Article Text

Abstract
Background As the most powerful T cell agonists known, superantigens (SAgs) have enormous potential for cancer immunotherapy. Their development has languished due to high incidence (60%–80%) of seroreactive neutralizing antibodies in humans and tumor necrosis factor-α (TNFα)-mediated cardiopulmonary toxicity. Such toxicity has narrowed their therapeutic index while neutralizing antibodies have nullified their therapeutic effects.
Methods Female HLA-DQ8 (DQA*0301/DQB*0302) tg mice expressing the human major histocompatibility complex II (MHCII) HLA-DQ8 allele on a high proportion of PBL, spleen and lymph node cells were used. In the established tumor model, staphylococcal enterotoxin G and staphylococcal enterotoxin I (SEG/ SEI) (50 µg each) were injected on days 6 and 9 following tumor inoculation. Lymphoid, myeloid cells and tumor cell digests from tumor tissue were assayed using flow cytometry or quantitated using a cytometric bead array. Tumor density, necrotic and viable areas were quantitated using the ImageJ software.
Results In a discovery-driven effort to address these problems we introduce a heretofore unrecognized binary complex comprizing SEG/SEI SAgs linked to the endogenous human MHCII HLA-DQ8 allele in humanized mice. By contrast to staphylococcal enterotoxin A (SEA) and staphylococcal enterotoxin B (SEB) deployed previously in clinical trials, SEG and SEI does not exhibit neutralizing antibodies in humans or TNFα-mediated toxicity in humanized HLA-DQ8 mice. In the latter model wherein SAg behavior is known to be ‘human-like’, SEG/SEI induced a powerful tumoricidal response and long-term survival against established melanoma in 82% of mice. Other SAgs deployed in the same model displayed toxic shock. Initially, HLA-DQ8 mediated melanoma antigen priming, after which SEG/SEI unleashed a broad CD4+ and CD8+ antitumor network marked by expansion of melanoma reactive T cells and interferon-γ (IFNy) in the tumor microenvironment (TME). SEG/SEI further initiated chemotactic recruitment of tumor reactive T cells to the TME converting the tumor from ‘cold’ to a ‘hot’. Long-term survivors displayed remarkable resistance to parental tumor rechallenge along with the appearance of tumor specific memory and tumor reactive T memory cells.
Conclusions Collectively, these findings show for the first time that the SEG/SEI-(HLA-DQ8) empowers priming, expansion and recruitment of a population of tumor reactive T cells culminating in tumor specific memory and long-term survival devoid of toxicity. These properties distinguish SEG/SEI from other SAgs used previously in human tumor immunotherapy. Consolidation of these principles within the SEG/SEI-(HLA-DQ8) complex constitutes a conceptually new therapeutic weapon with compelling translational potential.
- immunotherapy
- melanoma
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
While immunotherapy of cancer has achieved several notable successes using both antigen specific and antigen non-specific approaches, of paramount concern to investigators is that a substantial fraction of patients fail to respond to these measures.1–3 This has fueled a universal search for conceptually new tools and strategies to eradicate solid tumors. To this end, we examine bacterial superantigens (SAgs) the most powerful T cell agonists known with intrinsic T cell receptor (TCR) and costimulatory activating properties in an effort to harness their vast hidden potential for cancer treatment.
The enterotoxins of Staphylococcus aureus (SEs) comprise a group of globular proteins with diverse sequences capable of activating up to 20% of the T cell repertoire.4 These molecules, known as SAgs, further form a bridge between the α- and/or β-chains of major histocompatibility complex II (MHC II) molecules and the variable component of the β-chain of TCRs leading to T cell activation.5 6 This canonical MHCII-SAg-TCR model has been recently revised following the revelation that SAgs possess intrinsic ligands that engage homodimeric sites on B7-2 and CD28 costimulatory molecules that regulate CD4+ T cell cytokine output.7–9 While SAgs induce non-specific, polyclonal expansion of CD4+ and CD8+T cells along with cytokines such as interferon-γ (IFNγ) and tumor necrosis factor-α (TNFα) they also activate recall T cell responses against viral, bacterial, autoimmune and tumor targets.10–13 Despite these shared properties, individual SAgs exhibit broad diversity in the strength of their T effector cell, T regulatory cell (Tregs) responses and cytokine output largely due to their differential topology and affinity for MHCII haplotypes and TCR.14–17
With respect to cancer treatment, canonical SAgs such as SEA and SEBalone or fused to tumor targeted antibodies have demonstrated antitumor effects in animal models.18 19 Their application to human cancer, however, has been hampered by hemodynamic toxicity and the presence of pre-existent seroreactive neutralizing antibodies.20 21 In human trials, these antibodies nullified the therapeutic effect of SAgs, contributed to their toxicity, and narrowed the number of human cancer patients eligible for treatment.20 22 Indeed, tumor remissions in response to canonical SAgs occurred predominantly in patients with minimal to absent levels of such neutralizing antibodies.21 In addition, the T cell-mediated tumor cytotoxic effects generated by wild type SAgs in humans have been invariably accompanied by TNFα-mediated hemodynamic toxicity.20 These findings spawned a quest to identify SAgs that exhibit minimal levels of neutralizing antibodies while conserving T cell effector and silencing TNFα activity. Our search led us to the staphylococcal enterotoxin growth cluster (egcSEs) of SAgs originally described by Lina and colleagues.23 This grouping is present in 80% or Staphyloococcus aureus isolates and consists of five functional enterotoxins situated in an operon outside of the core-genome .24 25 Unlike canonical SEA, sepsis associated with these SEs was attended by a significantly lower incidence of toxic shock.26 A functional assessment of individual egcSEs showed that while both SEG and SEI induced potent T cell activation, SEG produced the lowest levels of TNFα and SEI the highest levels of IFN-γ.14 SEG and SEI were further shown to exhibit low levels of pre-existing neutralizing antibodies in human sera largely ascribed to weak transcription and suboptimal toxin secretion by the parental Staphylococcus aureus.27 28 Their subdued toxicity coupled with minimal levels of neutralizing antibodies in human sera suggested that these agents might work together to produce a more favorable therapeutic index against human cancer than previously deployed SEs.
The quality and strength of SAg-induced T effector cell and cytokine responses are critically reliant on their functional interaction with endogenous MHCII. We thereby commenced a search for human MHC class II allele that could preserve SEG/SEI T effector function in vivo while silencing the disabling effects of toxicity-inducing TNFα. Kotb et al underscored the clinical relevance of SAg specificity for human MHCII alleles in governing the severity of toxicity induced by streptococcal pyrogenic exotoxin (SPEA) l in streptococcal sepsis. 16 17 To examine the effect of MHCII alleles on SEG/SEI behavior, we turned to transgenic murine models that express human MHC class II in the absence of murine MHCII on a large percentage of myeloid cells.29 Because human MHCII allotypes are more efficient at SAg presentation than their murine MHCII counterparts, these humanized models have been preferred by investigators to examine human-like T cell and cytokine responses against canonical SAgs such as SpeA and SEB.30 For these studies, we selected the MHCII transgenic model expressing the HLA-DQ8 allele (DQA*03:01, DQB*03:02) because of its broad expression in humans and structural features that include highly polymorphic α-chain and β-chain variants capable of engaging SAgs and a broad repertoire of peptides.31–33 Indeed, a group of human and murine melanoma neopeptides that binds selectively to MHCII and/or HLA-DQ alleles and activates CD4+ T cells was recently unearthed via high throughput epitope mining.34–37 To our knowledge, humanized MHCII transgenic mice including the humanized HLA-DQ8 model have not been previously exploited to examine the effect of SAgs and/or tumor neopeptides in antitumor therapy.
Our working hypothesis was that SEG/SEI’s unique properties deployed in humanized HLA-DQ8 tg mice could enable an antitumor response against established melanoma without undue toxicity. Remarkably, the humanized model unveiled the nature and scale of the tumoricidal response and long-term challenge-resistant survival induced by these SAgs. The tumoricidal response preserved T cell-mediated tumor killing while negating TNFα-induced toxicity. SEG/SEI in this model further unleashed a broad CD4+ and CD8+T cell mediated anti-tumor network marked by tumor antigen priming, chemotactic mobilization and recruitment of tumor reactive T cells culminating in the appearance of tumor specific memory.
