. In one clinical trial focused on HIV-positive relapsed/refractory lymphoma, investigators used the proteasome inhibitor, bortezomib, to activate latent gammaherpesviruses (GHVs) in tumor cells.21 The investigators hypothesized that bortezomib would induce lytic activation of the GHVs promoting lymphoma cell killing and might also inhibit HIV infection by restoring the APOBEC3G cytidine deaminase, a natural antiretroviral. The trial did confirm activation of Kaposi sarcoma herpesvirus and Epstein-Barr virus and reported responses in 17 of 22 patients, but treatment also allowed for chemotherapy and rituximab. Strategies to activate latent viruses may be an interesting approach for other cancers as well, and other drugs may be useful for viral reactivation. Panobinostat, a histone deacetylase inhibitor, was reported to reactivate latent HIV in aviremic adult patients, which was used as a way to more completely eradicate HIV with antiretroviral therapy.22 We also found a study that used OVs to infect tumor cells ex vivo as a strategy to optimize autologous cell vaccination.23 These studies suggest that other approaches with OVs are possible and may merit further development.
We hypothesized that most OV clinical trials would focus on melanoma and other skin cancers given the easy accessibility of tumors for local injection. While melanoma was the most common tumor targeted in clinical trials (n=30), and represented the largest number of patients (n=1000), this was skewed by the phase III OPTiM clinical trial of T-VEC which enrolled 436 patients. There was only one clinical trial reporting OV treatment in cutaneous squamous cell carcinoma, although other studies in non-melanoma skin cancer are currently in progress.24 We found that the next most common cancer being targeted with OVs was GI tumors, including colorectal, pancreas, gastric, esophageal and anal tumors (see table 1 and figure 3). We also found many clinical studies were open to any solid tumor and the heterogeneity of patient populations in OV clinical trials may provide challenges to better defining early signals of response. A minority of studies focused on pediatric tumors and patients with a variety of hematological malignancies. Further preclinical studies would be useful to understand potential difference in viral permissiveness and replication across tumor types by individual OV agents.
Another controversial issue in OV clinical development is the selection of the best route of administration. While OVs are ideally suited for direct IT injection, this may limit the number and location of tumors that can be directly treated. While intravenous delivery offers the potential to infect metastatic lesions in multiple locations, administration of OVs into the circulation is limited by dilution in peripheral blood and potential clearance by pre-existing antibodies and other serum proteins.11 There is much interest in identifying novel delivery mechanisms that avoid premature viral clearance while promoting drug biodistribution to tumor sites, including viral envelope modifications, nanodelivery vehicles, integration into stem cells and other cellular carriers.25 Our data suggest that while IT was the most common mode of OV delivery representing 48 clinical trials and 1482 patients (45.8%), there are a large number of studies that have used intravenous administration (34 clinical trials and 1147 patients; 35.5%). Other modalities included hepatic artery infusion, intraperitoneal delivery, intravesical delivery, intradermal injection, direct injection into the tumor bed and convection-enhanced delivery. In some studies, both IT and intravenous administration was used. Overall, there were no major differences in safety or bioshedding that could be distinguished between intravenous and IT (online supplemental table 2) but this retrospective review was not powered to identify differences. Further studies to understand how delivery impacts viral distribution to the tumor site, viral clearance, antitumor immunity and safety are needed. Clinical trials may also consider alternative delivery routes to help define how this may impact therapeutic outcomes.
Most of the clinical trials were early phase studies making analysis of clinical endpoints difficult. Nonetheless, we attempted to explore therapeutic responses and found that the overall objective response across the studies was 9% with an additional 12% of patients achieving stable disease as the best response. While these numbers are low, it is important to remember that most of the studies were early phase and not designed to detect clinical responses. It was notable that only 53% of the reported studies used standardized RECIST criteria to report objective responses making analysis of clinical responses challenging (figure 8A). Of the responses reported the largest numbers appear to be in studies using HSV-1, adenovirus, reovirus and vaccinia virus but this may reflect the larger number of trials being conducted with these OVs (figure 8C). Indeed, HSV-1 includes the only phase III clinical trial which enrolled 436 patients.4 We also examined whether the route of administration impacted antitumor activity. Not surprisingly, objective responses were higher with IT delivery likely related to more rapid viral clearance and dilution when administered by the intravenous route (figure 8D). It was interesting, however, that intravenous administration was associated with objective responses in 52 patients suggesting that further investigation and optimization of intravenous delivery of OVs is warranted.
