Article Text

Abstract
Background The inhibitory functions triggered by the programmed cell death-1 (PD-1) receptor following binding to its ligand (PD-L1) protect healthy organs from cytotoxic T cells, and neutralize antitumor T cell attack. Antibody-based therapies to block PD-1/PD-L1 interaction have yielded notable results, but most patients eventually develop resistance. This failure is attributed to CD8+ T cells achieving hyporesponsive states from which recovery is hardly feasible. Dysfunctional T cell phenotypes are favored by a sustained imbalance in the diacylglycerol (DAG)- and Ca2+-regulated transcriptional programs. In mice, DAG kinase ζ (DGKζ) facilitates DAG consumption, limiting T cell activation and cytotoxic T cell responses. DGKζ deficiency facilitates tumor rejection in mice without apparent adverse autoimmune effects. Despite its therapeutic potential, little is known about DGKζ function in human T cells, and no known inhibitors target this isoform.
Methods We used a human triple parameter reporter cell line to examine the consequences of DGKζ depletion on the transcriptional restriction imposed by PD-1 ligation. We studied the effect of DGKζ deficiency on PD-1 expression dynamics, as well as the impact of DGKζ absence on the in vivo growth of MC38 adenocarcinoma cells.
Results We demonstrate that DGKζ depletion enhances DAG-regulated transcriptional programs, promoting interleukin-2 production and partially counteracting PD-1 inhibitory functions. DGKζ loss results in limited PD-1 expression and enhanced expansion of cytotoxic CD8+ T cell populations. This is observed even in immunosuppressive milieus, and correlates with the reduced ability of MC38 adenocarcinoma cells to form tumors in DGKζ-deficient mice.
Conclusions Our results, which define a role for DGKζ in the control of PD-1 expression, confirm DGKζ potential as a therapeutic target as well as a biomarker of CD8+ T cell dysfunctional states.
- immunotherapy
- programmed cell death 1 receptor
- CD8-positive T-lymphocytes
- drug evaluation
- preclinical
- immune evation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- programmed cell death 1 receptor
- CD8-positive T-lymphocytes
- drug evaluation
- preclinical
- immune evation
Background
T cell inhibitory receptors help to sustain self-tolerance, limiting collateral tissue damage during physiological immune responses. These negative regulatory molecules, known as immune checkpoint receptors, represent the main mechanisms of tumor immune evasion.1 The potentiation of immune responses to tumors through immune checkpoint targeting (ICT) represents a very promising therapeutic approach.2 In particular, blocking programmed cell death-1 (PD-1) interaction with its ligands (PD-L1/2) has yielded notable success for the treatment of melanoma and several solid tumors.3–5 Despite these remarkable results, clinical studies show that more than 40%–65% of melanoma patients present primary resistance to ICT directed to the PD-1/PD-L1 axis, and approximately 43% of initial responders develop acquired resistance by 3 years.6 7 The greatest challenge to full exploitation of anti-PD-1/PD-L1 blocking therapies is to understand and to develop effective combinations to overcome the mechanisms that trigger resistance. Positive initial responses to anti-PD-1/PD-L1 treatment are associated with increased CD8+ T cell tumor infiltration. Acquired resistance is attributed mainly to the progressive, sustained promotion of negative signals that eventually drive T lymphocytes into dysfunctional states that closely resemble viral-induced T cell exhaustion.8 Although PD-1 expression can thus be used to identify antigen-reactive tumor-infiltrating T lymphocytes (TIL),9 reaching certain levels of PD-1 expression might indicate terminally differentiated dysfunctional T cells.10
PD-1 limits T cell receptor (TCR) downstream signaling through recognition of its ligands, expressed on the surface of antigen-presenting cells.11 Following ligand recognition, tyrosine phosphorylation of the PD-1 cytoplasmic domain by Src family of kinases leads to recruitment of Src homology region 2 domain-containing phosphatases 1 and 2,12 13 which dephosphorylate signaling molecules downstream of the TCR.13–15 In healthy cells, robust PD-1 upregulation in response to self-antigens and its signaling via PD-1/PD-L1 interactions contributes substantially to the early cell fate decisions of CD8+ T lymphocytes between tolerance and differentiation into cytotoxic T cells (CTL).16 In cancer, PD-1/PD-L1 triggering facilitates upregulation of additional receptors that limit T cell responses, as well as cytosolic molecules with inhibitory functions.1 Identifying and targeting key regulatory switches could help to reverse these loops, improving the response to existing therapies.
Anergy and exhaustion are two distinct metabolic T cell states imposed by different mechanisms,17 but both depend on reduced Ras activation and on facilitated transcription of nuclear factor of activated T cells (NFAT)-regulated genes.18–20 NFAT-dependent transcription in the absence of activator protein-1 (AP-1) facilitates expression of negative receptors such as TIM3 or LAG3 (18–20), and also upregulates that of E3 ubiquitin ligases, phosphatases, and diacylglycerol (DAG) kinases (DGK), that act at different steps to further limit activation of the pathways that ultimately trigger AP-1 transcription.21 DGKα and ζ isoforms, abundantly expressed in naïve T cells, act as negative regulators of Ras activation downstream of TCR and costimulatory receptors.22 DGKα and ζ are upregulated in tolerant T lymphocytes,23 24 and their abundance in TIL and engineered CAR T cells correlates with hyporesponsive, anergic states that limit cytotoxic functions.25 26 Compared with DGKα, DGKζ has additional functions that limit the PKCθ/PDK-1/AKT axis, that in turn regulates nuclear factor κ-light-chain-enhancer of activated B cells (NFκB) activation and mTOR metabolic control downstream of CD28 activation.27 DGKζ-deficient mice show partial but clear resistance to orthotopically implanted tumors,28–30 suggesting that DGKζ inhibition help to re-invigorate exhausted TIL and engineered CAR T cells.31 32
In spite of its potential therapeutic value, there is limited information on the effects of DGKζ targeting in human T cells, partially as a result of the lack of isoform-specific inhibitors.31 32 Here, we used the triple parameter reporter (TPR) cell line33 to investigate the effects of short hairpin interfering RNA (shRNAi)-mediated silencing of DGKζ on the regulation of the T cell transcriptional program in conditions of full stimulation or PD-1-dependent inhibition. We confirm that DGKζ silencing promotes robust NFκB and AP-1 activation and identify its contribution to NFAT-regulated transcription. Depletion of DGKζ does not fully restore PD-1-mediated inhibitory actions in TPR cells that constitutively bear this negative receptor, but greatly limits the extent of PD-1 expression at the early stages of T cell responses. This control of PD-1 expression by DGKζ is conserved in mouse CD8+ T cells, and explains the reduced ability of MC38 colon adenocarcinoma cells to induce tumors when engrafted in DGKζ-deficient animals. Our experiments suggest that genetic, and probably pharmacological, manipulation of DGKζ alter CD8+ T cell fate, tipping the balance between effector and tolerant/exhausted cells in the tumor milieu. These data strongly support the therapeutic potential of DGKζ and the need for isoform-specific inhibitors to add to current immunotherapy tools.
Methods
Antibodies and reagents
For pharmacological stimulation, we used phorbol 12-myristate 13-acetate (PMA; Sigma) and ionomycin (Calbiochem). For antibody (Ab)-based stimulation, we used mouse antihuman or hamster antimouse monoclonal anti-CD3 and -CD28 Ab (555336, 555725, 553058, 553295; all from BD Pharmingen). For flow cytometry analyzes, we used LIVE/DEAD fixable red cell death staining kit (L23102, Invitrogen) and antimouse CD45-APC (L23102, eBioscience) or Percp-Cy5.5, CD8-BV610 and CD25-PCy7 (103132, 100744, and 102016, respectively; all from Biolegend), TCRβ-FITC (732247, Beckman Coulter) and PD-1-APC780 (47998582, eBioscience), antihuman PD-1-APC (17-9969-42, eBioscience) and CD25-PE (A07774, Beckman Coulter) and paraformaldehyde (TedPella). For western blot analysis, we used anti-DGKα (11 547–1-AP, ProteinTech), DGKζ (105195, Abcam), α tubulin (T5168, Sigma), GAPDH (G-9, Santa Cruz), horseradish peroxidase-conjugated antimouse and rabbit IgG (P0447, P0260, Dako), antirabbit IgG Dylight 800 (SA5-35571, Invitrogen) and mouse IgG AlexaFluor 680 (175775, AbcamLife). IKKβ inhibitor PS-1145 was from Sigma-Aldrich, MEK inhibitor PD98059 was from Calbiochem, and calcineurin (CaN) inhibitor FK506 was from Merck Millipore. Anti-PD-1 (nivolumab) humanized Ab was from BioVision. For TIL isolation, we used collagenase type I (Worthington), dispase II and DNase I (both from Roche). Trypsin was from Biowest.
