Article Text

Abstract
Background Well-characterized preclinical models are essential for immune-oncology research. We investigated the feasibility of our humanized mouse model for evaluating the long-term efficacy of immunotherapy and biomarkers.
Methods Humanized mice were generated by injecting human fetal cord blood-derived CD34+ hematopoietic stem cells to NOD-scid IL2rγnull (NSG) mice myeloablated with irradiation or busulfan. The humanization success was defined as a 25% or higher ratio of human CD45+ cells to mice peripheral blood mononuclear cells.
Results Busulfan was ultimately selected as the appropriate myeloablative method because it provided a higher success rate of humanization (approximately 80%) and longer survival time (45 weeks). We proved the development of functional T cells by demonstrating the anticancer effect of the programmed cell death-1 (PD-1) inhibitor in our humanized mice but not in non-humanized NSG mice. After confirming the long-lasting humanization state (45 weeks), we further investigated the response durability of the PD-1 inhibitor and biomarkers in our humanized mice. Early increase in serum tumor necrosis factor α levels, late increase in serum interleukin 6 levels and increase in tumor-infiltrating CD8+ T lymphocytes correlated more with a durable response over 60 days than with a non-durable response.
Conclusions Our CD34+ humanized mouse model is the first in vivo platform for testing the long-term efficacy of anticancer immunotherapies and biomarkers, given that none of the preclinical models has ever been evaluated for such a long duration.
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Intoduction
Cancer is the world-leading cause of mortality,1 and the development of more effective and safer anticancer therapies is an unmet medical need. In recent years, immune checkpoint inhibitors (ICIs), such as programmed cell death-1/ligand-1 (PD-1/PD-L1) and cytotoxic T lymphocyte antigen-4 inhibitors, have been approved for use in many types of cancer.2 The main advantage of ICIs is durability of their effectiveness.2 3 For instance, pembrolizumab as a first-line treatment for the treatment of advanced non-small cell lung cancer demonstrated a 5-year overall survival rate of around 23% in the KEYNOTE-001 study, compared with a historical control of ~5%, prior to the introduction of PD-1 inhibitor therapy.4 Despite the advantage of ICIs, there are limitations of ICIs in terms of toxicity and efficacy; ICIs sometimes provoke serious immune-related adverse events including skin disorder, endocrinopathy, colitis, hepatitis, nephritis and pneumonitis, which should be carefully monitored during ICI treatment.5 Furthermore, only a proportion of patients show good and durable efficacy to ICIs. Consequently, researchers are working to enhance the efficacy of ICIs in patients with immune cold or immune exclusive tumor microenvironment.6
In the era of immuno-oncology, a preclinical model for basic or translational research is an urgent unmet need. Historically, syngeneic mouse models have been mainly used for this purpose due to advantages including easy establishment,7 and the interaction between tumor cells and a fully competent immune system.8 However, the major caveat of syngeneic models is the rapid growth of murine tumors, which does not facilitate the development of the chronic inflammatory environment that is characteristic of human tumors.9 10 Finally, murine tumors do not normally reflect the genetic complexity of human tumors because they have lower mutational loads.10 11
Owing to these limitations, humanized mouse models have been developed, whereby mice are ‘engrafted with functional human cells, tissues, or organs’. Humanized mouse models can be categorized into three types according to the source of human immune cells and engrafting methods: human peripheral blood lymphocyte (hPBL) mouse model, hCD34+ (also named human HSC for ‘hematopoietic stem cell’) mouse model and human bone marrow-liver-thymus (hBLT) mouse model.12 13 In more detail, the hPBL mouse model is built by intravenous or intraperitoneal injection of peripheral blood mononuclear cells (PBMCs),14 and represents the simplest, fastest, and most economical model. However, the inevitable occurrence of Graft-versus-host disease (GvHD) could be an obstacle to cancer research.15 Thus, the window of immuno-oncology research using the hPBL model is restricted to 4–6 weeks after PBMC injection before the development of GvHD.11 14 16 17 The hCD34+ model is established by the injection of hCD34+ HSCs,18 which can be sourced from the human umbilical cord11 19 and adult bone marrow,20 and are relatively easy to obtain compared with when building the BLT model. Advantages of the hCD34+ model also include the multilineage differentiation of the immune system, which allows more specific mechanistic studies.21 22 However, this model has disadvantages in that it takes a long time for cell differentiation, up to 10 weeks, and has a variable success rate.23 The BLT model is generated by the transplantation of human fetal liver and thymus tissue into the subrenal capsule alongside the intravenous injection of autologous hCD34+ cells from the same fetal liver.22 This model allows a complete and fully functional human immune system by providing the microenvironment of human thymus.24 However, if during the process of T cell selection in the implanted thymus, some T cells with affinity for mouse major histocompatibility complex (MHC) are not eliminated, GvHD consequently occurs at a higher incidence than in other hCD34+ models.23 Most importantly, in terms of the generation of the BLT model, the availability of fetal liver and thymus is very low and this model also requires difficult surgical implantation of tissues.
Overall, the hCD34+ model has longer immunological persistence than the hPBL model,25 and is easier to establish than the hBLT model, based on the higher availability of human HSCs without the need for a complex surgical technique.26 Therefore, we have reasonably selected hCD34+ model for further development as a preclinical in vivo model to study immuno-oncology drugs. Considering the notable characteristics of ICIs, such as durable response and long-term survival, a long-lasting preclinical in vivo model may be more appropriate to evaluate efficacy of ICIs. Thereby, we investigated our own hCD34+ HSC humanized mouse model to evaluate the efficacy of ICIs, focusing on the durability of the response and simultaneous immune monitoring.
Materials and Methods
Mice
3-week female NOD.Cg-PrkdcscidIl2rγtm1Wjl/SzJ (NOD-scid IL2rγnull , NSG) mice were purchased from Jackson Laboratory (ME, USA, #005557). Mice were housed in a specific-pathogen free animal facility at CHA University (Seongnam, Korea).