Results
SEG/SEI induce robust T cell proliferation and production of T cell effector molecules along with diminished Treg differentiation in splenocytes from humanized MHCII mice
Although a fundamental feature of SAgs is the ability to induce robust T cell mitogenesis, individual SAgs often exhibit differential proliferative strength.21 22 26 In initial studies, we therefore compared the mitogenic activity of SEG, SEI, SEG/SEI with canonical SAgs SEB and SEA using naïve splenocytes from MHCII HLA-DQ8 humanized mice and C57BL/6 mice (MHCII I-Aα b I-Aβ b). In l both strains, SEG, SEI, SEG/SEI, induced proliferation in CD4+ and CD8+T cells comparable to that of canonical SEB and SEA (figure 1A,B). This finding affirmed the potent T cell mitogenic ability of SEG, SEI and SEG/SEI. Notably, T cell mitogenesis induced by all SEs in splenocytes from HLA-DQ8 humanized mice was more potent relative to that of C57BL/6 splenocytes (figure 1A,B). The subdued proliferative response of C57BL/6 mice has been attributed to reduced binding affinities of SAg to murine MHCII alleles.38 Next, we evaluated the ability of SEG, SEG and SEG/SEI to activate key T cell effector molecules, IFNγ and perforin-granzyme-B, in murine CD4 +and CD8+T cells. SEG, SEI and SEG/SEI stimulated increased levels of IFNγ in CD4 +splenocytes from HLA-DQ8 tg and C57BL/6 mice but comparable levels of IFNγ in CD8+ cells from both strains (figure 1C,D). CD4+ and CD8+T splenocytes from HLA-DQ8 tg mice showed significantly higher levels of perforin-granzyme-B levels relative to splenocytes from C57BL/6 mice (figure 1E,F). We next evaluated the capacity of SEG/SEI to induce differentiation of CD4 +CD25+Foxp3+ (Tregs) in T cells from HLA-DQ8 tg and C57BL/6 mice. In both strains, SEG, SEI and SEG/SEI induced significant reductions of the Treg population relative to the untreated controls (figure 1G). Collectively, these findings indicate that SEG, SEI and SEG/SEI presented by HLA-DQ8 splenocytes from humanized HLA-DQ8 tg mice possess robust T cell proliferative function comparable to that of canonical SEA and SEB. SEG, SEI. SEG/SEI further demonstrate an ability to stimulate key T effector cell molecules, IFNγ and perforin-granzyme-B while diminishing the Treg population. The reduced proliferation and T effector response to SEG/SEI noted in C57BL/6 splenocytes expressing murine MHCII relative to splenocytes from HLA-DQ8 tg mice displaying human MHCII accords with previous reports.38

Evaluation of mitogenic strength, T cell effector and Treg differentiation in response to SEG, SEI and SEG/SEI in vitro. CD4+ and CD8+ splenocytes from HLA-DQ8 tg, and C57BL/6 mice were incubated with SEA, SEB, SEG, SEI, (1 µg/mL) and SEG/SEI (0.5 µg/of each) for 72 hours and assayed for proliferation (A, B). CD4+ and CD8+T cells from HLA-DQ8 TG, and C57BL/6 mice were incubated with SEG, SEI (1 ug/mL) or SEG/SEI (0.5 µg/mL of each) for 72 hours and assayed for IFNγ (C, D) or granzyme and perforin (E, F). CD4 +T cells from HLA-DQ8 TG, and C57BL/6 mice were incubated with SEG, SEI, (1 µg/mL) or SEG/SEI (0.5 µg/mL of each) for 72 hours and FOXP3 expression was quantified (G). Data expressed as mean±SD. ANOVA with Tukey post test, n=3–5. *P<0.05, **p<0.01, ***p<0.001, ****p<0.001.
SEG/SEI-treated HLA-DQ8 tg mice display long-term survival of established B16-F10 melanoma and resistance to tumor rechallenge
In preliminary studies, we examined the genetic compatibility of HLA-DQ8 tg and C57BL/6 mice. As expected from their shared genetic background39 mitomycin treated C57BL/6 splenocytes failed to stimulate HLA-DQ8 cells in mixed lymphocyte culture (online supplemental figure 1A). Predictably, the B16-F10 melanoma indigenous to the C57BL/6 mice was lethal in both strains 20 days after receiving a tumorigenic dose of B16-F10 melanoma cells (figure 2A,B). HLA-DQ8 tg mice y were not immunologically impaired since they eradicated the genetically unrelated 4T1 carcinoma of Balb/C origin (online supplemental figure 1B). We also established that within 6 days after injection of B16-F10 melanoma cells both C57BL/6 and HLA-DQ8 tg mice exhibited omental metastases of comparable degree along with established histopathologic stroma and an angiogenic network (online supplemental figure 2). Taken together, these data indicate genetic compatibility of the C57BL/6-derived B16-F10 melanoma in HLA-DQ8 tg mice and further show that the B16-F10 tumor was broadly established in both strains at the time SEG/SEI treatment was initiated.
Supplemental material
Supplemental material
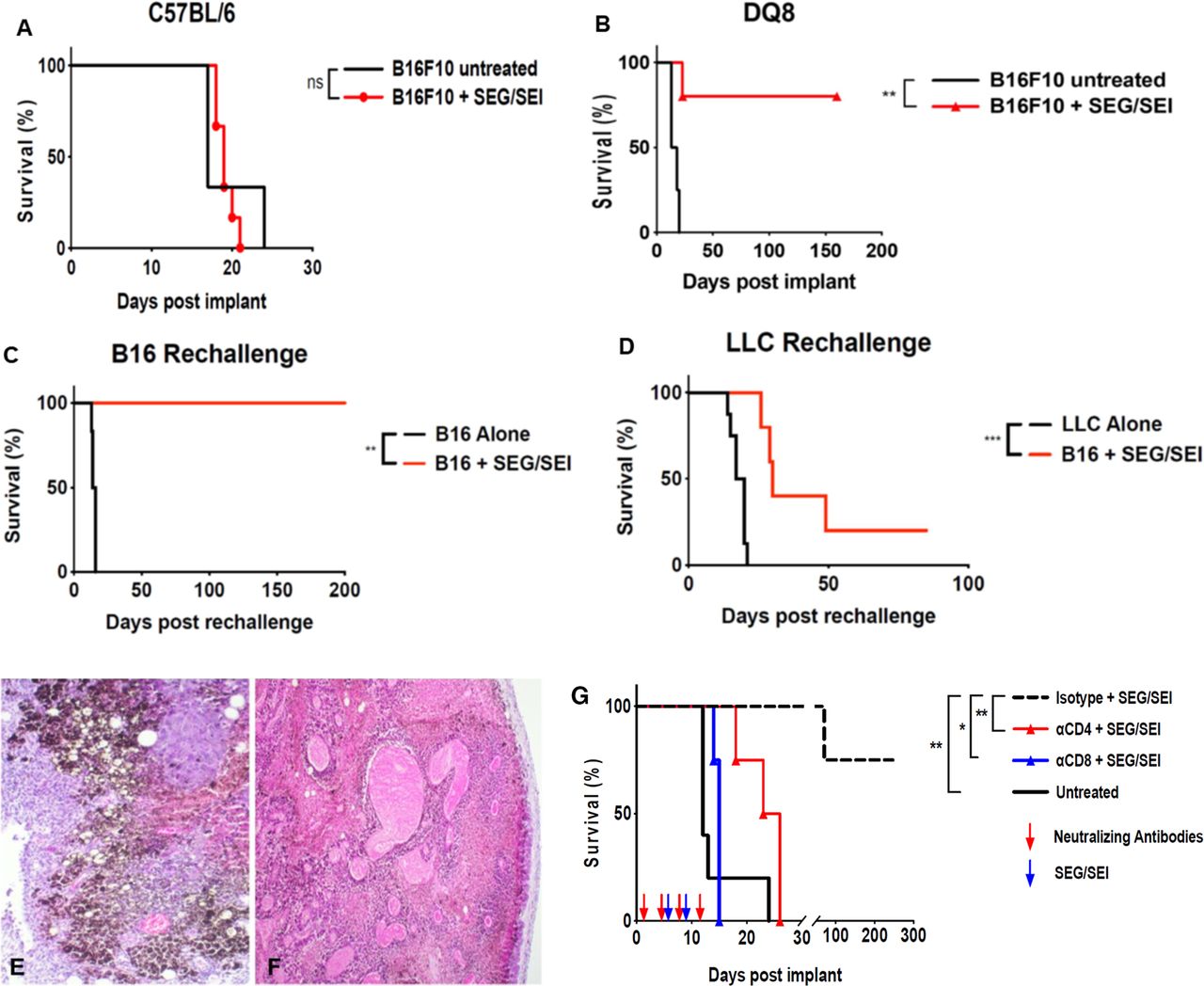
Survival of mice bearing established B16-F10 melanoma treated with SEG/SEI is shown. (A) C57BL/6 or (B) HLA-DQ8 tg mice received 2.5×105 B16-F10 cells IP on day 0. SEG/SEI, 50 µg of each, was administered ip on days 6 and 9. Controls received B16-F10 tumor cells but no treatment (Kaplan-Meier with log-rank n=10–11). (C) On day 160, 4 surviving mice from (B) were rechallenged with 2.5×105 B16-F10 cells ip. Untreated controls received B16-F10 tumor alone. (D) an additional five mice from (B) that survived 160 days were challenged with 2.5×105 Lewis lung carcinoma cells (LLC). Untreated controls received LLC cells alone (Kaplan-Meier with Log-rank). Histopathology analysis of B16-F10 omental tumors on day 13 after tumor inoculation from (E) untreated and (F) SEG/SEI -treated HLA-DQ8 TG mice is shown (H&E ×10 magnification). (G) HLA-DQ8 tg mice received 2.5×105 live B16-F10 cells ip on day 0, followed by the treatment with SEG/SEI, 50 µg of each, on days 6 and 9 and ip injections of anti-CD4, anti-CD8 and anti-lgG2b isotype control as shown (red arrows) (Kaplan-Meier with Log-rank, n=4–5). *P<0.05, **p<0.01.