Cell lines, culture and stimulation
Human Jurkat JE6.1-derived TPR cells, transduced to express NFAT-GFP, NFκB-CFP (cyan fluorescent protein) and AP-1-Cherry, were generated and selected as reported.33 TPR cells retrovirally transduced with vectors encoding PD-1 were described.33 Human leukemic Jurkat cells were from ATCC. T cell stimulator (TCS) cells, a murine thymoma cell line (Bw5147) engineered to stably express an antihuman CD3 single-chain fragment anchored to the cell membrane via a human CD14 stem, have been described.34 TCS cells expressing no human costimulatory molecule were used as control TCS (TCS-control) and compared with cells retrovirally transduced to express human CD86 (TCS-CD86), either alone or in the presence of PD-L1 (TCS-CD86/PD-L1). Murine MC38 adenocarcinoma cell line was a kind gift from Prof. Santos Mañes (CNB-CSIC). All cells were maintained in complete RPMI medium (cRPMI) consisting of RPMI (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen), 1% HEPES (Sigma), 2 mM L-glutamine and penicillin and streptomycin (100 U/mL; Biowest) (37°C, 5% CO2).
C57BL/6J-DGKζ-deficient mice were reported previously.35 Lymph nodes were isolated from 6 to 12 weeks mice. Cells were maintained in cRPMI supplemented with 50 mM β-mercaptoethanol (Merck) (37°C, 5% CO2).
For pharmacological/Ab-based stimulation, TPR cells (5×104) in 50 µL were stimulated in cRPMI medium with PMA (50 µM) and ionomycin (1 µM; Sigma-Aldrich) for indicated times, or with anti-CD3/CD28 mAb (1 µg/mL each) for 24 hours.
For experiments with mouse cells, T lymphocytes were isolated from lymph nodes according to standard protocols and maintained in complete medium throughout the assay. Cells (2.5×106) were cultured on anti-CD3 mAb-coated plates with medium containing anti-CD28 mAb (72 hours, 37°C).
For stimulation with TCS cells, TPR or Jurkat cells (5×104) were cocultured with TCS cells (2×104) in 100 µL medium in a 96-well round-bottom plate for the indicated times (37°C, 5% CO2). Where indicated, 30 µM PS-1145 (IKKβ inhibitor), 50 µM PD98059 (MEK inhibitor), 20 nM FK506 (CaN inhibitor) or 0.5 µg/mL anti-PD-1 (nivolumab) was added.
Flow cytometry analysis
TPR/TCS or Jurkat/TCS cocultures were stained with anti-mouse CD45-APC or Percp-Cy5.5 to exclude TCS cells from analysis. Live cells were gated using forward and side scatter parameters within the CD45- subset. Expression of NFAT-GFP, NFκB-CFP and AP-1-Cherry, was determined by flow cytometry analysis of the live cell population using a CytoFLEX S flow cytometer (Beckman Coulter). Mean and SD of the geometric mean fluorescence intensity (gMFI) of the whole population and the percentage of positive cells were determined from triplicate wells. Transcription factor activity was calculated by normalizing data to the maximal activation for each experiment.
Jurkat and LN mouse T cells were washed after stimulation and stained with LIVE/DEAD fixable red cell death staining kit, and stained with a mixture of Ab. Cells isolated from tumors were subjected to the same procedure, in this case CD45 staining was used to exclude MC38 cells from analysis. CD8+ T cells were identified among total T cells (CD45+ TCR+) and the expression of CD25 and PD-1 evaluated with the corresponding Ab.
Data were analyzed using FlowJo software (V.10.2, FlowJo LLC, Ashland, Oregon, USA).
Western blot analysis
Cells were lysed and processed as in27 and western blots probed with specific antibodies. Protein bands were visualized by enhanced chemiluminescence (ECL detection, Amersham Bioscience) or with an Odyssey scanner (LI-COR). Densitometric analysis of proteins was performed using ImageJ.
Plasmids and transfections
TPR or Jurkat cells in logarithmic growth (4–5 x 105 cells/mL) were transfected with 15–20 µg plasmid DNA by electroporation using Gene Pulser II (BioRad) (270 mV, 975 µF). For DGKζ silencing experiments, a validated sequence for the DGKζ isoform was selected to generate the shRNAi pSUPER construct.27 Equivalent murine sequences were cloned and used as negative controls.36 Expression was evaluated from days 1 to 4 post-transfection by western blot. Plasmids encoding the interleukin 2 (IL-2) promoter region were reported.23 The IL-2 promoter was obtained from Addgene. Renilla luciferase vector was from Promega.
Dual-luciferase reporter assay
TPR cells were transfected with 15 µg of the IL-2 promoter construct, with 200 ng renilla as internal control. After 24 hours, cells were washed, allowed to recover (6 hours) and stimulated using TCS cells for 24 hours (37°C, 5% CO2). Cells were harvested and assayed for luciferase activity using the Dual-Luciferase Reporter assay (Promega). Luciferase activity was reported relative to renilla luciferase activity.
IL-2 detection in culture supernatants
Supernatants from TPR/TCS cocultures were collected and IL-2 levels measured using the ELISA MAX Deluxe Set Human IL-2 kit (Biolegend).
Tumor experiments and TIL isolation
MC38 cells were trypsinized from subconfluent monolayers, washed, and injected subcutaneously in 100 µL (5×105) into one flank of female 6- to 8-week-old C57BL/6J wild type (wt) or DGKζ-deficient mice. Tumor growth was monitored in a blind manner with calipers, and area and volume were estimated according to the formulas: area=a x b/volume = (a2 x b)/2, where a is tumor width and b is tumor length in mm. Mice were sacrificed when wt tumors reached 1 cm3, at ~19 days postinjection, and tumors were excised, measured and weighed.
For TIL isolation, tumors were fragmented into 1 mm3 pieces using a scalpel. Fragments were suspended in DMEM culture medium (Invitrogen) supplemented with 20 mM HEPES, with 2 mg/mL collagenase type I, 2.5 mg/mL dispase II and 0.1 mg/mL DNase I, and incubated with gentle shaking (15 min, 37°C). The resulting suspension was filtered with a 70 µm filter, washed with PBS+5% FBS and centrifuged (5 min, 300 X g, 4°C). Resulting pellets were processed for flow cytometry analysis.
Statistical analysis
Flow cytometry data were analyzed with GraphPad Prism V.6 software. Data are shown as mean±SEM Samples were assumed to fit normality. When more than two conditions were analyzed, we applied analysis of variance and Bonferroni post-test analysis. If not applicable, parametric unpaired t tests were performed. In all cases, differences were considered statistically not significant (ns) for p>0.05, and significant for p values *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Results
The TPR cell model is a useful cell platform to examine the contribution of DAG-regulated signals to functional T cell activation
The TPR cell model allows the concurrent flow cytometry analysis of NFAT, NFκB and AP-1 transcriptional activation.33 These three transcription factors classically represent the end-point activation of Ca2+-dependent CaN activation, as well as of Ras/extracellular signal-regulated kinase (ERK)- and protein kinase C (PKC) θ/ΙκΒ kinase (IKK) β-regulated pathways. Flow cytometry analysis of fluorescent proteins coupled to transcription factors enables simultaneous quantification of the signal intensity as determined by the reporter gene induction on a per cell basis (gMFI). The percentage of responding cells reflects the digital characteristics of TCR-delivered signals that ensures scaled T cell responses according to dose and affinity for the antigens encountered.37 Stimulation of TPR cells with phorbol 12-myristate 13-acetate (PMA) and the Ca2+ ionophore ionomycin evidenced a strong, uniform cell response with distinct kinetics for the different reporters (figure 1A). The early, robust NFAT induction correlated with its direct nuclear entry as the result of its CaN-dependent dephosphorylation.38 The induction of NFκB or AP-1, which require successive activation of small GTPases and kinases, accumulated over time (figure 1A).