Generation of humanized mice
4-week female NSG mice were used to generate humanized mice following myeloablation and transplantation of hCD34+ HSCs. First, bone marrow was suppressed by γ-irradiation (2.4 Gy) or busulfan (Korea Otsuka Pharmaceutical, Korea). Busulfan was dissolved in dimethyl sulfoxide (Sigma Chemical Co., Missouri, USA) and diluted with 0.9% saline. Busulfan was intraperitoneally injected into NSG mice at 3-dose levels (20, 30 and 40 mg/kg body weight) to determine the optimal dosage. After considering safety and efficacy, 30 mg/kg was selected as the ultimate dose level. Second, hCD34+ HSC, which were isolated from human umbilical cord blood (hUCB), were injected into myeloablated mice via the tail vein within 24 hours of myeloablation. To prevent infections, we prophylactically added ciprofloxacin (0.33 mg/mL) to the drinking water. hUCB was provided by the CHA Bundang Medical Center Cord blood Bank (icord, Korea). hUCB was provided in a frozen or fresh status. Mononuclear cells (MNCs) were separated from hUCB using Ficoll-Paque Plus density gradient centrifugation. hCD34+ cells were isolated from MNCs using a magnetic activated cell sorting (MACS) human CD34 MicroBead Kit (Miltenyi Biotec, Spain) according to the manufacturer’s instructions. The purity of hCD34+ cells was determined using a CytoFLEX flow cytometer. The same number of viable hCD34+ cells were injected into mice, irrespective of whether hCD34+ cells were isolated from frozen or fresh hUCB. Humanization success was defined as a 25% or higher ratio of hCD45+ cells to mice PBMCs.
Tumor xenograft models and treatment regimens
MDA-MB-231 cells (1×107) or triple negative breast cancer (TNBC) patient-derived tumor xenograft (PDTX) tissues were subcutaneously implanted into the right flank of NSG or humanized NSG mice. When tumors reached 80–100 mm3, mice were treated with either phosphate buffered saline (PBS) or anti-PD-1 monoclonal antibody (BE0188, clone J116, Bioxcell, New Hampshire, USA) of 200 µg/mouse by intraperitoneal injection, every 3 days, up to six times. The tumor size was measured every 2 or 3 days using a caliper, and tumor volumes were calculated using the modified ellipsoid formula (1/2 × (length × width2). ‘Tumor response’ was defined as a tumor volume smaller than the mean tumor volume subtracted by 2 SD by referring to control group. Calculations of tumor response and ‘non-response’ are represented as following equations;
Tumor response=Tumor vol in PD-1 treatment <PBS control (mean − 2 × SD)
Non-response=Tumor vol in PD-1 treatment ≥PBS control (mean − 2 × SD)
‘End of tumor response’ was defined as the tumor status whereby the volume increased 100% or more, taking as a reference the smallest tumor volume after showing a response since the start of treatment. ‘Durable response’ was defined as a response sustained over 60 days since the start of treatment, whereas a non-durable response was defined as the end of the tumor response recorded earlier than 60 days.
Flow cytometric analysis of mouse PBMC and tumor tissue
Peripheral blood was collected by retro orbital bleeding from mice. Cells were lysed by RBC lysis buffer (eBioscience, USA) and centrifuged before collecting the PBMC layer. The tumors, spleens, livers, lungs and blood cells were collected from humanized mice at sacrifice. Tumor tissues were minced prior to incubation for 1 hour at 37°C with shaking in a mixture of collagenase D (1 mg/mL, Roche, Basel, Switzerland) and DNase I (10 mg/mL, Roche). Cells were suspended by repeated pipetting, filtered through a 70 µm cell strainer, and lysed to remove RBCs. After washing with PBS, suspended cells were filtered through a nylon mesh. These single-cell suspensions from tumor tissues were blocked with an antibody against mouse CD16/32 and human TruStain FcX (Biolegend). For analysis of surface markers, cells were stained with the indicated antibodies. A list of antibodies is given in online supplemental table S1. Labeled cells were acquired using a CytoFLEX flow cytometer and analyzed using FlowJo v10 (FlowJo LLC, Oregon, USA).
Supplemental material
Multiplex human cytokine assay
To analyze multiple human cytokines, approximately 150 µL of mouse blood was collected by the retro orbital bleeding method and centrifuged for 10 min at 1000 × g. Each isolated serum sample was analyzed using the LEGENDplex Human Th1 panel’ bead-based immunoassay kit (BioLegend, California, USA) (including interleukin (IL)-2, IL-6, IL-10, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ) according to the manufacturer’s directions. Data were acquired with a CytoFLEX flow cytometer, and analyzed by LEGENDplex V.8.0 software (BioLegend, California, USA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism V.5.0 software (GraphPad Software, La Jolla, California) and IBM SPSS for Windows Release 19 (SPSS). Student’s t-test was used to compare the success rate of humanization and tumor volume between two groups. Analysis of variance (ANOVA) test was used to compare the immune cells, cytokines and tumor volume among three groups. The LSD test was used for post-hoc analysis. Values are represented as mean±SD unless otherwise indicated. All p values were two tailed, and p values<0.05 were regarded as significant.
Results
Generation of humanized mouse model using hCD34+ HSCs
Humanized mice were generated by myeloablation and transplantation of hCD34+ HSCs. Detailed procedures are described in the Method section. Flow cytometry demonstrated that hCD45+ cells started to appear 8 weeks after injection of hCD34+ HSCs, and differentiation of human T and B cells occurred at 11 weeks (figure 1A).