Next, we compared the ability of SEG/SEI to prolong survival of mice with established B16-F10 melanoma in HLA-DQ8 tg and C57BL/6 mice. In initial studies, C57Bl/6 mice received a tumorigenic dose of B16-F10 cells on day 0 and were subsequently injected with SEG and SEI, 50ug of each, on days 6 and 9. As predicted from their muted T cell proliferative response and diminished ability to generate T effector molecules in vitro, SEG/SEI did not prolong survival in C57BL/6 mice (figure 2A). In a further effort to unveil the antitumor properties of SEG/SEI we turned to HLA-DQ8 tg mice which faithfully induce more potent human-like responses to SAgs relative to wild type murine models. Strikingly, 9 of 11 HLA-DQ8 tg mice inoculated with B16-F10 melanoma and treated as above with SEG/SEI survived for 160 days whereas all untreated HLA-DQ8 tg controls died within day 20 after tumor inoculation (figure 2B). Next, we determined whether the long-term surviving mice could mount a specific antitumor memory response against the parental melanoma. Thus, on day 160, five of the nine surviving mice were rechallenged with a tumorigenic dose of Lewis lung carcinoma (LLC) cells while the remaining four mice were rechallenged with a tumorigenic dose of parental B16-F10 melanoma cells. The group rechallenged with B16F10 melanoma survived for an additional 200 days (figure 2C) whereas four of five mice challenged with LLC died within 50 days (figure 2D). Histology of untreated and SEG/SEI-treated tumors in HLA-DQ8 tg mice 13 days after initial tumor inoculation showed disseminated islands of melanoma cells in the untreated samples whereas sections from SEG/SEI-treated mice displayed broadly propagated tumor necrosis engulfing the core and tumor periphery (figure 2E,F). Hence, while SEG/SEI treatment was ineffective against melanoma in C57BL/6 mice by turning to the HLA-DQ8 model we brought to light the potent ability of SEG/SEI to eradicate the tumor and induce long-term survival. This finding points to a key role of the HLA-DQ8 allele in enabling the tumoricidal effect of SEG/SEI. The striking capacity of HLA-DQ8 mice to reject a tumorigenic melanoma rechallenge but not rechallenge with LLC suggests that the SEG/SEI treatment induced specific anti-tumor memory in these mice. The tumor specificity of the memory response further suggested a role for melanoma neoantigens in processing and priming a tumor reactive T cell population that was subsequently expanded and enriched by SEG/SEI treatment.
Anti-CD4 or anti-CD8-specific antibodies abrogate the anti-tumor effect of SEG/SEI against established melanoma
Having shown that SEG/SEI could stimulate both CD4 +and CD8+T cells in splenocytes from HLA-DQ8 tg mice and induce long-term survival against established melanoma, we next determined the relative roles of CD8+ and CD4+T cells in the tumoricidal response. B16-F10 melanoma was inoculated into HLA-DQ8 tg mice on day 0 and SEG/SEI, 50 ug of each, injected ip on days 6 and 9. Individual groups received anti-CD4 or anti-CD8 specific antibodies while controls received isotype matched IgG2b antibodies. Results shown in figure 2G demonstrate attenuated survival of the SEG/SEI-treated mice receiving anti-CD4 or anti-CD8 antibodies whereas 80% of mice receiving isotype control antibodies survived for 200 days after tumor inoculation. These data indicate a significant role for both CD4 +and CD8+T cells in the SEG/SEI-induced antitumor response in HLA-DQ8 tg mice.
SEG/SEI-treated HLA-DQ8 tg mice show increased T effector cells along with minimal Tregs and myeloid cells in the tumor microenvironment (TME)
In search of the T cell population that generated the tumoricidal response, we next quantitated CD4+, CD8 +T cells, T effector cells, Tregs and myeloid cells in tumors of mice treated with SEG/SEI compared with untreated controls. HLA-DQ8 tg mice were inoculated with B16-F10 melanoma on day 0 and treated with SEG/SEI, 50 ug of each, on days 6 and 9. Tumors were removed and analyzed on day 13. Untreated tumor bearing HLA-DQ8 tg mice served as controls. Tumors from SEG/SEI-treated HLA-DQ8 tg mice showed a striking increase of CD8+ and granzyme-B +T effector cells relative to tumors from untreated HLA-DQ8 tg mice (figure 3A,B). By contrast, myeloid cells in tumors from SEG/SEI-treated HLA-DQ8 tg mice were significantly reduced while Tregs were unchanged compared with the untreated controls (figure 3C,D). Ratios of T effector cells to Tregs or T effector cells to myeloid cells in SEG/SEI-treated HLA-DQ8 tg mice vs untreated HLA-DQ8 tg controls were significantly increased (figure 3 legend). Collectively, these findings point to CD8 +T effector cells as key mediators of the acute tumoricidal response and suggest that their antitumor effectiveness in the TME may be enhanced by the minimal presence of myeloid cells and suppressive Tregs.

Evaluation of T cells and myeloid cells in B16-F10 tumors from HLA-DQ8 tg mice treated with SEG/SEI is shown. HLA-DQ8 tg mice received 2.5×105 live B16-F10 cells IP on day 0, followed by the treatment with SEG/SEI, 50 µg of each, on days 6 and 9. Controls were inoculated with tumor but received no treatment. On day 13, tumors were excised and assayed for (A) CD3 +CD8+cells, (B) CD8+granzyme B+ cells, (C) CD25+FoxP3+ cells, (D) CD11b+Gr1+ cells. ANOVA with Tukey post test. n=3. *P<0.05, **p<0.01,. Ratio of CD8+granzme B+ cells to CD25+Foxp3+ cells ≥10:1 (n=4, p=0.01) and ratio CD8+ granzyme B+ cells to CD11b+GR1+cells=3.9:1 (n=4, p=0.03). Data expressed as mean±SD, Student’s t test, two tailed. ANOVA, analysis of variance; ns, not significant.
SEG/SEI-treated HLA-DQ8 tg mice display increased Th-1 cytokines in serum and TME
Having shown that SEG/SEI activated IFNγ in vitro, we now examined the effect of SEG/SEI treatment on production of IFNγ and other cytokines in vivo in both serum and the TME. HLA-DQ8 tg mice were inoculated with a tumorigenic dose of B16-F10 cells on day 0 and subsequently injected with SEG and SEI, 50 ug of each, on days 6 and 9. Serum levels of TH-1 and TH-2 cytokine levels were evaluated 24 hours after each treatment on days 7 and 10. On day 7, HLA-DQ8 tg mice exhibited a striking surge of IFNγ with minimal changes in TNFα levels (figure 4A,B). On day 10, however, following the second SEG/SEI treatment, the IFNγ response was attenuated along with minimal levels of TNFα and interleukin-6 (IL-6) (figure 4A–C). By contrast TH-2 cytokines IL-4, IL-10 and IL-13 were not significantly elevated in HLA-DQ8 tg mice after either SEG/SEI treatment (figure 4E–F). Notably, the sharp increase in IFNγ was unattended by any evident toxicity in HLA-DQ8 tg mice. The minimal toxicity was likely due to the muted levels of TNFα following both SEG/SEI treatments . TNFα has been shown to be causative in toxic shock in response to canonical SAgs, SpeA and SEB in HLA-DQ8 mice at doses significantly lower than d SEG/SEI as used here.30 40–43 SEG/SEI in HLA-DQ8 mice therefore displayed a remarkable ability to induce potent IFNγ and T effector cell responses while sharply curtailing TNFα production and preventing the appearance of toxic shock.