The majority of studies reported were designed to evaluate safety of OV treatment, and our data confirm previous statements that OV treatment is tolerable across many different viral agents, combination strategies and delivery routes. As shown in table 2, the most common adverse events were low-grade constitutional symptoms, including fever, fatigue, chills/rigors, nausea/vomiting, non-specific pain and flu-like symptoms. Other common adverse events were low-grade injection site pain. We evaluated high-grade treatment emergent events and found most events related to disease progression or other drugs used in combination treatment regimens (online supplemental tables 2 and 3). Although there was one case of a grade 4 infusion reaction associated with intravenous delivery of a reovirus, no major differences were noted between intravenous and IT delivery or with specific viruses. Thus, OV therapy does appear to have an overall highly tolerable safety profile with largely non-overlapping toxicity with other cancer therapeutics. While most of the local and constitutional symptoms were low grade, occasional high-grade events were reported and attention to premedicating with acetaminophen and other analgesics might be considered to prevent or ameliorate these reactions.
Given the safety profile and mechanisms of action associated with OV, they have been suggested as good agents for combination studies. In fact, preclinical data and emerging clinical data support the combination strategy, perhaps most notable for OV and immune checkpoint blockade.17 An initial high rate of response of 50% in patients with advanced melanoma was reported for T-VEC in combination with ipilimumab in a small phase I study.5 This was later confirmed in a larger phase 2 study in which 198 patients with melanoma were randomized to receive T-VEC and ipilimumab or ipilimumab alone.26 In this study, the response rate was doubled from 18% for ipilimumab alone to 38% for patients treated with the combination. In another small phase I study, T-VEC and pembrolizumab was associated with a 62% objective response rate in patients with advanced melanoma, with a third achieving a complete response.6 A larger randomized clinical trial evaluating T-VEC and pembrolizumab versus pembrolizumab alone has completed enrollment and is awaiting follow-up (NCT02965716). Thus, we hypothesized that most of the combination studies would be OV and immunotherapy. However, in review of the 97 clinical studies, only 36 included a combination agent and only five were with immunotherapy (figure 4). Surprisingly, most of the combination studies were evaluating OV and cytotoxic chemotherapy. Prodrugs and radiation therapy were also being investigated in combination with OV; and there is clinical data supporting these concepts.27–29 The use of chemotherapy may be a reflection of the tumors being studied and indeed the most common regimens were not specified with the selection of the chemotherapy left to investigator choice or carboplatin/paclitaxel regimens, commonly used in studies of patients with non-small cell lung cancer. Chemotherapy administration did appear to be associated with expected adverse events but it did not appear to be significantly worse with associated OV treatment. Preclinical studies of how therapeutic responses are improved with specific OVs and other agents should be a high priority with more rapid translation of promising combinations into the clinic.
An important correlate of OV administration is to ensure delivery of virus to tumor cells and assess the degree and kinetics of viral shedding in body fluids and compartments. While we found that viral bioshedding was assessed in 71 studies, most limited this to evaluation of blood or serum in 56 studies, urine in 36 studies and tumor tissue in only 26 studies. Only a few studies included more extensive tissue evaluation, including saliva, fecal samples, skin, injection sites and sputum. Further, while detection of virus is important, detection of replication competent virus is more meaningful, and this requires plaque assays or other methods that can detect viral replication. In the studies included in this report, most investigators used PCR (n=58) to detect viral genome while 12 studies used both PCR and confirmatory plaque assay with one study relying solely on plaque assay for viral detection. Of note, there were no reports in any study of household or close contact transmission of OVs. These data suggest that biodistribution is not being reported routinely or in a standardized manner across most OV studies. An assessment of tumor biopsies for the presence of replicating virus should be considered as part of trial designs, especially in early phase clinical studies. Alternatively, dedicated bioshedding studies could be contemplated to provide a more comprehensive assessment of viral biodistribution and kinetics, and only one trial of T-VEC was dedicated to such an assessment.30
Since the outcome of OV treatment likely depends on the balance between antiviral and antitumor immunity, we assessed whether the OV clinical trials were reporting data on these responses. While 58 studies did report antiviral antibody titers, only 27 included specific assessment of neutralizing antibody titers. Analysis of cell-mediated viral responses was less common and was reported in only 10 clinical trials, although all were able to detect an increase in viral-specific T cells after treatment. Less than half of the clinical trials (n=48) attempted to evaluate antitumor immune responses with most focusing on more general descriptive analyses, such as immunohistochemical staining of tumor biopsies for immune cells, serum cytokine levels, and analysis of numbers and activation status of peripheral blood T cells (see table 3). Only seven studies directly assessed tumor-specific T cells, largely by interferon-gamma ELISPOT assay. Clearly, more efforts should be made to include biomarkers of both humoral and cellular virus-specific and tumor-specific immune responses in clinical OV studies.