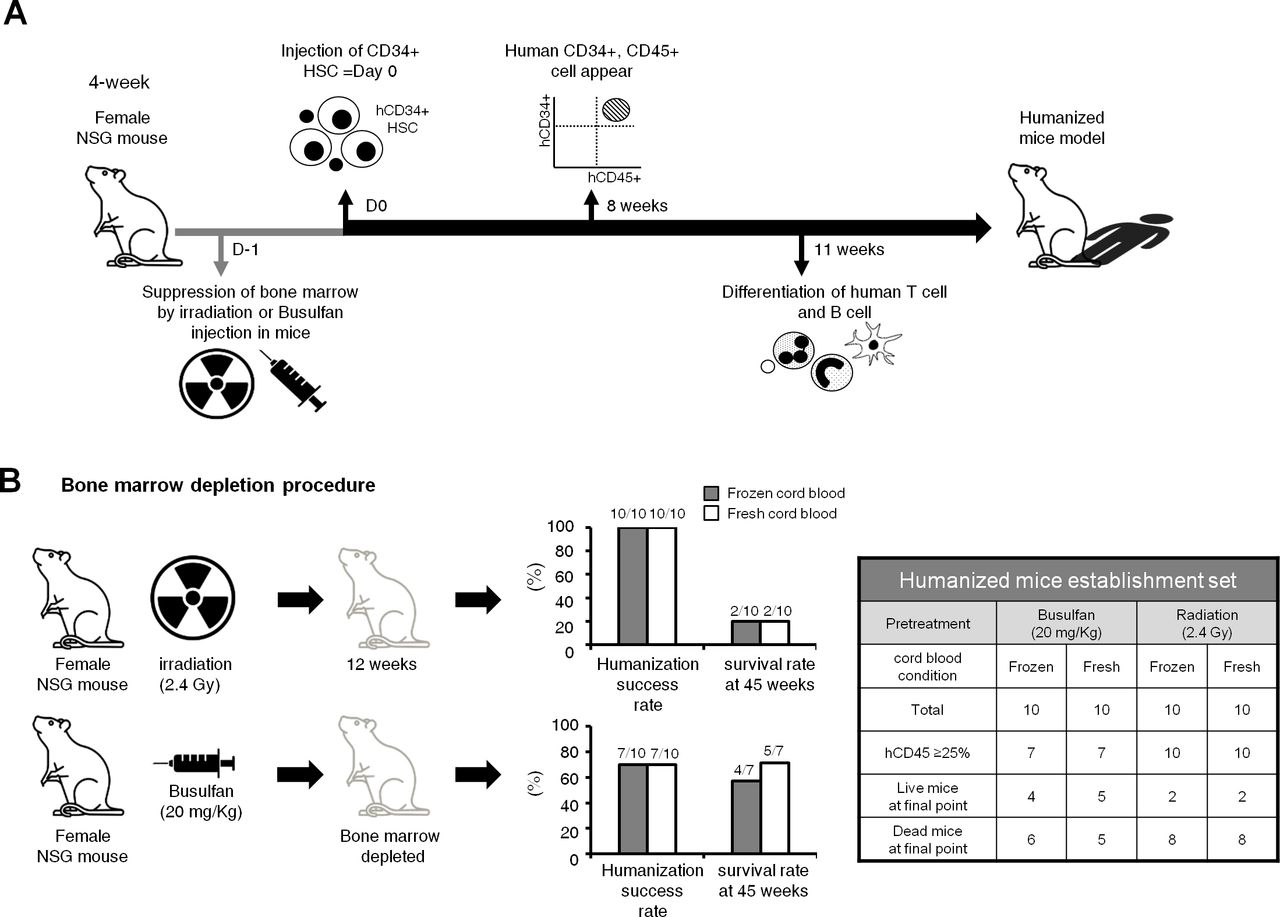
Scheme for the generation of the humanized mouse model. (A) 4-week female NOD-scid IL2rγnull (NSG) mice were prepared to generate humanized mice. Humanized mice were generated following myeloablation and transplantation of hCD34+ hematopoietic stem cells (HSCs). NSG mice were irradiated or injected with busulfan to suppress bone marrow function. Then, hCD34+ HSCs, isolated from human umbilical cord blood, were injected to myeloablated mice via the tail vein within 24 hours of myeloablation. 8 weeks after the injection of hCD34+ HSCs, hCD45+ cells started to appear, and differentiation of human T and B cells occurred at 11 weeks. (B) In order to optimize the humanization process in terms of efficiency and durability, irradiation versus busulfan were compared as myeloablative methods, and fresh versus frozen status of cord blood were compared as the source of human HSCs. Although the humanization success rates were higher in the irradiation group than in the busulfan group, the 45-week survival rate was higher in the busulfan group (64.3%, 9/14 humanized mice) than in the irradiation group (20%, 4/20 humanized mice). The fresh versus frozen status did not significantly affect the humanization success rate or the 45-week survival rate.
To optimize the humanization process, irradiation (2.4 Gy; 20 mice)27 28 versus busulfan (20 mg/kg; 20 mice)29 were compared as myeloablative methods in the humanized mice establishment set in terms of both humanization efficiency and durability. To determine initial testing dosage of busulfan, we referred to the Choi’s study in which they compared the survival rates of mice according to busulfan dosages of 20, 30 and 40 mg/kg for myeloablation. A 40 mg/kg of busulfan treatment was highly toxic, but a 20 mg/kg of busulfan treatment was sufficiently safe.29 Therefore, the test was started at a 20 mg/kg in this study. Moreover, 20–35 mg/kg of busulfan was mostly used for myeloablation in other previous studies.28 30–32
The humanization success rate, which was defined as a 25% or higher ratio of hCD45+ cells to mice PBMCs was higher in the irradiation group (100%, 20/20) than in the busulfan group (70%, 14/20) at 12 weeks from the injection of hCD34+ HSCs (figure 1B). Regarding the level of hCD45+ cells, it was higher in the irradiation group than in the busulfan group at the starting point for immune cell monitoring (12 weeks), but the statistical difference in the level of hCD45+ cells in both groups disappeared overtime at 21 weeks and 45 weeks (figure 2A). However, in terms of long-term survival, the 45-week survival rate was higher in the busulfan group (64.3%, 9/14 humanized mice) than in the irradiation group (20%, 4/20 humanized mice) and Kaplan-Meier survival analysis also showed a trend toward longer survival in busulfan group than radiation group (figure 2B; p=0.071). For this reason, busulfan treatment was selected as a myeloablative method for further humanization experiments in order to establish an appropriate preclinical model for long-term evaluation of immuno-oncology drugs. In the next attempt of humanization, we planned to increase the dosage of busulfan up to 30 mg/kg in order to enhance the engraftment of hCD34+ cells. As a result, the humanization success rate was 85% (17/20) and all 17 humanized mice survived until the final time point (4 months).