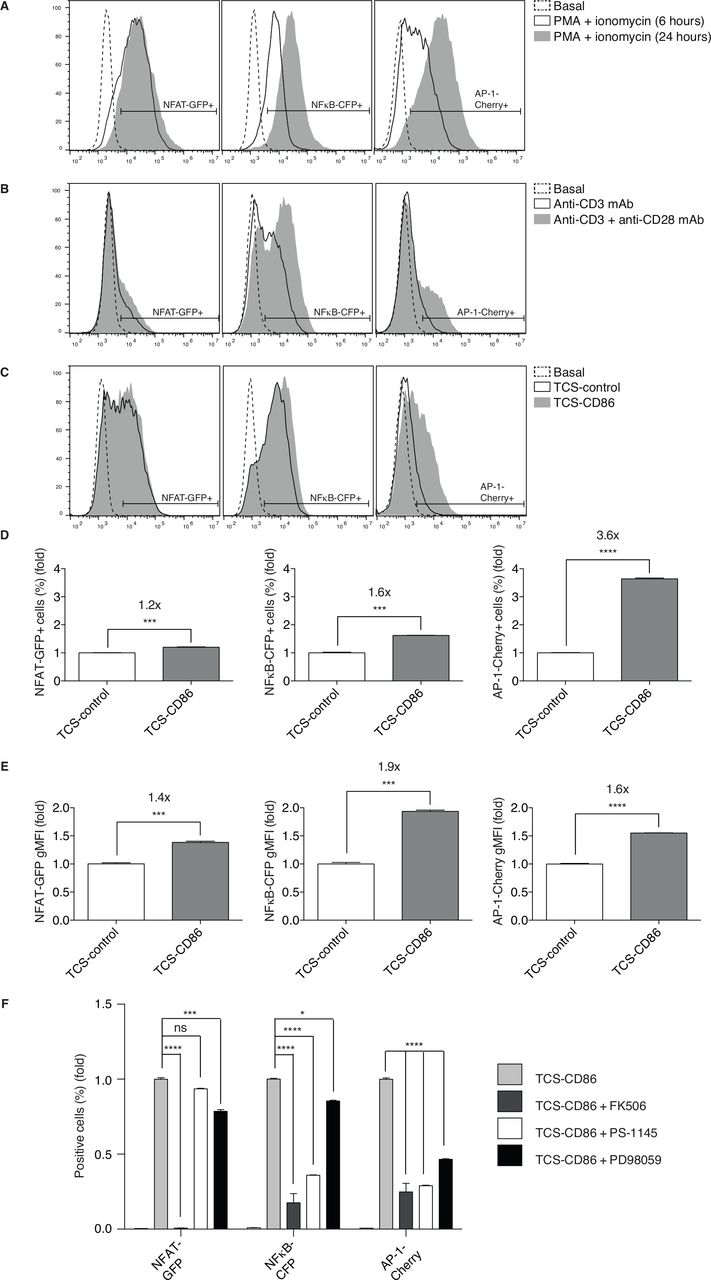
Functional evaluation of the TPR cell model in response to pharmacological and physiological stimuli. (A–C) NFAT-GFP (left), NFκB-CFP (middle) or AP-1-Cherry (right) induction was analyzed. (A) TPR cells were stimulated using PMA and ionomycin for the indicated times. (B) TPR cells were stimulated using anti-CD3 or anti-CD3/28 mAb for 24 hours. (C) TPR cells were stimulated using TCS-control or TCS-CD86 cells for 24 hours. (D, E) Fold induction of response to TCS-CD86 cells. NFAT-GFP (left), NFκB-CFP (middle) or AP-1-Cherry (right) expressing cell percentage (D) or geometric mean fluorescence intensity (gMFI) (E) was analyzed. TCS-CD86/TCS-control ratios are shown above the graphs. Values are normalized to the TCS control-mediated stimulation condition=1.0. Data were analyzed using parametric unpaired t test; ***p<0.001, ****p<0.0001. (F) Fold induction of response to CaN (FK506), IKKβ (PS-1145) or MEK (PD98059) inhibition in TCS-CD86-stimulated TPR cells. NFAT-GFP, NFκB-CFP or AP-1-Cherry expressing cell percentage was analyzed. Values are normalized to the TCS-CD86-mediated stimulation condition in the absence of inhibitors=1.0. Data were analyzed using two-way ANOVA and Bonferroni post-test; ns *p>0.05, ***p<0.001, ****p<0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; AP-1, activator protein-1; NFAT, nuclear factor of activated T cells; NFκB, nuclear factor κ B cells; ns, not significant; TCS, T cell stimulator; TPR, triple parameter reporter.
At difference from the uniform response induced by pharmacological stimulation, 24 hours treatment of TPR cells with agonistic mAb to the TCR-CD3 complex resulted in a small percentage of NFAT-positive and AP-1-positive cells but a higher proportion of NFκB activation (figure 1B). Addition of anti-CD28 mAb increased the activity of the three reporters (figure 1B), especially that of NFκB and AP-1 (figure 1B, middle and right), in accordance with the specific contribution of DAG-regulated pathways downstream of CD28 costimulation.23
As described,33 TPR coculture with control TCS cells (TCS control) promoted strong induction of NFAT and NFκB, with lower AP-1 induction (figure 1C). CD28 stimulation with CD86-expressing TCS cells (TCS-CD86), as shown for the CD80 ligand,33 further increased expression of the three reporters. Individual transcription factor analysis revealed maximal increase in the frequency of AP-1-expressing cells after costimulation (figure 1D). The effect of CD28 stimulation on a per cell basis was also observed for the three transcription factors, with maximal effect for NFκB expression (figure 1E). Bi-parametric analyzes confirmed a marked increase in the percentage of NFAT/AP-1-expressing cells following incubation with TCS-CD86 cells (online supplemental figure 1). These results add new information to existing data on this model, showing that it accurately reproduces the strict requirement for CD28 engagement to reach the AP-1 transcription threshold.
Supplemental material
The activation of CaN in response to Ca2+ not only triggers NFAT nuclear translocation but also cooperates with PKCθ in NFκB and AP-1 regulation.39 In agreement, CaN inhibitor FK506 treatment prevented induction of the three transcription factors (figure 1F). NFAT transcription was independent on the PKCθ/IKKβ signals, as demonstrated by the minimal effect of the IKKβ inhibitor PS-1145. Pharmacological IKKβ blockade nonetheless markedly diminished NFκB and AP-1 activation, in accordance with results from primary human T cells40 (figure 1F). MEK inhibition by PD98059 limited AP-1 transcription but also partially decreased NFAT and NFκB induction (figure 1F), which confirmed the reported role for MEK, an intermediate kinase of the Ras/ERK axis, in the activation of these two transcription factors.41 42 Overall, these results confirm the critical requirement for the Ca2+/CaN axis, and reveal AP-1 transcriptional activation as the sensor most sensitive to signal limitation.
DGKζ limits transcriptional activation in TPR cells
Biochemical studies combined with luciferase-based assays demonstrate that DGKζ silencing in Jurkat T cells leads to enhanced Ras/ERK and PKCθ/AKT pathway activation, which translates into enhanced NFκB transcription.27 The TPR model allowed us to examine the result of DGKζ silencing on the simultaneous activation of NFAT, NFκB and AP-1. We used previously validated shRNAi sequences to attenuate DGKζ expression in TPR cells. DGKζ targeting achieved similar extent of silencing than that reported in Jurkat T cells,27 without affecting that of DGKα (figure 2A and online supplemental figure 2). Flow cytometry analysis of DGKζ-silenced TPR cells at different times after coculture with TCS-control or TCS-CD86 cells showed an enhanced percentage of NFAT-, NFκB- and AP-1-positive cells that was detected as early as 4–6 hours poststimulation (figure 2B). The effect of DGKζ silencing varied for each transcription factor, with increased NFAT and AP-1 induction at early times that contrasted with the sustained, enhanced activation of NFκB throughout the kinetic course. Analysis on a per cell basis showed no major effect of DGKζ depletion for NFAT induction (figure 2C, left), but exhibited increased NFκB and AP-1 expression (figure 2C, middle and right). As also reported for mAb-stimulated Jurkat cells,27 43 DGKζ silencing did not replace the CD28 requirement for AP-1 induction (figure 2B–C, right). Analysis of the percentage of cells that coexpressed the three reporters confirmed a marked elevation in DGKζ-silenced cells in response to TCR alone or to TCR/CD28 stimulation (figure 2D). Our results illustrate the utility of fluorescent protein-based TPR cells to demonstrate that, as shown in mice, DGKζ potently limits the threshold of responding T cells.