Evaluation of cytokines and chemokines in serum and tumor from HLA-DQ8 tg mice treated with SEG/SEI is shown. (A–H) HLA-DQ8 tg and C57BL/6 mice received 2.5×105 B16-F10 cells ip and were treated with SEG/SEI, 50 µg of each, ip on days 6 and 9. Controls were inoculated with r tumor but received no SEG/SEI treatment. Serum cytokines were analyzed 24 hours after each SEG/SEI treatment, n=4–5. (I, J) HLA-DQ8 tg mice received 2.5×105 live B16-F10 cells ip on day 0, followed by the treatment with SEG/SEI, 50 µg of each, on days 6 and 9. Controls were inoculated with B16-F10 cells Iip but received no treatment. On days 13 and 16 tumors were assayed for (I) cytokines and (J) chemokines, respectively. Data expressed as mean±SD. ANOVA with Tukey post-test. n=3. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance.
Next, we analyzed B16-F10 tumors obtained from SEG/SEI-treated HLA-DQ8 tg mice for the presence of cytokines on day 13 following tumor inoculation. HLA-DQ8 tg mice received a tumorigenic dose B16-F10 melanoma cells on day 0 and were treated with SEG/SEI, 50 µg of each, on days 6 and 9. Controls were inoculated with tumor but received no treatment. SEG/SEI-treated HLA-DQ8 tg mice showed striking increases in IFNγ, TNFα and IL-6 in the TME relative to untreated controls whereas TH-2 cytokines IL-4 and IL-10 were not significantly altered (figure 4I). The tumoricidal function of IFNγ coupled with its ability to upregulate MHCII expression on melanoma cells and prime cytolytic CD8 +T cells likely contributed significantly to the acute anti-tumor response.44 TNFα. also noted in the TME. has been shown to synergize with IFNγ in tumor cytolysis.45 The presence of IFNγ and TNFα s in the TME, not seen in untreated controls, suggests that these are the major cytokine mediators of to the acute tumoricidal effect in SEG/SEI-treated HLA-DQ8 tg mice.
SEG/SEI-treated HLA-DQ8 tg mice display increased chemokines in the TME
A hypothetical barrier to successful immunotherapy is inadequate recruitment of activated T cells and T effector cells into the tumor sites. An optimal chemokine profile in the melanoma TME may be critical for recruitment of activated T cells. While several chemokiness are known to be activated by SAgs,44 ,46 the ability of SAg to induce chemokine accumulation in tumors has not been demonstrated. To evaluate intratumoral chemokines, HLA-DQ8 tg mice were inoculated ip with a tumorigenic dose of B16-F10 melanoma and treated with SEG/SEI, 50 µg of each, on days 6 and 9. Controls received tumor cells but no treatment. Omental tumors were removed on day 16 and evaluated for chemokines. SEG/SEI-treated mice showed significant increases in CCL2,CCL3, CCL4, CCL5, CXCL9, CXCL10 chemokines relative to the untreated controls (figure 4J). Interestingly, Harlin et al, identified the very same chemokine expression profile in treatment-responsive metastatic melanomas and further showed that each of them was efficient in chemotaxis of activated T cells.47 Each of these chemokines displayed a significantly higher level than CXCL5 and CXCL1 and CCL22 chemokines that have been shown to recruit PMN/MDSCs, PMNs or Tregs, respectively (figure 4J).48–50 Collectively, these findings suggest that the intratumoral chemokine constellation identified here after SEG/SEI treatment contributed to the recruitment T effector cells in the TME.
SEG/SEI-treated HLA-DQ8 tg mice recruit T effector cells to the site of tumor injection
Untreated tumors generally contain only small numbers of T effector cells tumor site. While SAgs have been shown to recruit activated T cells to their injection site,51 they have not been shown to recruit T cell to tumors, We, therefore, determined whether SAg injected into the same peritoneal site as tumor inoculation could induce robust trafficking of T cells to established peritoneal tumors. Effector T cells once situated in the peritoneum are known to traffic rapidly to tumor niches in the omentum.52 To test this concept, HLA-DQ8 tg mice were inoculated ip with a tumorigenic dose of B16-F10 melanoma cells and treated with SEG/SEI ip on days 6 and 9. Twenty-four hours after each treatment on days 7 and 10, tumors were removed and both peritoneal lavage and peripheral blood were assessed for the presence of T effector cells. Controls were inoculated with B16-F10 cells on day 0 but received no treatment. On days 7 and 10, SEG/SEI treated mice showed striking absolute and incremental increases in the number of activated (CD44+CD62L-) and CD8 +effector (granzyme-perforin+) T cells in both peripheral blood and peritoneal lavage compared with untreated controls (figure 5A–F). Analysis of omental tumor in SEG/SEI treated mice on day 10 showed a significantly greater degree of tumor necrosis relative to the untreated control encompassing a mean of 70% of the tumor area (figure 5G,K). These findings indicate that SEG/SEI recruited T effector cells to the site of tumor injection in the peritoneal cavity wherein they traffic to tumor niches in the omentum and likely constitute the major T cell population mediating the tumoricidal response. The SEG/SEI-mediated recruitment of T effector cells to the tumor site thus appears to convert the tumor from ‘cold’ to ‘hot’. The concurrent increase of effector T cells in both the peripheral blood and peritoneum after ip injection of SEG/SEI suggests that the peritoneal effector T cell population may have originated, in part, from an SEG/SEI-activated effector T cell population in the peripheral blood.

T cell recruitment to tumor injection site after administration of SEG/SEI to HLA-DQ8 TG mice is shown. HLA-DQ8 TG mice received 2.5×105 B16-F10 cells ip and were treated with SEG/SEI, 50 µg of each, IP on days 6 and 9. Controls were inoculated with tumor but received no treatment. Twenty-four hours after each SEG/SEI treatment peritoneal lavage and peripheral blood were assayed for (A, B) CD4 +and CD8+T cells, (C, D) perforin-granzyme B+T effector cells, and (E, F) CD44 +CD62L- cells. (G–J) Histopathology of omental tumor sections on day 10 from untreated and SEG/SEI-treated mice is shown. (H,E) ×10 magnification (G, I); x40 magnification (I, J). Quantification of histological necrosis in tumor sections from untreated and SEG/SEI-treated mice on days 7 and 10 is shown (K). n=3–4. Data expressed as mean±SD. *P<0.05, **p<0.01, ***p<0.001, ****p<0.001.