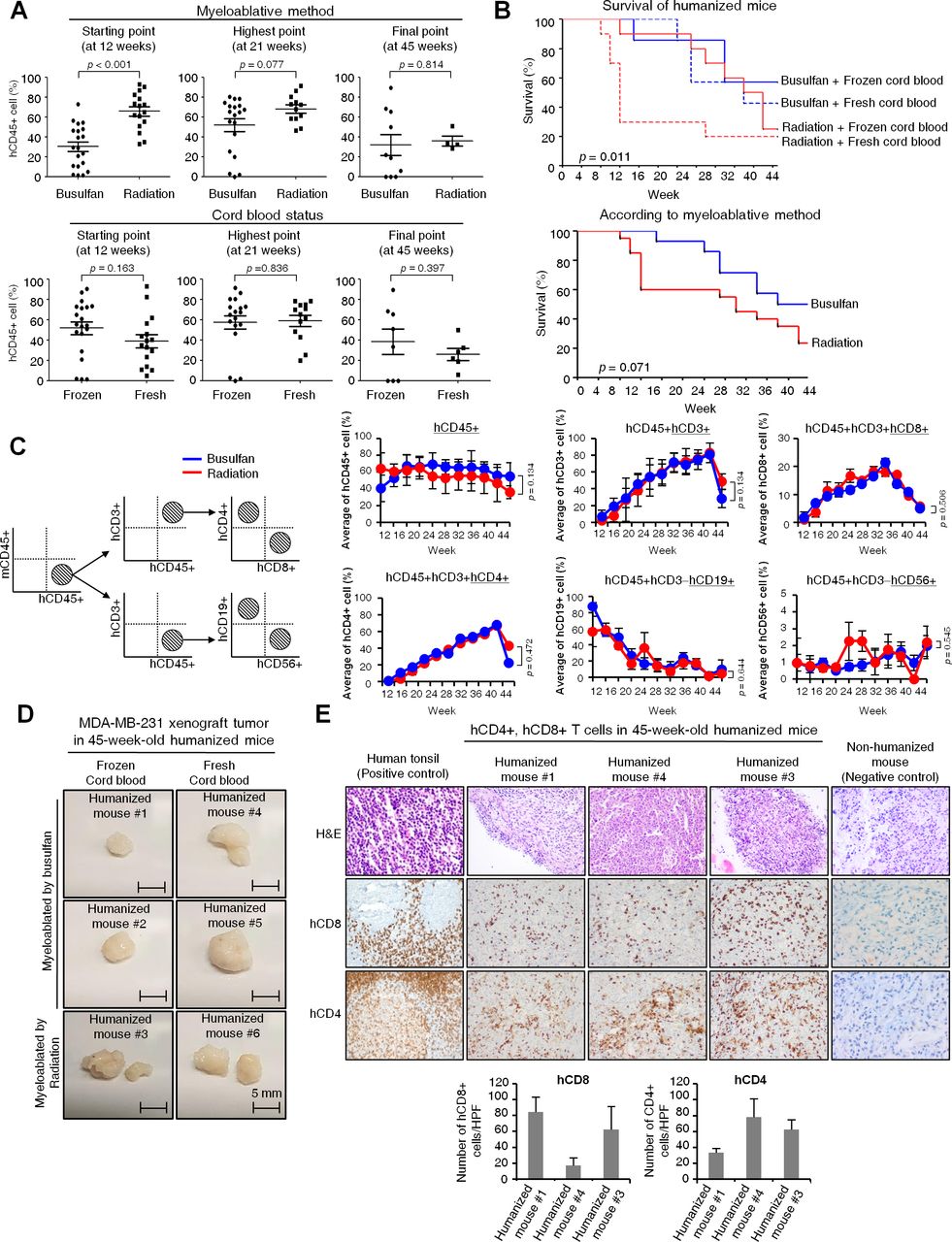
Feasibility of the humanized mouse model for long-term immune monitoring. (A) The proportion of cells in the peripheral blood was analyzed for the humanization marker hCD45+ by flow cytometry over 12–45 weeks from the injection of hCD34+ HSCs. Percentages of hCD45+ cells are demonstrated at three time points (starting point, highest point and final point) according to the myeloablative method or cord blood status. P values were calculated by Student’s t-test. Data are presented as mean±SD. (B) Kaplan-Meier survival curve of humanized mice. Survival of humanized mice was monitored up to 45 weeks according to the myeloablative method and cord blood status (upper). Survival curve was represented according to the myeloablative method (lower). P value was calculated by Breslow (Generalized Wilcoxon) test. (C) Gating strategy for flow cytometry analysis (left). Various immune cell markers, including hCD45, hCD3, hCD4, hCD8, hCD19 and hCD56 in the peripheral blood were analyzed by flow cytometry over 12 weeks to 45 weeks from the injection of hCD34+ HSCs by myeloablative method (right). When hCD45+ cells were taken as a whole (100%), percentages of hCD4+ or hCD8+ cells were generated from hCD45 and hCD3 double positive cells, whereas percentages of hCD19+ or hCD56+ cells were generated from hCD45+ and hCD3− cells. P values were calculated by Student’s t-test. Data are presented as mean±SD. (D) Tumorigenicity test of MDA-MB-231 cells in 45-week-old humanized mice. MDA-MB-231 cells were xenografted at 45 weeks post engraftment of hCD34+ HSCs. Mice were sacrificed 43 days after the implantation of MDA-MB-231 cells; the resultant tumors are demonstrated according to the myeloablative method and cord blood status. Scale bars, 5 mm. (E) Immunohistochemical staining of hCD8 and hCD4 in MDA-MB-231 xenograft tumors of 45-week-old humanized mice. Human tonsil was used for the positive control and non-humanized mouse tumor tissues were used for negative control. H&E images were collected at ×200 magnification and immunohistochemical images were collected at ×400 magnification. The bar graph represented the average of hCD8+ and hCD4+ cells in five random, non-overlapped fields at ×400 magnification. Data are presented as mean±SD.
We compared fresh (20 mice) versus frozen (20 mice) cord blood as the source of human HSCs in the humanized mice establishment set. The humanization success rates were comparable in both fresh and frozen cord blood groups, irrespective of the myeloablative method (figure 1B). The PD-1 inhibitor test set, in which fresh cord blood was used, showed a similar humanization success rate (84%, 32/38) to that in the previous experiment, in which frozen cord blood was used. That is to say, the cord blood status did not affect level of hCD45+ cells throughout the monitoring period (figure 2A, online supplemental figure 1A). In the humanized mice establishment set, the long-term survival rate at 45 weeks was not significantly different according to the cord blood status in either the irradiation or busulfan groups (figure 1B). The reason why the humanization outcome is similar using both groups cord blood is likely to be because the same numbers of viable hCD34+ cells (3×104 cells) were injected, even though cellular viability may have been different between the fresh and frozen cord blood.
Feasibility of the humanized mouse model for long-term immune monitoring for 11 months
We followed up the survival duration of humanized mice and monitored immune cells in the peripheral blood by flow cytometry from the injection of hCD34+ HSCs up until 45 weeks. As mentioned above, a significant portion (64.3%) of 14 humanized mice that were myeloablated with busulfan survived up to 45 weeks in the humanized mouse establishment set (figure 2B). Although the late phase hCD45+ cells were gradually decreased over time, the humanization status (hCD45+ cells ≥ 25% to mice PBMCs) was maintained up to 45 weeks, irrespective of the myeloablative method or cord blood status. Each step of FACS analysis for detection of immune positive cells was represented (figure 2C, online supplemental figure 1B,C). As shown in figure 2C, we assessed the dynamic alteration of human immune cells, such as T cells, B cells and natural killer (NK) cells, in peripheral blood up to 45 weeks. Expression of hCD3+ T cells gradually increased and then decreased rapidly at 45 weeks. The hCD4+ or hCD8+ T cells also showed the similar pattern with hCD3. Furthermore, hCD19+ cells (ie, human B cells) showed a sharp increase at the beginning of transplantation of hCD45+ HSCs, and then gradually decreased over time. The hCD56+ cells (ie, human NK cells) were maintained at around at 2% for 45 weeks (figure 2C).