Supplemental material
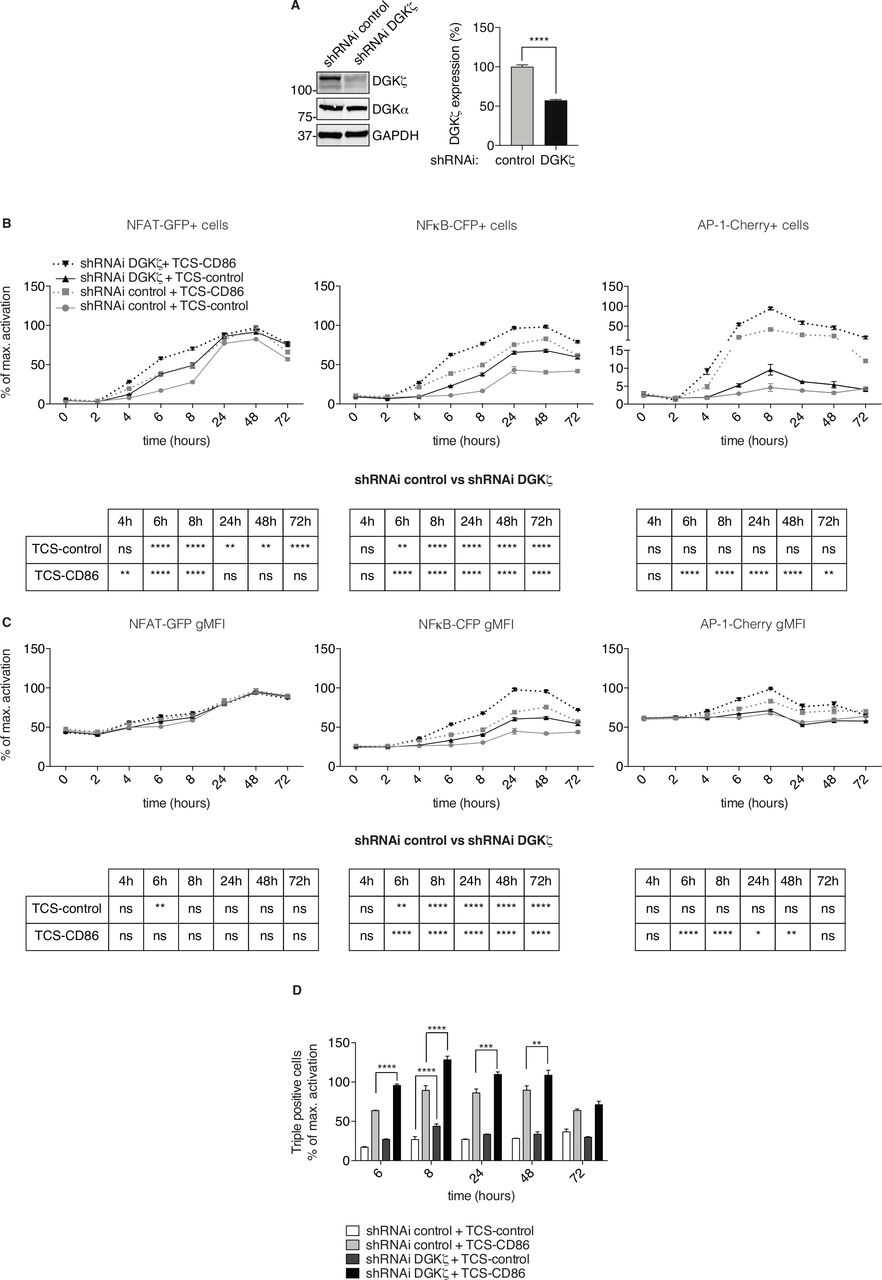
Effect of DGKζ silencing on the transcription factor activity of NFAT, NFκB and AP-1. (A) DGKζ was silenced in TPR cells and silencing confirmed by western blot (left), quantification of DGKζ expression in control and silenced TPR cells was determined in three independent experiments (right). Control and DGKζ-silenced TPR cells were stimulated with TCS-control or TCS-CD86 cells for the indicated times. NFAT-GFP (left), NFκB-CFP (middle) or AP-1-Cherry (right) expressing cell percentage (B) or geometric mean fluorescence intensity (gMFI) (C) was analyzed. (D) Control and DGKζ-silenced TPR cells were stimulated with TCS control or TCS-CD86 cells for the indicated times and the percentage of cells simultaneously expressing the three reporters was analyzed. Transcription factor activity was calculated by normalizing data to the maximal activation. Data were analyzed using two-way ANOVA and Bonferroni post-test; ns *p>0.05, **p<0.01, ***p<0.001, ****p<0.0001. The analysis of the experiments represented in B and C, are summarized in the tables below each figure. Results are representative of at least three independent experiments with similar results. AP-1, activator protein-1; DGK, diacylglycerol kinases; NFAT, nuclear factor of activated T cells; NFκB, nuclear factor κ B cells; ns, not significant; shRNAi, short hairpin interfering RNA; TCS, T cell stimulator; TPR, triple parameter reporter.
PD-1 triggering restricts transcriptional regulation and IL-2 secretion in TPR cells
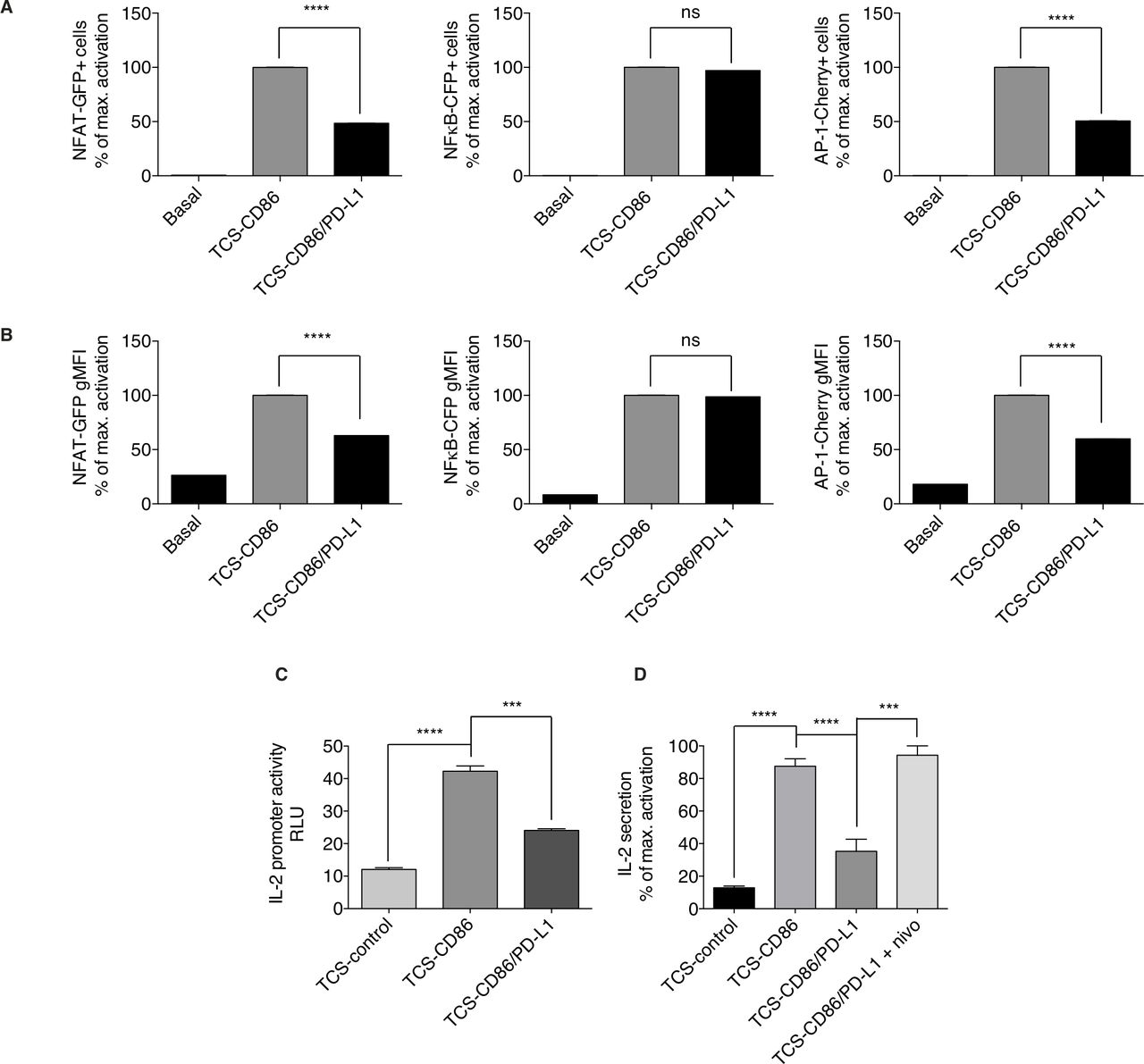
Previous studies used the TPR system to demonstrate that PD-1 ligation results in strong NFAT/NFκB downregulation with a minimal effect on AP-1.33 We used the same PD-1-expressing TPR (TPR-PD-1) cells, but here we incubated them with TCS cells expressing CD86 alone or in combination with PD-L1 (TCS-CD86/PD-L1) to study the effect of PD-1 ligation in the presence of costimulatory signals. At difference from the effect reported for TCR single stimulation, PD-1 triggering with CD28 costimulation reduced the percentage (figure 3A) and the expression (figure 3B) of NFAT and AP-1, but had no effect on NFκB activation. These results coincide with the inhibition of TCR-driven early signals by PD-1 engagement as well as with studies showing that, in human T lymphocytes, CD28 promotes NFκB activation in the absence of TCR triggering.44

Effect of PD-1/PD-L1 triggering on transcriptional regulation and secretion of IL-2. (A, B) PD-1 expressing TPR (TPR-PD-1) cells were stimulated using TCS-CD86 or CD86/PD-L1 bearing TCS (TCS-CD86/PD-L1) cells for 24 hours. NFAT-GFP (left), NFκB-CFP (middle) or AP-1-Cherry (right) expressing cell percentage (A) or geometric mean fluorescence intensity (gMFI) (B) was analyzed. Transcription factor activity was calculated by normalizing data to the maximal activation. (C) Luciferase activity of an IL-2 reporter construct was determined in TPR-PD-1 cells stimulated with TCS-control, TCS-CD86 or TCS-CD86/PD-L1 cells for 24 hours. Luciferase activity was corrected using an internal renilla luciferase control. (D) IL-2 secretion to medium was assessed in supernatants of TPR-PD-1 cells stimulated with TCS-control, TCS-CD86 or TCS-CD86/PD-L1 cells for 24 hours. When indicated, a humanized anti-PD-1 antibody (nivolumab (nivo)) was added. IL-2 secretion was calculated by normalizing data to the maximal activation. Data were analyzed using parametric unpaired t-test (A, B) or one-way ANOVA and Bonferroni post-test (C, D); NS ***p<0.001, ****p<0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; IL-2, interleukin 2; NFAT, nuclear factor of activated T cells; NFκB, nuclear factor κ B cells; ns, not significant; PD-1/PD-L1, programmed cell death-1/ligand 1; TCS, T cell stimulator; TPR, triple parameter reporter.