This report has several important limitations, including the retrospective nature of the review, the focus on including only peer-reviewed published papers identified on PubMed, the heterogeneity of the clinical trials and the long period of time (20 years) encompassed by the review. We recognize that many OV clinical trials have not been reported and were, thus, not included in this review. For example, many studies of an oncolytic coxsackievirus have been conducted with interesting clinical and biomarker results but these have only been reported in abstract form, to date and thus were not included in our analysis.31 Further, negative studies, such as a recent Phase III clinical trial of an oncolytic vaccinia virus, Pexa-Vec in combination with sorafenib, in hepatocellular carcinoma was reported through press releases but data was not published.32 Although disappointing, publication of negative studies would be helpful to generate discussion and provide important information on how to improve OVs, patient selection and study designs for OV therapy.
Despite the limitations of the current report, our data support the need for a more organized effort to standardize clinical trial design for OV clinical trials. Investigators should consider using standardized endpoint response criteria when reporting clinical responses. Since RECIST appears to provide a similar pattern of response to that seen in studies not using RECIST, this may be an appropriate tool for monitoring OV-treated patients. We would also support efforts to work with regulatory agencies to identify better methods for assessing injected lesions independently from patient-level responses as this may be informative in small, early phase clinical studies. Collecting data on viral bioshedding is important and attempts to monitor virus through measurement of replicative virus rather than PCR across multiple anatomic sites should be the standard in phase I studies, and this might be omitted from later phase development unless a specific transmission concern is seen. While most trials did evaluate antiviral antibody titers, additional efforts need to be made to evaluate antiviral T cell responses. Whenever possible, assessment of both antiviral and antitumor cellular immunity in early phase trials may provide important insights into the underlying mechanisms of antitumor immunity with OVs and guide late stage development. Finally, while OVs are well suited for combination regimens, few published studies have explored OV with other immunotherapy agents and this would be a high priority for further clinical investigation.
In conclusion, we summarized the clinical experience with OVs over the past 20 years. While the data are not exhaustive, it provides a snapshot into the types of OVs being used in the clinic, tumors being targeted, combinations in development and demonstrates the status of correlative studies being done. OVs represent a novel approach and may require additional research to optimize the viral vectors, promote better patient selection and improve viral delivery and biomarker analysis. The unique mechanism of action may also require a change in current methodology used for clinical endpoint assessments, and recently investigators have proposed new criteria for RECIST monitoring of OVs and other IT agents (itRECIST).12 Our data suggests that currently most OV studies use large DNA viruses with modifications and transgene expression has largely employed GM-CSF with most given by IT delivery although there is an increasing number of IV studies. While most studies have used monotherapy OVs, published combination studies have largely been with chemotherapy. There is a need for more preclinical studies to better define the underlying biological mechanisms that OVs use to mediate antitumor activity and clinical studies need to consider more standardized approach to defining viral distribution and integrating appropriate biomarker studies that provide information on both the antiviral and antitumor immune responses. Investigators in the field should also be encouraged to publish their data, which will speed clinical development and help optimize the full potential of OVs for the treatment of patients with cancer.
Acknowledgments
The authors wish to thank Gail Iodice and Praveen Bommareddy for useful conversations.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors HLK: conceptualization and research design. NM and HLK: conducting research, manuscript writing. All authors: data acquisition and analysis, review, editing and approval of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests HLK is an employee of Immuneering Corporation. DMM is a member of the scientific advisory board for Checkpoint Therapeutics, and has received honoraria from Pfizer, Merck Sharpe & Dome, Sanofi Genzyme and Regeneron. RH reports grant support from Bristol-Myers-Squibb and Novartis.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.