MDA-MB-231 cells, that is, TNBC cells were implanted in 45-week-old humanized mice following injection of hCD34+ HSCs in order to test the tumorigenicity of aged humanized mice for potential use as a preclinical model for long-term evaluation of immuno-oncology drugs. As a result, 6 of 9 MDA-MB-231-implanted mice developed tumors 70–160 mm3 by 43 days (figure 2D). In the tumor sections of 45-week-old humanized mice, many tumor infiltrating lymphocytes, including hCD4+ and hCD8+ T cells, were observed. This suggests that ICIs can be evaluated for a long duration, up to 45 weeks, using this humanized mouse model (figure 2E). In addition, normal organs, such as the spleen, liver and lung collected from 45-week-old humanized mice, demonstrated infiltrated hCD4+ and hCD8+ T cells by immunohistochemistry (IHC), suggesting that those human immune cells could be persistently viable for up to 45 weeks. In contrast, no hCD8 or hCD4+ T cells were observed on non-humanized mice tissues (online supplemental figure 3A,B).
Long-term antitumor efficacy of a PD-1 inhibitor in a MDA-MB-231 cell xenograft humanized mouse model
At first, we confirmed the inhibition efficiency of human PD-1 inhibitor by in vitro T cell proliferation assay. In online supplemental figure 2, a Ki67 expression on hCD3+ Jurkat cells increased with human PD-1 inhibitor treatment (24 hours; 31.12% and 48 hours; 56.23%) compared with untreated controls (24 hours; 6.86% and 48 hours; 44.26%), suggesting that PD-1 inhibitor used in the current study effectively enhances T cell proliferation by blocking binding of PD-1 on T cells to PD-L1 on tumor cells. However, a human PD-1 expression itself was not changed (online supplemental figure 2).
To demonstrate the effectiveness of our hCD34+ humanized mouse model as a preclinical platform for the long-term evaluation of immuno-oncology drugs, the antitumor efficacy of a PD-1 inhibitor was monitored for up to 120 days in a MDA-MB-231 cell xenograft humanized mouse model (figure 3A). As mentioned above, in this PD-1 inhibitor test set, busulfan was used at an increased dose of 30 mg/kg from 20 mg/kg in order to enhance the humanization success rate. In total, 32 (84.2%) of the 38 mice that received hCD34+ HSCs fulfilled the humanization criteria (hCD45+ cells ≥ 25% to mice PBMCs). The 32 mice that fulfilled the humanization criteria were selected for subsequent experiments regarding the PD-1 inhibitor efficacy; 17 humanized mice were implanted with MDA-MB-231 cells and 15 humanized mice were implanted with TNBC PDTX tumor (figure 3B,C). In the MDA-MB-231 xenograft experiments, of the 12 mice that received the PD-1 inhibitor, 11 (91.9%) showed a response. In 8 (72.7%) of the 11 responders, the response was durably sustained over 60 days (ie, durable responders), with a response duration of 79.3±34.4 (mean±SD) days. In 3 (27.3%) of the 11 responders, the tumors regrew and showed an end of response less than 60 days (ie, non-durable responder) and a response duration of 39.3±16.3 (mean±SD) days. In the later period, after around 100 days, the tumor volume curve was transiently downward in the PBS-control group; this was because the mouse with the biggest tumor died at that time and the tumor volume was only averaged in the surviving mice (figure 3D). One case did not respond to PD-1 inhibitor treatment (ie, non-responder) (online supplemental figure 4A), and showed no significant difference in the immune cell or cytokine profiling from the non-durable responders (data not shown).
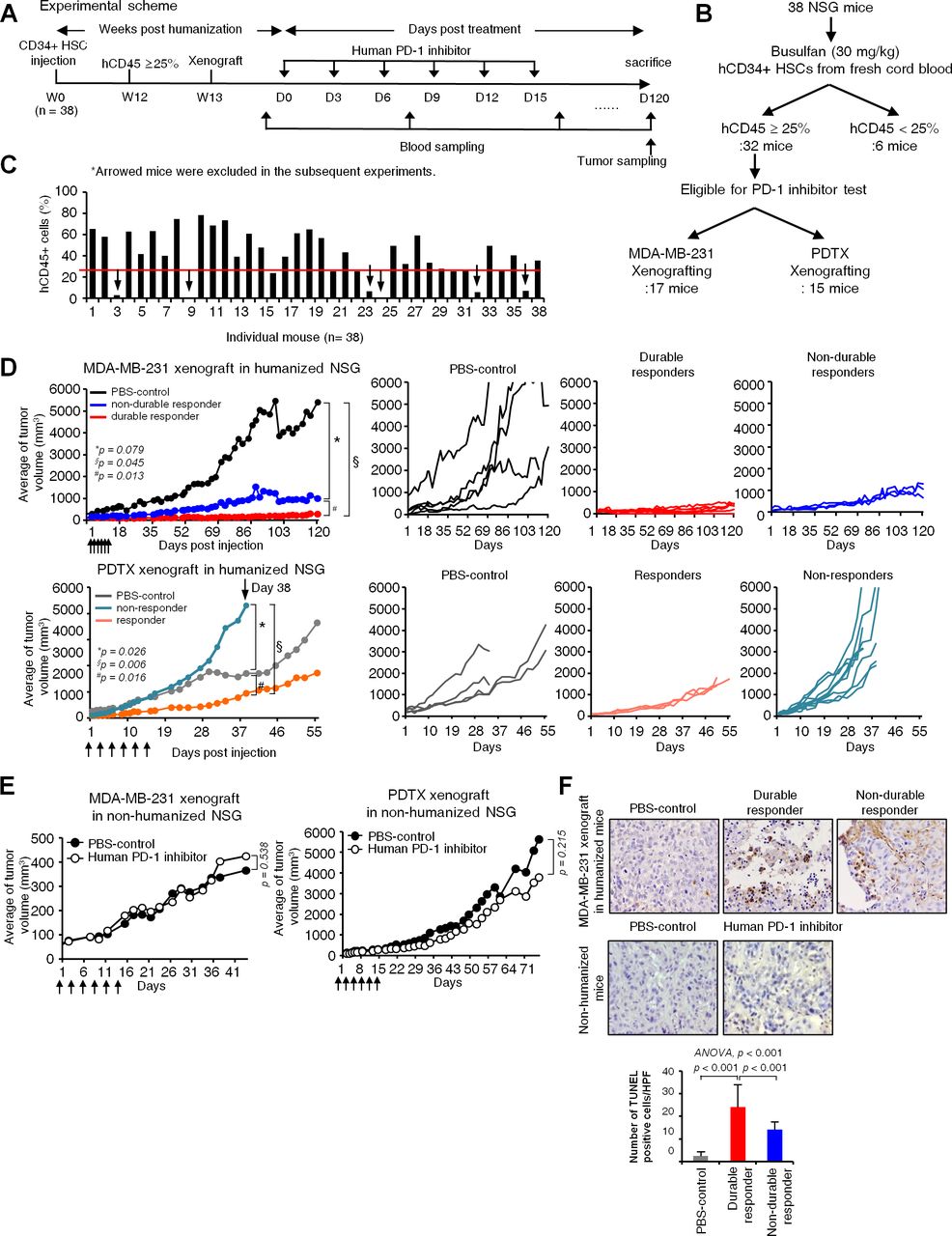
Long-term antitumor efficacy of the programmed cell death-1 (PD-1) inhibitor in the MDA-MB-231 cell xenograft humanized mouse model. (A) Scheme of the PD-1 inhibitor test set. Humanized mice were generated by injecting hCD34+ hematopoietic stem cells (HSCs) in mice, myeloablated with busulfan. MDA-MB-231 cells were xenografted 12 weeks after the injection of hCD34+ HSCs. When tumors reached 50–100 mm3, the PD-1 inhibitor was administered six times every 3 days. Tumor size was monitored up to 120 days from PD-1 inhibitor treatment, and blood and tumor samples were collected at indicated time points. (B) Schematic flow diagram of humanization experiment for PD-1 inhibitor test. Thirty-eight NOD-scid IL2rγnull (NSG) mice were myeloablated by busulfan and engrafted with CD34+ HSCs isolated from fresh cord blood. Only 32 mice that fulfilled the humanization criteria (hCD45+ cells ≥ 25%) were selected for the subsequent experiments regarding the PD-1 inhibitor efficacy. (C) Demonstration of the level of hCD45+ cells (Y-axis) in individual mouse (X-axis; 38 mice). Six arrows