Correct and timely coordinated activation of NFAT, NFκB and AP-1 is indispensable for transcription of IL-2, which orchestrates the ensuing clonal expansion of effector populations.45 Analysis of a luciferase-coupled IL-2 promoter in TPR-PD-1 cells demonstrated the need for CD28 stimulation, and confirmed the PD-1-driven limitation of IL-2 transcription (figure 3C). Determination of IL-2 secretion by stimulated cells fully reflected the observations in the promoter experiments; only those cells stimulated by TCS-CD86 cells showed robust secretion of this cytokine, which was greatly restricted by PD-1/PD-L1 ligation (figure 3D). These results confirm a strict requirement for CD28 triggering to reach the correct AP-1 threshold, which acts in concert with NFAT and NFκB,46 47 and the limitation on IL-2 production imposed by PD-1 engagement, even in the presence of costimulatory signals.48 TPR-PD-1 cells stimulated with TCS-CD86/PD-L1 cells restored IL-2 production after incubation with an anti-PD-1 humanized Ab (nivolumab, nivo) (figure 3D). Our analyzes confirm the utility of the TPR cell model to reproduce the specific PD-1 contribution to T cell hyporesponsive states, as well as the effect of blocking antibodies used in the clinic.
DGKζ contributes to PD-1 inhibitory effects on AP-1-dependent transcription
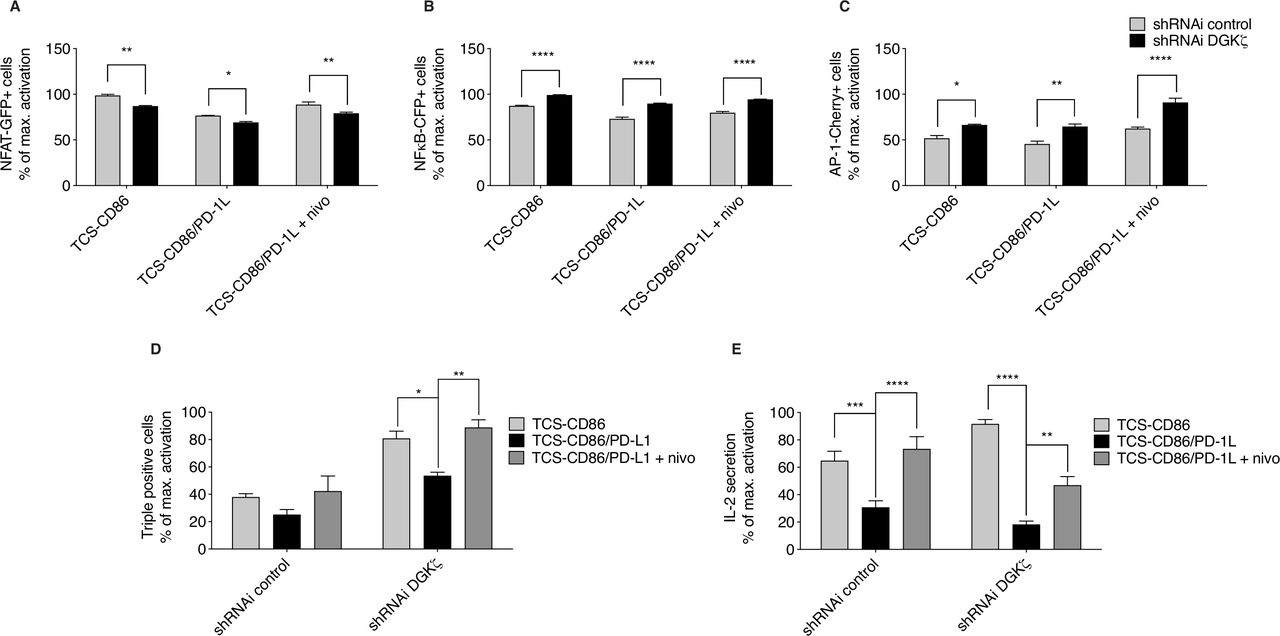
We investigated the effect of DGKζ silencing in the context of PD-1 inhibitory function. As observed in TPR cells, DGKζ silencing in TPR-PD-1 cells increased the frequencies of NFκB- and AP-1-expressing cells, but diminished that of NFAT, compared with control cells (figure 4A–C). PD-1 ligation decreased NFAT transcription both in control and DGKζ-silenced cells (figure 4A), and had no obvious effect on NFκB induction (figure 4B). Nivolumab-mediated blockade of PD-1/PD-L1 interaction restored NFAT in control and DGKζ-silenced cells (figure 4A). Compared with control cells, DGKζ-silenced cells showed less sensitivity to PD-1-dependent AP-1 inhibition and displayed further induction of AP-1 following nivolumab treatment (figure 4C). Although the effects of DGKζ silencing were small in the analysis of each independent transcription factor, the evaluation of percentage of TPR-PD-1 cells that coexpressed the three reporters confirmed a significantly higher proportion of responding cells following DGKζ silencing, which nonetheless decreased after PD-1 triggering and was fully restored by nivolumab treatment (figure 4D). The biological significance of these changes was reflected by the analysis of IL-2 secretion, which confirmed enhanced IL-2 production by DGKζ-silenced TPR-PD-1 cells (figure 4E). DGKζ silencing did not confer resistance to PD-1 engagement, resulting in severe impairment of IL-2 secretion, with partial but significant recovery following nivolumab addition (figure 4E).