Vaccination with irradiated melanoma cells followed by SEG/SEI affords protection of HLA-DQ8 tg mice from living melanoma cell challenge
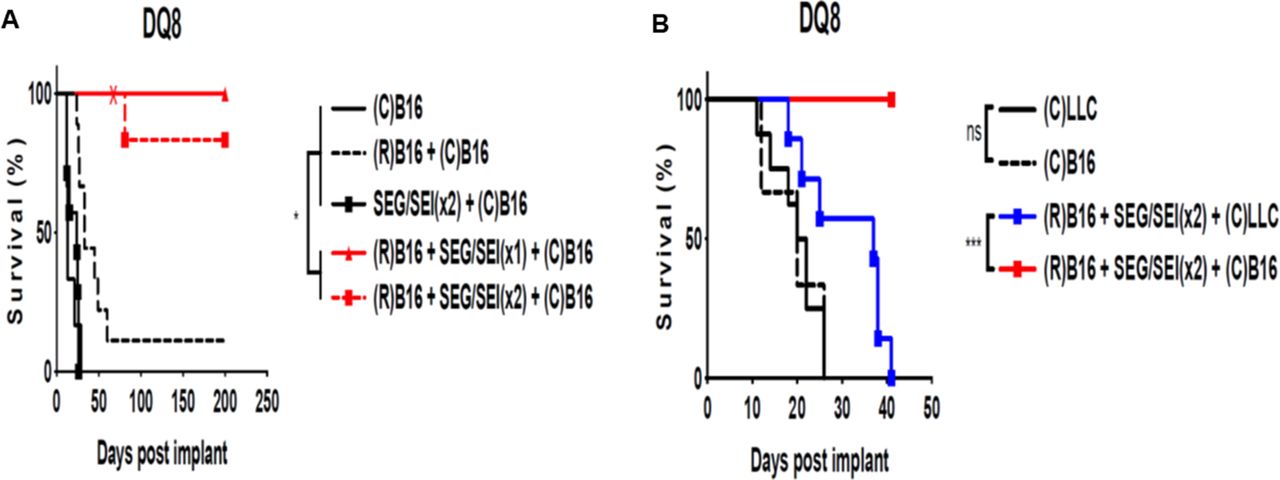
We next determined whether SEG/SEI could enhance an antitumor immune response to vaccination with radiation-inactivated B16-F10 melanoma cell and confer protection from live B16-F10 challenge in HLA-DQ8 tg mice. Tumor cells exposed to ionizing radiation often carry new mutations in genes encoding proteins involved in DNA-repair mechanisms and cell-cycle regulation some of which are likely to be immunogenic.53 To test this notion, HLA-DQ8 mice were vaccinated with 1x 106 irradiated B16-F10 melanoma cells on day −13, treated with SEG and SEI, 50 ug or each, ip on days −7 and day −3 and challenged with 2.5×105 viable B16-F10 cells on day 0. Control mice received viable B16-F10 cells alone, irradiated tumor cell vaccination on day -13 plus viable B16-F10 cells on day 0 0r SEG/SEI on days -7 and -3 plus viable B16-F10 cells on day 0. HLA-DQ8 tg mice receiving irradiated tumor cell vaccination followed by SEG/SEI survived significantly longer after live melanoma challenge than all controls (figure 6A). We next determined whether the antitumor protection induced by irradiated melanoma cell vaccination combined with SEG/SEI treatment could protect surviving mice against a challenge with the parental tumor. All HLA-DQ8 tg mice that survived for more than 60 days after tumor inoculation were rechallenged with a tumorigenic dose of melanoma cells and remained tumor-free for the succeeding 140 days (figure 6A). These results show that vaccination followed by SEG/SEI administration elicits anti-melanoma protection against primary and secondary B16-F10 melanoma challenge. Next, we examined the anti-tumor specificity of the response induced by vaccination with irradiated B16-F10 cells and SEG/SEI treatment. HLA-DQ8 tg mice were vaccinated with 1×106 irradiated B16-F10 cells on day −13, treated with SEG and SEI, 50 µg of each, on days −7 and −3 and challenged with 2.5×105 viable B16-F10 or LLC cells on day 0. All mice pretreated with irradiated B16-F10 cells plus SEG/SEI and challenged with viable B16-F10 cells demonstrated survival to day 40 whereas none of the mice similarly vaccinated and treated with SEG/SEI but challenged with LLC cells survived beyond 40 days (figure 6B). Hence, the protection induced by the vaccination regimen of irradiated tumor cells plus SEG/SEI was tumor specific. To better understand the immune basis of the recall response of vaccinated mice to tumor challenge HLA-DQ8 tg mice were vaccinated with 1×106 irradiated B16-F10 cells on day −13, treated with SEG/SEI, 50 µg of each. ip of each, on days −7 and −3 and challenged with 2.5×105 live B16-F10 cell on day 0. On day 40, they were rechallenged 2.5×105 live B16-F10 cells and 6 days later peritoneal lavage and omental tumors were examined. HLA-DQ8 mice tinoculated with B16-F10 tumor cells on day 0 and sacrificed 6 days later served as untreated controls. Evaluation of peritoneal lavage cells from vaccinated and tumor rechallenged mice showed significant elevations of activated (CD44+CD62L-) and memory (CD127+) T cells relative to the untreated controls (figure 7A–D).54 Total omental tumor density in vaccinated mice was 11-fold less than the untreated controls (figure 7E). Histologicanalysis of all omental tumor deposits in vaccinated and untreated control mice showed striking differences (figure 7F–H). Whereas control tumor cells were situated in discrete tumor nodules with well developed stromal matrix (figure 7F,G), tumor cells in the vaccinated mice were positioned amid large aggregates of mononuclear cells devoid of organized tumor matrix or distinct tumor nodules (figure 7H,I). These mononuclear cells aggregates likely constitute omental milky spots infiltrated by tumor cells. Vaccination, therefore, reduced the density of omental tumor cells and constrained the formation of organized tumor nodules relative to untreated controls. Collectively, these data indicate that vaccination with radiation-inactivated B16-F10 cells plus SEG/SEI affords tumor specific,and long-term anti-tumor protection against B16-F10 challenge. This protection appeared to be mediatedby f effector and memory T cell populations.
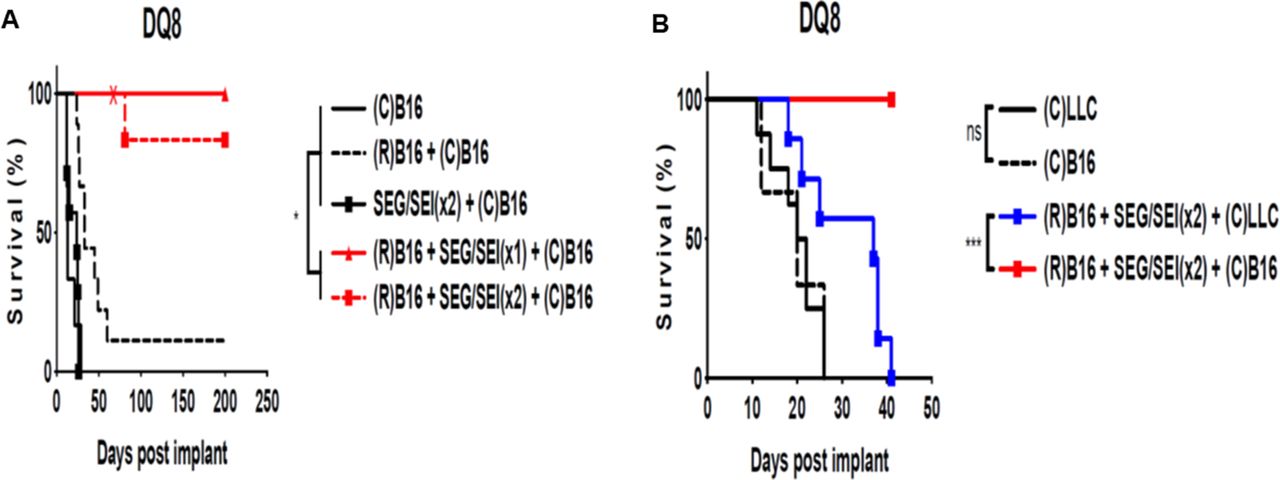
Vaccination with inactivated B16-F10 melanoma plus SEG/SEI in HLA-DQ8 tg mice induces long term survival. (A) HLA-DQ8 tg mice were treated with 1×106 irradiated (15 000 rads) B16-F10 melanoma cells ip on day −13 injected with SEG/SEI 50 µg of each ip on days −7 and −3 and challenged with 2.5×105 live B16-F10 cells IP day 0. Controls received live melanoma challenge alone on day 0 or irradiated B16-F10 cells on day −13 plus 2.5×105 live melanoma challenge on day 0 or SEG/SEI on days −7 and −3 plus 2.5×105 live melanoma cells.on day 0. HLA-DQ8 tg mice surviving >60 days (‘X’) were rechallenged with 2.5×105 live B16-F10 cells ip (X). (B) HLA-DQ8 Ttg mice were inoculated with 1×106 irradiated (15 000 rads) B16-F10 melanoma cells ip on day −13, injected with SEG/SEI, 50 µg of each, ip on days −7 and −3 and challenged with 2.5×105 live B16-F10 cells or Lewis lung carcinoma cells on day 0. Controls received 2.5×105 live B16-F10 cells or Lewis lung carcinoma cells alone on day 0. Kaplan-Meier curves with log-rank test, n=10. *P<0.05, ***p<0.001. Acronym ‘R’ denotes irradiated tumor cell vaccination on day −13 and ‘C’ indicates challenge with either B16-F10 melanoma or Lewis lung carcinoma tumor cells on day 0. ns, not significant.