indicate mice which did not fulfill humanization criteria. The humanization success rate was 84.2% (32/38). (D) In vivo efficacy test of the PD-1 inhibitor in the humanized mouse model. The average tumor growth inhibition curves of all mice in each group were drawn first. Then, the tumor growth inhibition curves of individual mice were drawn as durable responders versus non-durable responders of MDA-MB-231 xenografted mice, and responders versus non-responders of patient-derived tumor xenograft (PDTX) xenografted mice. P values were calculated by Student’s t-test. (E) In vivo efficacy test of the PD-1 inhibitor in the NSG mouse model without humanization. PD-1 inhibitors showed no significant anticancer efficacy in either the MDA-MB-231 xenograft or PDTX xenograft non-humanized NSG mice. P values were calculated by Student’s t-test. (F) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay using tumor sections at sacrifice. Representative images in each group are shown and bar graphs represent average numbers of apoptotic cells in each group in five random, non-overlapped fields at ×400 magnification. Data are presented as mean±SD. Apoptotic cells were increased in durable responders compared with PBS-treatment controls or non-durable responders. Non-humanized mouse tumor tissues were used for negative control. P values were calculated by analysis of variance test (post-hoc p values; LSD).
In the TNBC PDTX model (15 humanized mice), only 3 (27%) of the 11 mice that received the PD-1 inhibitor showed a response to PD-1 inhibitor treatment (figure 3D). This low response rate may reflect a response rate of around 20%–25% in the clinical setting of metastatic TNBC.33 34 The existence of PD-1 inhibitor efficacy suggests that positive and negative selection of human T cells probably occurred and led to the development of functional T cells in the humanized mice.
To confirm that the non-humanized NSG mouse tumor model cannot be used for testing the efficacy of ICIs, we implanted MDA-MB-231 cells or TNBC PDTX tissue in NSG mice without humanization (ie, lacking human immune cells). In contrast to the humanized mouse model, in the non-humanized NSG mouse tumor model, the PD-1 inhibitor showed no antitumor activity (figure 3E, online supplemental figure 4B); this finding confirmed that a preclinical model loaded with human immune cells is necessary to properly evaluate ICIs in vivo. In addition, a TUNEL assay, using tumor sections at sacrifice, demonstrated increased apoptotic cells in durable responders compared with non-durable responders (p<0.001) and PBS-treatment controls (p<0.001; figure 3F). Also, apoptotic cells were hardly observed in PBS-control, irrespective of whether it was assessed in humanized or non-humanized mice (figure 3F), which suggests the absence of unusual cytotoxic effects in humanized mice.
We then investigated if the level of hCD45+ cells in peripheral blood affects PD-1 inhibitor response. In the MDA-MB-231-xenograft experiments, the levels of hCD45+ cells were not significantly different among PBS-control, durable responders and non-durable responders before and after PD-1 inhibitor treatment in humanized mice (online supplemental figure 4C).
T cell profiles associated with a durable response to PD-1 inhibitors in humanized mice
To investigate T cell profiles, which may be associated with durable responses to PD-1 inhibitors, we compared the expression of hCD4+ or hCD8+ T cells between durable and non-durable responders. Using flow cytometry to analyze tumor tissues, there was a trend for increased hCD8+ T cells in durable responders compared with non-durable responders (ANOVA, p=0.010 among three groups; LSD, p=0.007 between durable and non-durable responders; figure 4A). Similarly, IHC analysis of tumor-infiltrating T lymphocytes (TILs) using tumor tissues at sacrifice also showed a trend for increased hCD8+ T cells in durable responders compared with non-durable responders (ANOVA, p=0.104 among three groups; LSD, p=0.041 between durable and non-durable responders) (figure 4C), although such differential expression of hCD8+ T cells was not observed in the peripheral blood (figure 4B). Our finding of a persistent increment of hCD8+ T cells in durable responders is consistent with the previous report that demonstrated that immune-inflamed tumors, characterized by the presence of sufficient immune cells, shows better response to ICIs.35 Contrary to hCD8+ T cells, the expression of hCD4+ T cells was not significantly different according to the response durability in both the peripheral blood (figure 4B) and tumor tissues (figure 4A,C) by flow cytometry or IHC.