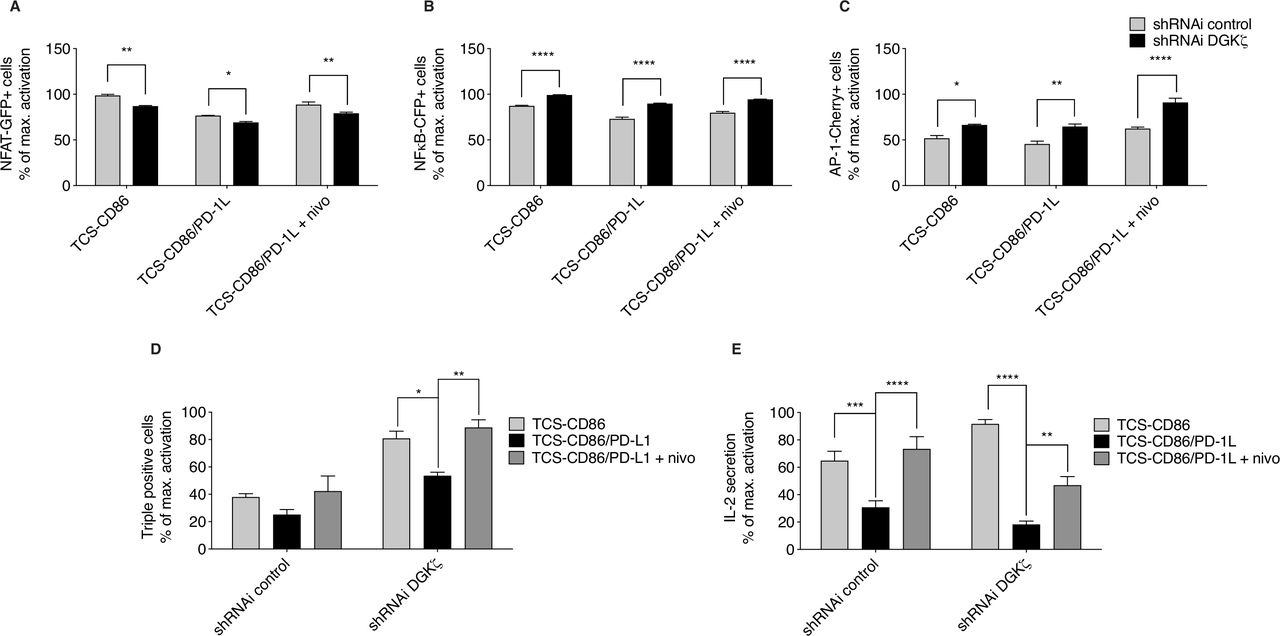
Effect of DGKζ silencing on the transcriptional restrictions imposed by PD-1/PD-L1 triggering. (A–D) Control or DGKζ-silenced TPR-PD-1 cells were stimulated with TCS-CD86 or TCS-CD86/PD-L1 cells for 24 hours. (A–D) NFAT-GFP (left), NFκB-CFP (middle) or AP-1-Cherry (right), or triple expressing cell percentage (D) was analyzed. Transcription factor activity was calculated by normalizing data to the maximal activation. (E) IL-2 secretion to medium was assessed in supernatants of control or DGKζ-silenced TPR-PD-1 cells stimulated with TCS-CD86 or TCS-CD86/PD-L1 cells for 24 hours. When indicated, a humanized anti-PD-1 antibody (nivolumab (nivo)) was added. IL-2 secretion was calculated by normalizing data to the maximal activation. Data were analyzed using one-way ANOVA and Bonferroni post-test; *p<0.5, **p<0.01, ***p<0.001, ****p<0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; DGK, diacylglycerol kinases; IL-2, interleukin 2; NFAT, nuclear factor of activated T cells; NFκB, nuclear factor κ B cells; ns, not significant; PD-1/PD-L1, programmed cell death-1/ligand 1; shRNAi, short hairpin interfering RNA; TCS, T cell stimulator; TPR, triple parameter reporter.
DGKζ promotes PD-1 expression by limiting IL-2 in activated T cells
The previous experiments demonstrate the utility of the TPR model for studying the contribution of DGKζ-regulated pathways in the activation of positive and negative receptors in human T cells, and suggest that this platform could be used to profile isoform-specific DGK inhibitors. TPR cells are nonetheless an artificial system in which receptors can be analyzed at a fixed level of expression. Naïve T cells do not constitutively express PD-1, which is induced following activation.49 PD-1-mediated inhibition depends on TCR signal strength, with greater inhibition at low levels of TCR stimulation.50 In line with this fact, PD-1-delivered inhibition can be overcome by CD2851 or IL-2.48 IL-2 production in response to CD28 engagement is central to CD28 ability to rescue from PD-1-driven negative regulation.48 IL-2 leads to cell expansion by ensuring expression of CD25, which constitutes the high affinity receptor for this cytokine. We studied the kinetics of CD25 and PD-1 induction in Jurkat T cells stimulated with PMA and ionomycin or TCS-CD86 cells. As shown, both proteins were rapidly induced as early as 6 hours in response to both stimuli (figure 5A–C). In agreement with IL-2-dependent regulation of both proteins,52 by 24 hours CD25 expression was further increased, whereas PD-1 expression was reduced (figure 5A–C).