Recall response of vaccinated HLA-DQ8 mice to live melanoma challenge is shown. HLA-DQ8 tg mice were vaccinated with 1×106 irradiated B16-F10 cells on day −13, treated with SEG/SEI, 50 µg of each, on days −7 and −3 and challenged with 2.5×105 live B16-F10 cells on day 0. On day 40, they were rechallenged 2.5×105 live B16F10 cells and 6 days later (day 46) omental tumor and peritoneal lavage cells were examined. Untreated HLA-DQ8 tg miceinoculated with B16-F10 tumor cells on day 0 and sacrificed 6 days later served as controls. Analysis of peritoneal lavage cells from untreated and vaccinated mice for the presence of CD127+, and CD44+CD62L- T cells is shown (A–D). Quantitation of omental tumor density in sections from vaccinated and untreated mice using the ImageJ software is shown (E). Sections of omental tumors from untreated (F, G) and vaccinated mice (H,I) are shown. (H,E) × 5 magnification (F, H) and X40 magnification (G, I). Data expressed as mean±SD; . n-3-4, **p<0.01, ***p<0.001.
SEG/SEI toxicity in HLA-DQ8 tg mice
SEG/SEI treatment in HLA-DQ8 tg mice was well tolerated. There were no acute deaths and mice showed no signs of inanition, anorexia or disability during or after treatment. Arthritis susceptible HLA-DQ8 tg mice55 treated with SEG/SEI also exhibited no clinical signs of arthritic joint swelling, ambulatory impairment, polydipsia or weight loss during 360 days of observation.
Discussion
In a discovery-driven effort to unearth new anticancer drugs, we introduce a heretofore unrecognized binary complex comprizing non-canonical SEG/SEI SAgs together with endogenous MHCII HLA-DQ8 allele in humanized mice. This pairing displayed striking tumoricidal strength and long-term melanoma survival (>360 days) entirely devoid of toxicity in both vaccinated mice and mice with established melanoma. SEG/SEI were chosen over canonical SAgs because they are devoid of sero-reactive neutralizing antibodies that have nullified the efficacy of canonical SAgs in human cancer trials. The HLA-DQ8 allele was selected for its large antigen groove and ability to functionally engage both SAgs and melanoma neoantigens. In humanized mice, the SEG/SEI (HLA-DQ8) complex orchestrated an acute tumoricidal response marked by robust T effector cell and IFNγ generation while asymmetrically silencing TNFα. This constrained TNFα response was surprizing since canonical SAgs in this very same model induce TNFα-mediated toxic shock. In addition, the complex unleashed a broad tumoricidal network featuring chemotactic mobilization and recruitment of CD8 +effector T cells to the TME while quelling the appearance of suppressive Tregs and myeloid cells. The anttumor response culminated in specific antitumor memory whereby long-term melanoma survivors rejected the parental melanoma but succumbed to challenge with the LLC. The novel revelation that SEG/SEI and HLA-DQ8 humanized model can unleash a striking tumoricidal response and tumoricidal network devoid of toxicity is slated to reinvigorate the field and forge a pathway to clinical translation.
We bring forth a broad range of evidence in support of the contention that the key mediators of the antitumor response induced by SEG/SEI in HLA-DQ8 mice are granzyme-perforin producing CD8 +T cells and IFNγ as follows: (1) Deletion experiments showed that the anti-tumor response induced by SEG/SEI in HLA-DQ8 mice was CD4 +and CD8+T cell dependent (figure 2G). (2) Analysis of T cell phenotypes in tumor lysates from the TME of SEG/SEG treated mice at day 10 (figure 5C,D and G,H) and day 13 (figures 2B,E,F and 3B) showed significant infiltration by CD8 +T granzyme-perforin +T effector cells and IFN γ . This was associated with extensive tumor necrosis indicative of a major antitumor pathological effect. These changes were not seen in the untreated controls. (3) SEG/SEI produced a sharp surge of serum levels of IFNγ in SEG/SEI-treated mice not seen in the untreated controls (figure 4A). IFNγ was also identified as the predominant cytokine in lysates of regressing tumors at day 16 (figure 4I). (4) In vitro studies confirmed the ability of SEG/SEI to mount a robust T effector cell and IFNγ responses in naïve HLA-DQ8 T cells (figure 1C–F). (5) The SEG/SEI-mediated anti-tumor response was specific for melanoma since these very same mice succumbed to challenge with the LLC (figure 2B,C,D). (6) In HLA-DQ8 mice vaccinated with irradiated melanoma cells and rechallenged with live melanoma cells, both T effector cells and T memory cells were identified in the TME but not seen in the untreated controls (figure 7A–D).
Moreover, the tumoricidal response in SEG/SEI-treated HLA-DQ8 mice cannot be attributed to cytokine storm since these mice exhibited no evident toxicity or toxic shock that typifies such storm. Remarkably, elevated TNFα levels which are the hallmark of ‘toxic shock’ were subdued in SEG/SEI-treated mice (figure 4B). Furthermore, the specificity of antitumor effect after melanoma rechallenge in SEG/SEI-treated mice supports our contention that a non-specific ‘cytokine storm’ is not the basis of the tumoricidal response. If cytokine storm were the cause of the anti-tumor response then long-term melanoma survivors would have been expected to resist challenge with both live melanoma and LLC cells. That only the long-term melanoma survivors rejected the live tumor challenge strongly suggests that specific T effector memory cells were involved in the anti-tumor effect. Indeed, such memory T cells were identified in the TME of mice after rechallenge with live melanoma.
The muted toxicity of SEG/SEI mice in the presence of the potent antitumor effect appeared to be based on the constrained TNFα response in HLA-DQ8 mice (figure 4B). This response was reliant on SEG/SEI since other SAgs deployed in HLA-DQ8 mice at lower doses induced TNFα-mediated toxicity.30 41–43 Diverse cytokine reactions and toxicity in humanized MHCII transgenic mice have been noted in response to SAgs and ascribed to their divergent affinities and binding conformations with human MHCII molecules.14 56 57 Such differential cytokine responses also appear to be dependent on interactions of conserved SAg motifs with the B7-CD28 axis.13 14 Indeed, cytokine-based toxicity of SAgs was abrogated in humanized MHCII mice deficient in CD28 or by treatment with anti-CTLA-Fc antibody.42 58 Taken together, these findings indicate that SEG/SEI works uniquely with HLA-DQ8 allele and possibly the B7-CD28 axis to preserve melanoma specific anti-tumor T cell response while silencing cytokine-based toxicity.