T cell profiles associated with a durable response to the programmed cell death-1 (PD-1) inhibitor. To investigate the T cell profiles that may be associated with a durable response to the PD-1 inhibitor, we compared the proportions of hCD4+ or hCD8+ T cells between durable and non-durable responders. (A and B) hCD4+ or hCD8+ T cells were analyzed using tumor tissue (A) and blood (B) by flow cytometry. Representative flow cytometry plots from a mouse in each group are shown. Dot plots represent average numbers of hCD8+ and hCD4+ cells in PBS group, durable responders, or non-durable responders. P values were calculated by analysis of variance (ANOVA) test, and post-hoc p values were calculated by least significant difference (LSD) test. Data are presented as mean±SD. ns, not significant. (C) Immunohistochemistry analysis of tumor-infiltrating T lymphocytes (ie, hCD8+ or hCD4+ T cells) using mouse tumor tissues. H&E images were collected at ×200 magnification and immunohistochemical images were collected at ×400 magnification. P values were calculated by ANOVA test after counting positive cells in five random, non-overlapped fields and post-hoc p values were calculated by LSD test. Data are presented as mean±SD.
Serum human cytokine profiles associated with durable responses to PD-1 inhibitors in humanized mice
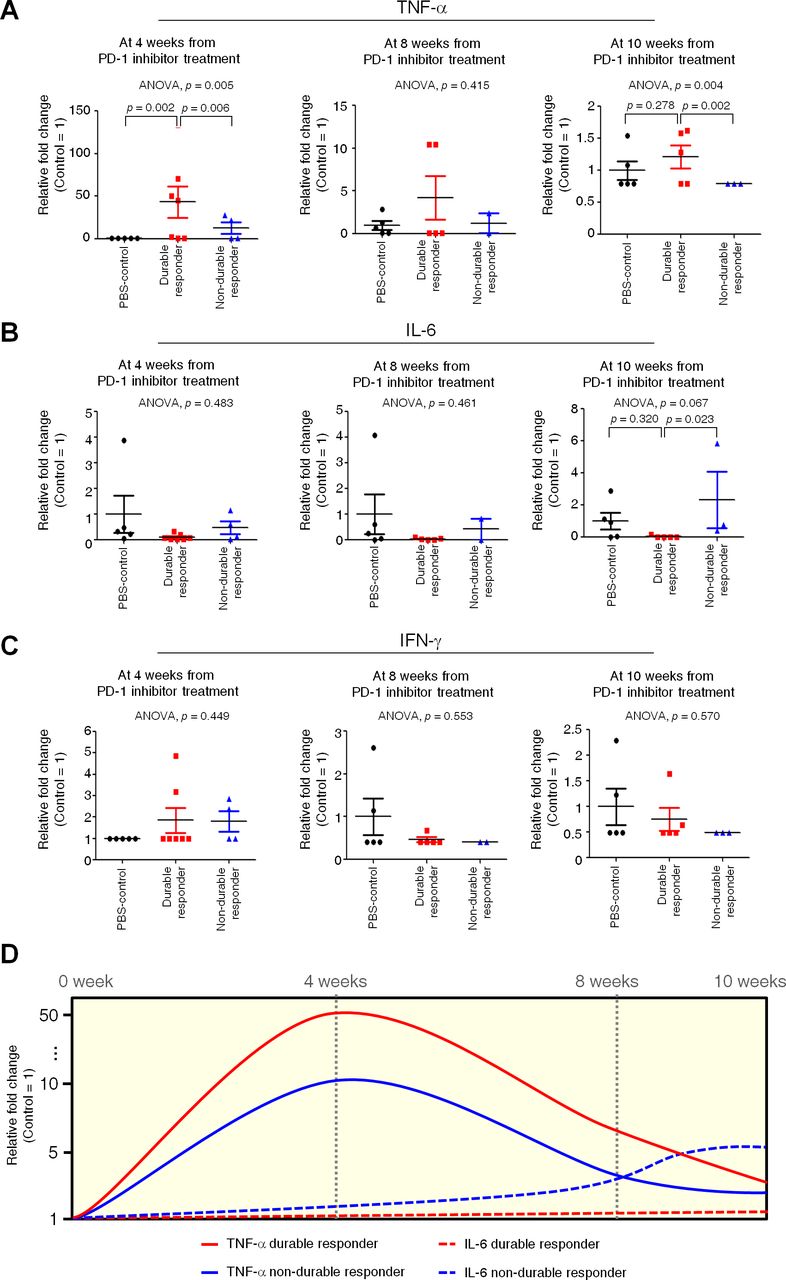
We analyzed serum human cytokine concentrations to determine evidence that early immunological reactions since early release of serum cytokines could predict durable responses to PD-1 inhibitors. Serum human cytokines were analyzed with the bead-based assay LEGENDplex from the 4th week from PD-1 inhibitor treatment (figure 5). Increase in serum TNF-α (figure 5A) was observed in durable responders compared with non-durable responders from the 4th week from PD-1 inhibitor treatment (ANOVA, p=0.005; LSD, p=0.006) and the pattern was maintained through weeks 8–10 (ANOVA, p=0.004; LSD, p=0.002). This correlation between an early increase in TNF-α and a durable response is supported by the growing evidence that TNF-α is involved in immune adverse events, such as immune colitis, which may more often occur in responders to ICIs.36 37 In contrast, serum IL-6 (figure 5B) was decreased in durable responders compared with non-durable responders at week 10 (ANOVA, p=0.067; LSD, p=0.023), and was accompanied by a similar pattern, although without statistical significance, during the previous weeks. This is in line with a previous study that reported the proinflammatory cytokine IL-6 enhanced cancer progression and was associated with a poor response to ICIs.38 In short, an early (4th week from PD-1 inhibitor treatment) increase and maintenance of serum TNF-α or a late (10th week from PD-1 inhibitor treatment) decrease of serum IL-6 may be potential predictive biomarkers of durable response (figure 5D). However, serum IFN-γ (figure 5C), IL-2 and IL-10 (data not-shown) did not change significantly between durable and non-durable responders throughout the study period.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serum human cytokine profiles associated with a durable response to the programmed cell death-1 (PD-1) inhibitor. Serum human cytokines, including TNF-α (A), IL-6 (B), and IFN-γ (C) were analyzed with the bead-based assay LEGENDplex at weeks 4, 8 and 10 from PD-1 inhibitor treatment in order to determine if an early immunological reaction could predict a durable response to the PD-1 inhibitor. P values were calculated by analysis of variance (ANOVA) test, and post-hoc p values were calculated by LSD test. Data are presented as mean±SD. (D) Response of serum cytokine by PD-1 inhibitor according to the time point. Cytokine level with time was represented as relative fold change (0 week; 1).
Discussion
In the current study, we successfully established an hCD34+ humanized mouse model, the advantages of which include a high success rate of humanization (approximately 80%), even with injection of a lower number of hCD34+ HSCs (3×104 cells) than those (1×105 ~ 1×106) in previous studies,25 27 29 an easy myeloablative method with busulfan, and feasible long-term immune monitoring (11 months). Furthermore, we confirmed the development of functional T cells by demonstrating a differential anticancer effect of PD-1 inhibitor in our humanized mouse model that was not apparent in non-humanized NSG mice.