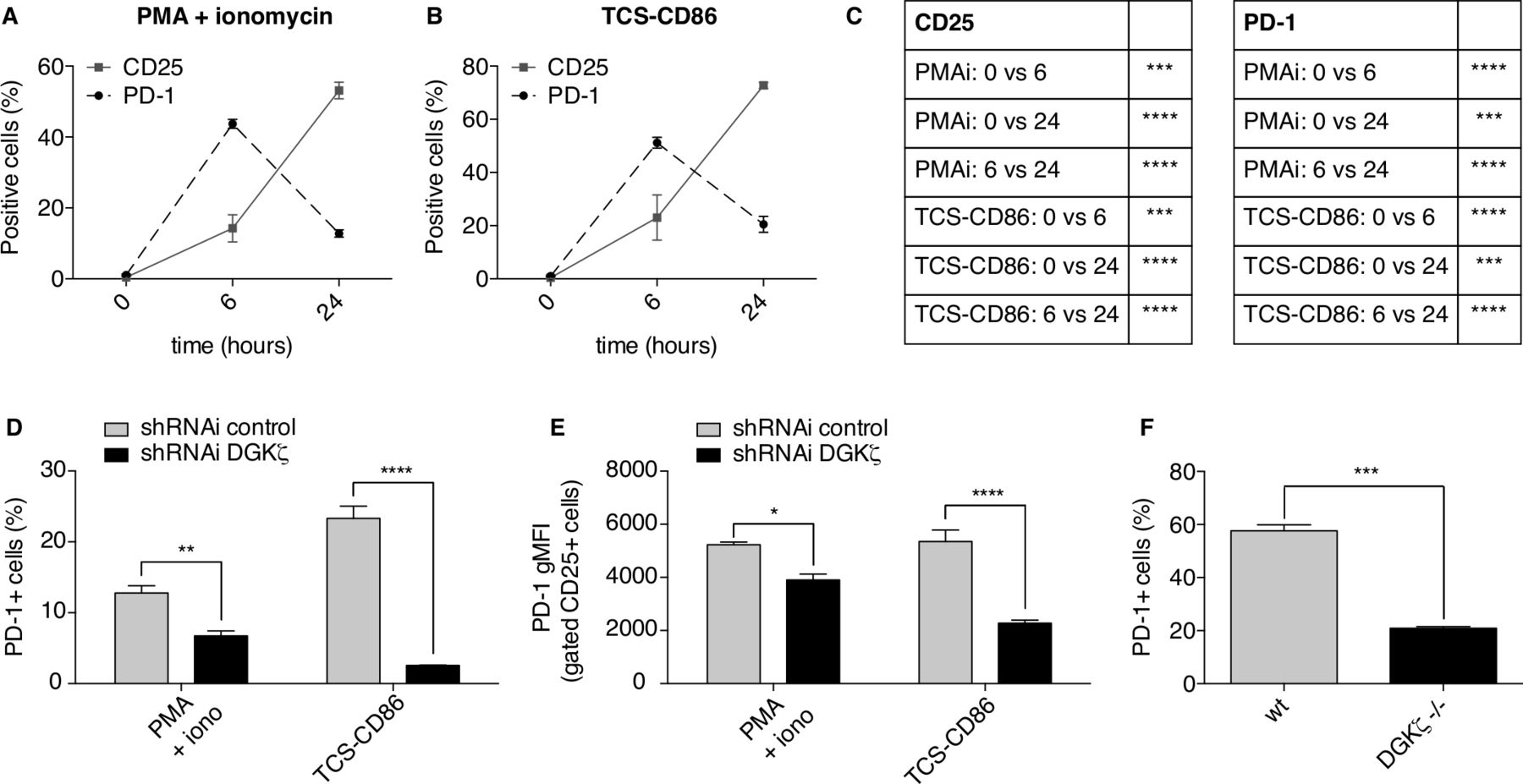
Stronger IL-2-dependent signals in DGKζ-deficient CD8+ T cells limit PD-1 expression. (A–C) Parental Jurkat cells were stimulated with PMA and ionomycin (PMAi) (A) or TCS-CD86 (B) for the indicated times. CD25 or PD-1 expressing cell percentage was analyzed. In C, the results of the comparisons are summarized in the table. D–E. DGKζ was silenced in Jurkat cells. Control or DGKζ-silenced Jurkat cells were stimulated with PMA and ionomycin (iono) or TCS-CD86 for 24 hours. PD-1 expressing cell percentage (D) or gMFI within CD25+ cells (E) was analyzed. (F) wt or DGKζ-deficient primary CD8+ T cells were stimulated with plate-bound anti-CD3 mAb and soluble anti-CD28 mAb for 72 hours. PD-1 expressing cell percentage was analyzed. Data were analyzed using two-way ANOVA and Bonferroni post-test (A–E) or parametric unpaired t test (F); *p<0.1, **p<0.01, ***p<0.001, ****p<0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; DGK, diacylglycerol kinases; gMFI, geometric mean fluorescence intensity; IL-2, interleukin 2; PD-1, programmed cell death-11; shRNAi, short hairpin interfering RNA; TCS, T cell stimulator; wt, wild type.
Concurring with its function as a negative regulator of CD28 signals, DGKζ silencing further reduced the percentage of PD-1+ Jurkat cells (figure 5D). The decrease in PD-1 expression was particularly marked in the CD25+ T cell population (figure 5E). Consistent with observations in the Jurkat model, stimulation of mouse T cells from DGKζ-deficient mice also led to a notable reduction in the percentage of PD-1+ CD8+ T cells compared with wt mice (figure 5F). These results indicate conservation in human and mouse T lymphocytes of a positive feedback by which DGKζ promotes PD-1expression and negative function.
DGKζ deficiency reduces in vivo growth of MC38 colon adenocarcinoma cells, a PD-1-related tumor model
Antigen recognition facilitates PD-1 upregulation in TIL, whereas PD-L1 expression by tumor or stromal cells increases the probability of PD-1 ligation.10 This leads to a scenario in which IL-2 production is reduced and the expansion of CD8+ CTL populations is limited.13 Our previous observations showing a correlation between enhanced IL-2 production and reduced PD-1 expression as a result of DGKζ depletion might be a plausible mechanism to explain the enhanced control of engrafted tumors observed in DGKζ-deficient mice.28–30 Although the Jurkat cell line is a good platform in which to study cytokine production, these cells lack cytotoxic abilities. We, thus, moved to mouse models that facilitate the examination of the DGKζ modulation of antitumor T cell functions.
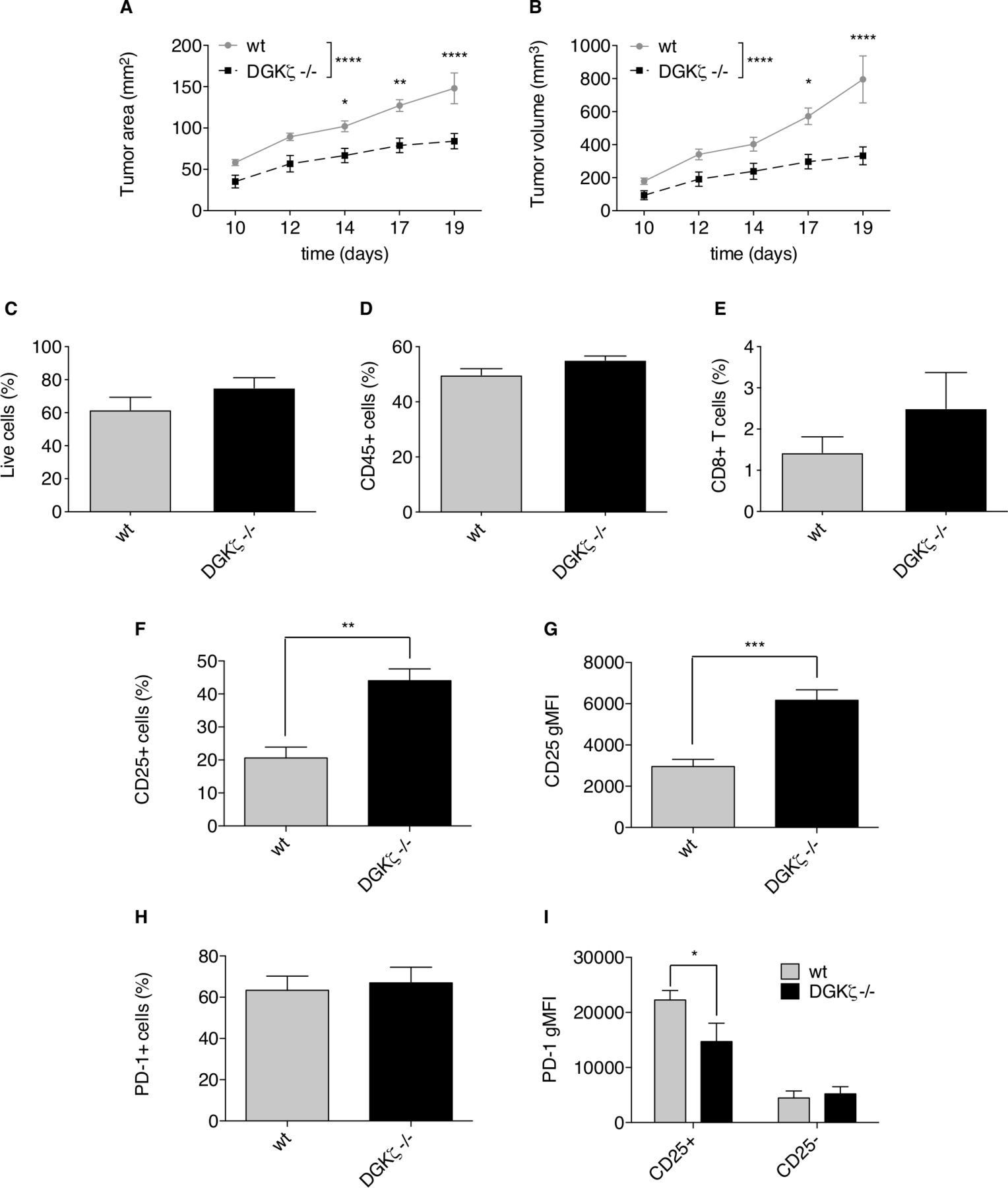
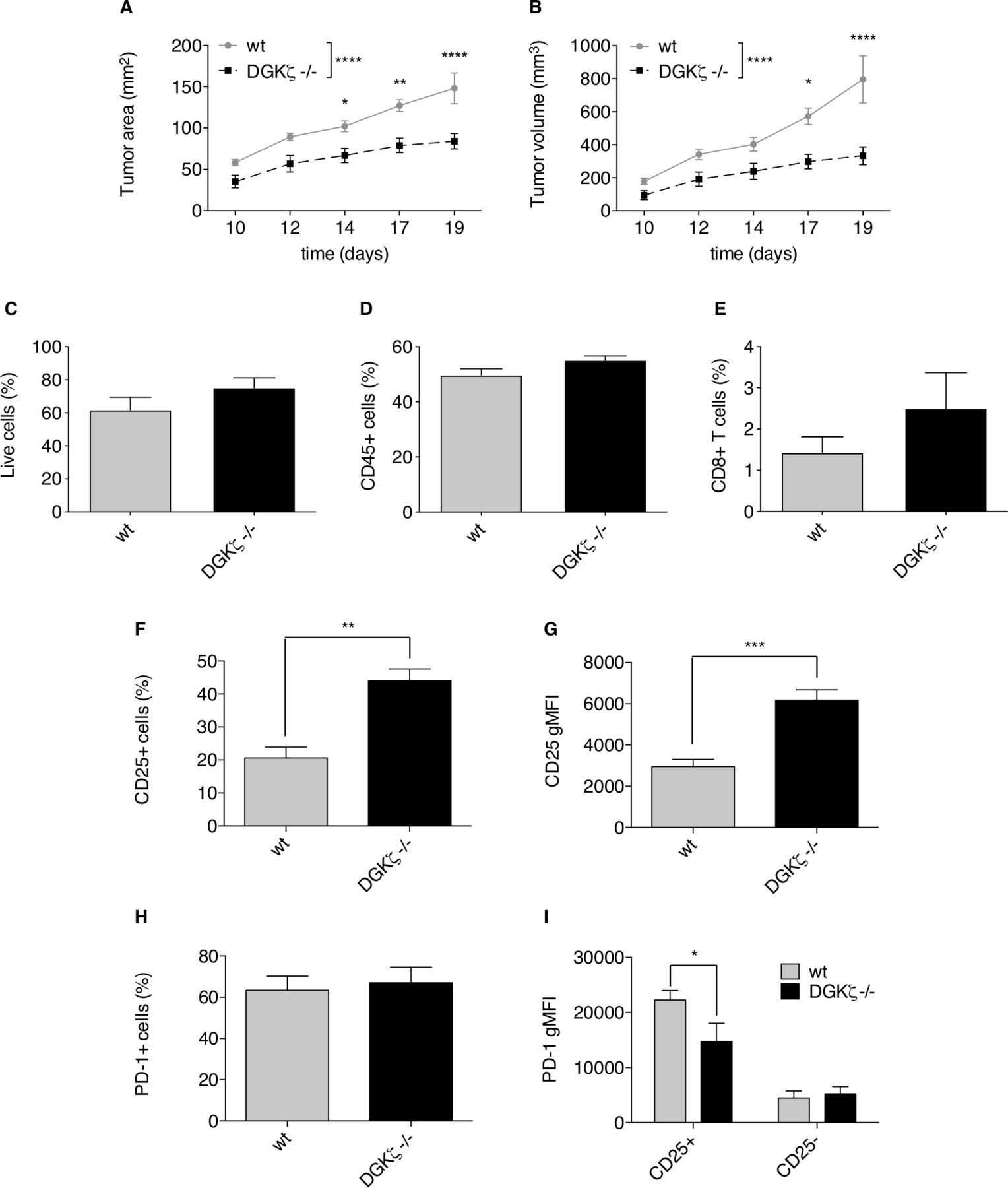
At difference from findings in human colorectal tumors, which are in most cases PD-1-resistant, the induction of orthotopic tumors in mice by subcutaneous injection of MC38 adenocarcinoma cells is a classical model of sensitivity to PD-1/PD-L1 immune checkpoint blockade.53 We selected this tumor model to test whether the observed modulation of the CD25/PD-1 axis by DGKζ could affect tumor development. Analysis of growth kinetics of MC38 engrafted cells showed a notable growth lag in DGKζ-deficient mice, which developed considerably smaller tumors than those in wt animals by the time at which they had to be sacrificed (figure 6A–B). We explored existing differences in CD25+ and PD-1+ CD8+ T cell populations in TIL isolated from tumors implanted in wt and DGKζ-deficient mice. TIL analysis showed similar percentage of live (figure 6C) and CD45+ cells (figure 6D) with a tendency, not statistically significative, of higher CD8+ T cell percentage in TIL isolated from tumors in DGKζ-deficient mice (figure 6E). TIL isolated from tumors grown in DGKζ-deficient mice showed