Our deletion experiments showing that CD4 +in addition to CD8 +T cell are required for the SEG/SEI-mediated induced antitumor response supports key role of the HLA-DQ8 allele in the antitumor response. (figure 2G). CD8 +T effector cells recruited to tumor sites and found together with IFNγ in the TME appear to the major effectors of the antitumor response.MHCII molecules, however, have also been shown to play an important role in processing tumor neoantigens, activating CD4 +T cells, priming tumor specific CD8 +T effector cells and collaborating with them in an effective antitumor response 59 60 Recent reports suggest a role of HLA-DQ8 in binding melanoma neoantigens and presenting them to CD4 +cells. 34 ,35 36 The notion that these tumor neoantigens are involved in the SEG/SEI-mediated anti-tumor response 61 ,62 is supported by tumor specific memory exhibited by long-term melanoma survivors that resisted autologous tumor rechallenge for more than 160 days while succumbing to the LLC (figures 2B and 6A). Indeed, B16-F10 melanoma-derived neopeptides with high affinity for MHCII have conferred protection against established melanoma when delivered on cationic liposomes.36 We surmise that shortly after tumor inoculation, HLA-DQ8 processed melanoma neopeptides and primed CD4 +T cells. Subsequent SEG/SEI administration expanded these antigen-primed T-cells in accord with SAg’s ability to initiate T cell recall responses.10–13 HLA-DQ8 allele is thereby repurposed in a hitherto unappreciated therapeutic role of priming a population of melanoma specific T cellsand serving as a scaffold for an SEG/SEI-mediated anamnestic T-cell-mediated anti-tumor response
SEG/SEI also generated T cell chemotactic molecules (figure 4J) that enabled recruitment of CD8 +T effector cells to the site of tumor injection and the TME (figure 3A,B). Such effector cell recruitment and consequent tumor cell necrosis indicates that SEG/SEI can convert the tumor from ‘cold’ to ‘hot’ (figure 5A–J). SEG/SEI further stimulated a surge of IFNγ output from both CD4 +and CD8+T cells (figures 1C, D and 4A). By days 13–16 after tumor implant, the TME was infiltrated by CD8 +T effector cells along with IFNγ and TNFα that appear to constitute the major tumoricidal effectors (figures 3A, B and 4I). The acute tumoricidal response (figure 5G,H) was therefore likely mediated by clones of primed melanoma-specific effector T-cells while a memory cell population derived from this lineage provided tumor specific protection against late melanoma challenge at day 160 (figure 2C). The latter feature constitutes a critical distinction from T cell agonist anti-PD-1 whose long-term T-cell memory response is epigenetically constrained.63
IFNγ produced by CD4 +and CD8+T cells in response to SEG/SEI emerges in a major role as a tumoricidal effector of its own while also priming tumor reactive CD8 +T cells.44 Following SEG/SEI injection on day 6 SEG/SEI-treated mice displayed robust IFNγ production (figure 4A). This likely upregulated MHCII expression on melanoma cells enabling them to bind SEG/SEI and activate CD8 +T effector cells.45 64 The same IFNγ surge was likely responsible for constraining Treg differentiation to account for the minimal Treg presence noted in the TME after SEG/SEI treatment (figures 1G and 3D).65 Such Treg reduction in the TME would also likely support the CD8 +effector T cell response. Melanoma specimens from subjects with improved prognosis exhibited an assemblage of chemokines that were strikingly similar to the chemokine profile we detected in tumors after SEG/SEI treatment47 (figure 4J). Several of these have been shown to be IFNγ and SAg inducible and are known to recruit effector T cells akin to those we noted in the TME of SEG/SEI-treated mice (figure 5A–F).
Despite the subdued serum TNFα response sufficient to silence toxic shock (figure 4B), SEG/SEI was still able to induce ample TNFα to contribute to the tumoricidal response. Indeed, we identified TNFα together with IFNγ in the TME of SEG/SEI-treated HLA-DQ8 tg mice (figure 4I). TNFα is reported to synergize in SAg-mediated tumoricidal responses and has been implicated in a self-sustaining feed-forward inflammatory process.45 66 The unique persistence in time of high affinity, zinc-binding SAgs such as SEI to MHCII on the surface of antigen-presenting cells may also enable a self-perpetuating cycle that generates T effector cells, IFNγ and T cell memory.67 Collectively, the combined presence of IFNγ, TNFα and T effector cells in the TME strongly supports their role as key mediators of a dynamic tumoricidal process that culminates in long-term memory and challenge-resistant melanoma survival. A schematic representation of the proposed chain of events is shown in figure 8.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Antitumor effect and network induced by SEG/SEI in HLA-DQ8 tg mice resulting in long-term antitumor memory and melanoma survival is shown. Following melanoma inoculation on day 0, HLA-DQ8 expressing antigen presenting cells (APCs) recognize melanoma neoantigens and present them to T cells resulting in priming of tumor reactive CD4+ and CD8+T cells. This priming alone is insufficient to induce an anti-tumor response. Introduction of SEG/SEI on days 6 and 9, however, results in expansion of both CD4+ and CD8+T cells. Following SEG/SEI treatment the primed CD4 +T cell population likely d promotes CD8 +T cell differentiation into T effector cells. The superantigenic T cell activation also generates T cell chemotactic molecules that enables recruitment of T effector cells to the tumor. SEG/SEI further initiates a surge of IFNγ from both CD4 +and CD8+T cells. By days 13–16, the TME is infiltrated by CD8 +T effector cells along with IFNγ and TNFα that appear to constitute the major tumoricidal effectors. CD8 +T effector cell tumor killing is likely enhanced by the relative paucity of Tregs and myeloid cells in the TME. Long-term surviving SEG/SEI-treated mice exhibit specific anti-tumor memory by resisting late challenge with melanoma cells while succumbing to inoculation with Lewis lung carcinoma. The long-term antitumor response suggests a self-sustaining feed forward mechanism possibly mediated by persistence of SEI in vivo in a high affinity, Zn++ dependent interaction with the HLA-DQ8 allele. IFNγ, interferon-γ; IL-6, interleukin 6; TNFα, tumor necrosis factor-α.
Investigators have long recognized that SAg behavior in humanized HLA-DQ8 mice is highly predictive of toxic shock in humans.41–43 Hence, the potent antitumor effects of SEG/SEI without toxicity in humanized HLA-DQ8 mice portend their successful clinical translation. In support of this concept, SEG and SEI as part of a staphylococcal filtrate were administered parenterally during treatment of advanced lung cancer and exhibited no significant constitutional or hemodynamic toxicity.68 69 In clinical translation, it may be desirable to shelter SEG/SEI from engaging other MHCII alleles in vivo. This may be accomplished ex vivo by linking SEG-SEI to HLA-DQ8 molecules and covalently coupling the complex to the surface of nanoparticles. Such SEG/SEI-HLA-DQ8 loaded nanoparticles may then be administered to the host. Strong binding affinities of SEG and SEI to MHCII should minimize their dissociation in plasma and absence of neutralizing antibodies against SEG/SEI in human sera should enable them to target t host TCRs.24 25 Highly developed cationic liposomes, T cell targeted and MHCII derivatized nanoparticles loaded with biologics or tumor neopeptides have already been shown to activate T cells in vivo and initiate therapeutic responses in autoimmune disease or melanoma bearing mice.37 70 71 Likewise, dendritic cells and macrophages loaded with small amounts of SAgs have been shown to initiate robust T cell responses in wild type mice.72 73 Using similar principles, the long-lived tumor specific protection exhibited by SEG/SEI in our vaccination studies has clear application to susceptible human populations.
Collectively, these findings unveil a hitherto unrecognized SEG/SEI-(HLA-DQ8) complex that activates, directs, and drives a tumoricidal network culminating in tumor specific memory and long-term tumor regression devoid of TNFα-mediated toxicity. SEG/SEI are also devoid of neutralizing antibodies in humans thereby surmounting an additional barrier to clinical application. These properties distinguish SEG/SEI from other SAgs used previously in human tumor immunotherapy. More broadly, consolidation of these powerful principles within the SEG/SEI-(HLA-DQ8) complex constitutes a conceptually new anti-tumor weapon with compelling translational potential.
Methods
Mice
All research performed, including animal and tissue collection, was conducted in accordance with the Animal Welfare Act and with the approval of the University of North Dakota’s Institutional Animal Care and Usage Committee g. Female HLA-DQ8 (DQA*0301/DQB*0302) tg breeding pairs were a gift from Dr. Chella David (Mayo Clinic, Rochester, Minnesota, USA) and their generation was described previously.38 The HLA-DQ8 phenotype was expressed on high proportion of peripheral blood lymphocytes (PBL), spleen and lymph node cells.29 Breeding pairs of C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, Maine, USA). Mice were bred and maintained in specific pathogen-free conditions within the Center for Biological Research at the University of North Dakota.
Cells
B16-F10 murine melanoma cells, LLC and 4T1 mammary carcinoma were obtained from American Type Culture Collection. All cells were maintained in complete Dulbecco’s Modified Eagle’s Medium (cDMEM) containing 10% heat inactivated fetal bovine serum (FBS) (Atlanta Biologicals) and 50 IU/mL Penicillin/Streptomycin (MP Biologicals) in T-75 flasks (CytoOne). Single cells were isolated from spleens and lymph nodes by passing organs through a 70 µm strainer (Falcon) with a 5 mL syringe plunger. All cells were washed with HBSS, RBCs were lysed with ACK lysis buffer for 5 min at 37°C washed again with cDMEM and filtered through a 70 µm strainer.
Superantigens
SEG and SEI, were produced in Escherichia coli M15 as His-tagged recombinant toxins and purified by affinity chromatography on a nickel affinity column as previously described.74 Protein purity was verified by sodium dodecyl sulfate-polyacrylamide gel electropheresis (SDS-PAGE) and TCR vBeta profiles established for each SE.74 75 LPS was removed from toxin solutions by affinity chromatography (Detoxi-GEL endotoxin Gel, Pierce Rockford, USA). The QCL-1000 Limulus amebocyte lysate assay (CambrexBioWhittaker, Walkersville, USA) showed that the endotoxin content of the recombinant SAg solutions was less than 0.005 units/mL. SEA and SEB were obtained from Toxin Technology (Sarasota, Florida, USA). All reagents were kept at either 4°C or −20°C and subject to no more than two freeze-thaw cycles.