Similarly to allogeneic HSC transplantation, myeloablation is an essential step to provide appropriate immunosuppression and prevent graft rejection before bone marrow is reconstituted with hCD34+ HSCs in humanized mice.14 Total body irradiation/cyclophosphamide,39 busulfan/cyclophosphamide,40 and busulfan/fludarabine41 are the three most commonly used myeloablative regimens in clinical practice. In the current study, we compared irradiation (2.4 Gy) versus busulfan (20 mg/kg or 30 mg/kg) as a myeloablative method. Although both irradiation and busulfan were effective in generating humanization, survival was longer in the busulfan group (45-week survival rate, 64.3%) than in the irradiation group (45-week survival rate, 20%). The shorter survival in the irradiation group was probably attributable to GvHD,42 which can be induced by the higher level of human immune cells in the earlier phase.15 23 In addition, busulfan may be more favored than irradiation in terms of easy accessibility. As a result, busulfan at 30 mg/kg may be considered to be an appropriate myeloablative method for research in which humanized mice needs to be used for long duration. However, the adequate dosage of busulfan might be different according to the strain and age of recipient mice.
The long-term persistence of humanization has not been demonstrated in previous humanized mouse models. A representative company of mouse model, Jackson laboratory has reported that their commercial hCD34+ humanized mouse model showed the presence of hCD45+ cells in mouse peripheral blood until 24 weeks after engraftment of CD34+ cells.43 Other researchers also reported that durability of CD34+ humanized mouse model ranged from 4 weeks to 20 weeks (online supplemental table 2).44–48 In contrast, our humanized mouse model showed long-term survival and long-lasting humanization status (hCD45+ cells ≥ 25% to mice PBMCs) that was maintained up to 11 months. When the human cancer cell line or PDTX tissues were implanted in our humanized mice, persistent tumorigenicity was confirmed from the early period of 12 weeks up to 11 months from the injection of human HSCs. In addition, the tumor immune microenvironment is continually constituted in our humanized mouse model, as evidenced by existence of tumor infiltrating T cells and serum cytokines even at 11 months after the injection of human HSCs. Taken together, our model could be an appropriate preclinical model to evaluate immuno-oncology drugs in terms of both long-term efficacy, one of representative features of ICIs, and biomarkers. To the best of our knowledge, this is the first in vivo platform, in which long-term immune monitoring is feasible (almost 1 year) in the immuno-oncology field. The discovery of biomarkers associated with the efficacy of ICIs is an urgent unmet need in the era of immuno-oncology. Indeed, PD-L1 expressions, microsatellite instability, tumor mutation burden, TILs, serum cytokines and so on have been studied as potential biomarkers.49 We tested our humanized mouse model as a biomarker research platform. In our study, increment of hCD8+ T cells was proved to be a biomarker associated with durable response to PD-1 inhibitor as expected. To further predict the efficacy of ICIs in vivo model, monitoring FoxP3+ Tregs, which was not assessed in our study, would be also helpful, because FoxP3+ Tregs were previously reported to decrease responding to ICIs.31 50 51 In our cytokine analyses using murine serum, except for associations of TNF-α or IL-6 with durable response to PD-1 inhibitor, an association of IFN-γ with durable response was not observed, even though IFN-γ was reported to be a strong biomarker of immune response.52 We suppose that inconsistent result might be attributable to 4 weeks or later time point when we checked IFN-γ because IFN-γ increased at early time of less than 4 weeks, but the change became insignificant after 4 weeks in previous studies.50–53 In a view of biomarker research platform, it was feasible to draw a small volume of blood from the humanized mice periodically every 2–4 weeks, and then to perform flow cytometry and multiplex cytokine analysis using this volume of blood. It was also feasible to obtain xenografted tumors at the point of sacrifice for flow cytometry and IHC in order to determine immune cell infiltration. Furthermore, the humanized mouse model allows more complex and invasive biomarker studies to be performed than those permitted in human subjects.
One of the limitations of this study is that HLA match test between the human HSC donor and implanted tumors or cells was not performed. Although it is considered that HLA matching may be important for successful xenografting, full HLA matching is difficult in reality for in vivo studies. Instead, in the Jackson Laboratory’s previous hCD34+ humanized mouse study, partially HLA-matched PDTX tumors were implanted and the tumor growth was not significantly affected in humanized NSG mice engrafted with hCD34+ HSCs compared with in non-humanized NSG mice.54 In another previous hCD34+ humanized mouse study, approximately 60–80% of PDTXs implanted in humanized mice developed into tumors, regardless of donor HLA type.55 Also, in those two studies, the HLA matching status did not affect the response rate to ICIs.54 55 That is, pembrolizumab showed a significant antitumor response in humanized mice, independent of HLA matching status, whereas no response to pembrolizumab was observed in non-humanized PDTX-bearing mice, just like in our study. Taken together, although in previous studies, xenografted tumor growth and antitumor response to ICIs in humanized mice were not significantly affected by HLA matching status, underlying mechanisms of this are unclear. Thus, in future humanization experiments, at least partial HLA matching may be needed until more experimental data are accumulated and mechanisms are revealed.
In conclusion, we acknowledge that the immune responses in humanized mice may not fully represent those in humans. However, humanized mouse models have the advantage of permitting more detailed and invasive experiments than are possible in human subjects. We successfully established an hCD34+ humanized mouse model, even with injection of a lower number of hCD34+ HSCs to NSG mice myeloablated with busulfan. More notably, using this model, we also demonstrated the feasibility of long-term immune monitoring for 11 months. None of the preclinical models has ever been evaluated for such a long duration. Therefore, our hCD34+ humanized mouse model provides the first in vivo platform for testing the long-term efficacy of anticancer immunotherapies and biomarkers.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conceptualization: NP and YWM. Methodology: all authors. Investigation: NP, KP, YBC, JH, GWS, JMJ, and YWM. Formal analysis: NP, KP, YBC, JH, NBK, GWS, JMJ, and YWM. Writing: NP and YWM. Visualization: NP, KP, A-YK, YBC, JH, GWS, JMJ and YWM. Supervision and project administration: YWM.
Funding This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number : HI16C1559).
Competing interests The corresponding author received research funds from several pharmaceutical companies including the AstraZeneca, Eisai, Dong-A ST, Chong Kun Dang, and Celltrion.
Patient consent for publication Not required.
Ethics approval All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC, #170062) of CHA University and were carried out in accordance with the approved protocols.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data are available. No data are available.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.