Flow cytometry
Cells were washed with Hanks balanced salt solution (HBSS), stained with Ghost Dye (TONBO) for viability, FC blocked (BioLegend) and stained for extracellular antigens. Cells were fixed and permeabilized using FoxP3 staining buffer kit (TONBO) for intracellular cytokine and transcription factor analysis. Fluorescence minus one (FMO) and single stained controls were used for gating and compensation. Gating strategies are indicated within each experiment. In general, doublets and cell debris were excluded, and only Ghost Dye negative cells were used for analysis. Samples were analyzed using a BD LSRII or Symphony A3 flow cytometer at the North Dakota Flow Cytometry and Cell Sorting Core. Data were analyzed using FlowJo software.
Assessment of T cells
Splenocytes were isolated by passing mouse spleens through a 70 µm nylon strainer (Falcon) using the plunger from a 5 mL syringe. Cells were washed with HBSS (Gibco), lysed for 5 min in ACK lysis buffer and subsequently washed and resuspended in DMEM (Gibco) containing 10% heat inactivated FBS (Atlanta Biologicals) and Pen/Strep (1000 IU) (cDMEM) for downstream analysis. T cell proliferation was evaluated using Cell Proliferation Dye eFluor 450 (eBiosciences) dye. Briefly, splenocytes were stained with proliferation dye after which, 2×105 cells/well were seeded in 96-well round-bottom tissue culture plates (Becton Dickinson) in cDMEM and stimulated with SAgs for 72 hours (37°C, 5% CO2 and humidity) in 200 µL volume. Concanavlin A, (Sigma) 1 µg/mL was used as a conrol. After 3 days, cells were processed for flow cytometry by using FC Block (BioLegend) and stained with 1:1000 dilution of TONBO ghost dye for 15 min. Cells were then washed with HBSS containing 2% FBS and stained for surface expression with 1:200 dilution of antibodies against CD3(17A2), CD4(RM4-5) CD8(53–6.7) and anti-CD127 (SB/199) (BioLegend) for 1 hour at 37°C. For detection of intracellular perforin (S16009A), granzyme B(QA16A02) and IFNγ(XMG1.2) (BioLegend) and FoxP3 (MF23) (TONBO), cells were fixed and permeabilized using Foxp3 staining buffer kit (TONBO) then stained for 1 hour at 37°C. FMO and single stained controls were used for gating and compensation.
Assessment of tumor cells
Tumors were isolated, rinsed with HBSS, minced with scissors and placed in 3 mL digestion solution containing 100 mg/mL type IV collagenase (Sigma) and 2000 IU/mL type IV DNase (Sigma) in DMEM (Gibco) for 1 hour (37°C, 5% CO2 and humidity) on a plate rocker. Tumor digests were passed through a 70 µm nylon strainer (Falcon) and cells were washed with HBSS (Gibco) and RBCs lysed for 5 min in ACK lysis buffer. Tumor cells were blocked using FC Block (BioLegend) and stained with 1:1000 dilution of TONBO ghost dye for 15 min. Cells were then washed with HBSS containing 2% FBS and stained with 1:200 dilution of antibodies against CD45(30-F11), CD3(17A2), CD4(RM4-5), CD8(53–6.7), CD44(IM7), CD62L(MEL-14), CD25(PC61), CD11b(M1/70), Gr-1(RB6-8C5) (TONBO and BioLegend) for 1 hour at 37°C. Cells were fixed and permeabilized using Foxp3 staining buffer kit (TONBO) then stained for 1 hour at 37°C for granzyme B(QA16A02), IFNγ (XMG1.2) (BioLegend), FoxP3 (MF23) (TONBO). FMO and single stained controls were used for gating and compensation. Gating strategies are indicated within each experiment.
Assays of cytokines and chemokines
For measurement of cytokines in blood, samples were collected from the submandibular vein mice into EDTA tubes. Plasma cytokine concentrations were processed via flow cytometry and measured using LEGENDplex kit (BioLegend). For measurement of cytokines and chemokines in tumor tissue, tumors were weighed and lysed for 5 min using 2 mm stainless steel beads (Next Advance) and a Bullet Blender (Next Advance) in 300 µL HBSS. Lysates were centrifuged for 5 min at 5000 x g. Supernatants were subjected to one freeze thaw cycle and cytokines and chemokines were measured using a TH cytokine cytometric bead array (Biolegend). Results are reported as (pg/mL)/mg of tumor tissue.
Mixed lymphocyte assays
Mitomycin C-treated C57BL/6 splenocytes (red) were cocultured with splenocytes from HLA-DQ8 tg mice for 3 days at 37°C in 5% CO2. CD3 +T cell proliferation (green) was measured by dye dilution via flow cytometry and reported as per cent proliferation. SEB was used as a positive control for T cell proliferation
In vivo tumor experiments
In vivo tumor experiments were carried out using an established model of peritoneal and omental tumor metastases for evaluation of intracavitary therapeutics.76 In the vaccination model, mice were injected with 1×106 irradiated (15 000 rads) B16-F10 melanoma cells intraperitoneally (ip) in 100 µL HBSS (Life Technologies) on day −13. On day −7 and day −3, mice received 100 µL injections ip of SEG and SEI (50 µg each). Mice were challenged on day 0 with 2.5×105 B16-F10 cells ip, evaluated daily and sacrificed when moribund. In the established tumor model, 2.5×105 B16-F10 melanoma cells were injected ip in 100 µL PBS on day 0. On days 6 and 9 post tumor inoculation, mice were injected with SEG and SEI (50 µg each) ip in 100 µL of PBS. The same protocol was used to test the relative roles of T cell subsets in the antitumor response.,Individual groups of HLA-DQ8 tg mice were injected ip on days 2,4,7,9,12 with rat anti- CD4+ (200 µg), rat anti-CD8+ (500 µg) or isotype matched rat IgG (300 µg) (BioXCell) in 100 µL PBS. Mice were evaluated daily and sacrificed when moribund.
Radiation
Tumor cells were irradiated at 15 000 rads using Gammacell 3000 (MDS Nordion, Canada).
Histology and quantification of tumor density and necrosis
Tumors were fixed with formalin and embedded in paraffin. Serial sections 5 µm thick were cut floated onto charged glass slides (Super-Frost Plus, Thermo Fisher Scientific) and dried overnight at 60°C. Sections were stained with H&E. Slides were scanned at 1200 dpi and photographed as a TIFF image. The necrotic and viable areas of these sections were identified histologically with a Leitz Diaplan microscope and corroborated using ImageJ software. Necrotic tumor area was expressed as the percentage of pixels in the total tumor area minus the area of the viable tumor cells in the section.77 For determination of tumor density, all visible omental tumors were excised, and sections were stained, scanned, and photographed as TIFF images as described above. Tumor bearing areas were confirmed histologically and quantitated using the ImageJ software. Tumor density was expressed as total number of pixels in tumor bearing regions in each tumor section. Statistical analysis was based on a mean of 4 tumors per group.
Data analysis
One-way analysis of variance with Bonferroni’s posttest and student’s t test were performed where indicated. Kaplan-Meier curves and Mantel-Cox test were used to evaluate survival data. Statistical analysis was performed using GraphPad Prism software V.7.0a (La Jolla, California, USA).
Supplemental material
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
DT and DB contributed equally.
Contributors Conceived of study: DT, DB; Designed the experiments: DT, DB, PK; Performed the investigations: PK, NR, TA, RL, DT; Analyzed the data: DT, DB, PK; Provided key materials: GL, CB, JB; Wrote the paper: DT; Edited the paper: DT, DB, PK.
Funding Supported by Center for Biomedical Research (NIH P20GM113123), the Dakota Cancer Collaborative on Translational Activity (DaCCoTA), CTR grant (NIH U54GM128729), the ND INBRE (NIH P20GM103442), Azore Cancer Imperative (non-profit).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All research performed, including animal and tissue collection, was conducted in accordance with the Animal Welfare Act and with the approval of the University of North Dakota’s Institutional Animal Care and Usage Committee g(IACUC).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data from this study is available on request. Please contact DB at david.s.bradley@und.edu.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.