a significant increase in CD25+ CD8+ T cell frequency (figure 6F) and in CD25 expression per cell (figure 6G) compared with those from wt mice. TIL analysis showed no marked differences in the percentage of PD-1+ CD8+ T cells (figure 6H) but, in agreement with findings in Jurkat and mouse T cells, PD-1 expression specifically in the CD25+ CD8+ T cell population was lower in TIL from DGKζ-deficient than in those from wt mice (figure 6I). These results confirm a role for DGKζ in limiting effector functions, and suggest DGKζ targeting as a potential means to avoid exhausted T cell phenotypes, even in the context of immunosuppressive milieus.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DGKζ deficiency limits MC38 tumor growth favoring expansion of CD8+CD25+ T cell populations. wt or DGKζ-deficient mice received subcutaneous injections of 5×105 MC38 colon adenocarcinoma cells. (A, B) Tumor progression was evaluated daily until wt animals had to be sacrificed following standards imposed by CNB-CSIC Ethics Committee for Animal Experimentation were reached by day 19. Tumor area (A) or volume (B) was analyzed. Tumors from wt or DGKζ-deficient mice were excised and cells analyzed by flow cytometry. (C–I) The percentage of live (C), CD45+ (D) and CD8+ T (E) cells were compared. CD25 expressing cell percentage (F) or gMFI (G) within the whole CD8+ T cell population was analyzed. PD-1 expressing cell percentage within the whole CD8+ T cell population (H) was analyzed I. PD-1 gMFI was analyzed within the CD8+CD25+ or CD8+CD25- T cell subsets. Data were analyzed using two-way ANOVA and Bonferroni post-test (A,B, I) or parametric unpaired t-test (C–H); *p<0.5, **p<0.01, ***p<0.001, ****p<0.0001. Results are representative of at least three independent experiments with similar results. ANOVA, analysis of variance; DGK, diacylglycerol kinases; gMFI, geometric mean fluorescence intensity; PD-1, programmed cell death-1; wt, wild type.
Discussion
DGKζ-dependent transformation of DAG into phosphatidic acid limits the activation of the RasGRP1/Ras-regulated and PKCθ-regulated pathways. This reaction maintains correct spatiotemporal coordination of these signaling pathways that ultimately guarantees an appropriate immune response.54 Abnormal upregulation of DGKζ in primary T cells and/or engineered CAR T cells that infiltrate solid tumors leads to hyporesponsive states that facilitate tumor immune evasion during tumor escape.55 Targeting DGKζ activity thus offers interesting therapeutic possibilities for cancer immunotherapy, but design of successful approaches demands identification of its functional interactors. In this study, we establish that DGKζ and PD-1 operate in a positive feedback loop in which DGKζ contributes to sustain PD-1 expression, leading to the onset of inhibitory functions that ultimately facilitate immune evasion.
Using the TPR cell model, we corroborated previous mouse data that linked DGKζ with control of NFκB and AP-1 activity,27 and found that DGKζ also negatively controls NFAT transcription. Although the mechanisms that support this last connection remain to be explored, our observations in cells in which DGKζ expression is only partially attenuated provide additional clues to explain the strong potentiating effects observed in genetically deficient mice. The use of CD86-expressing TCS cells confirms the relevance of CD28-driven signals for stabilization of AP-1 induction, in agreement with earlier observations.56 Together with NFAT and NFκB, AP-1 regulates the transcription of cytokines and cell surface receptors that facilitate T cell cycle entry, clonal expansion, and anergy avoidance.57 58 Accordingly, only TPR cells cocultured with TCS-CD86 showed noticeable IL-2 transcription and secretion. The TPR model thus provides a reliable, easy to manipulate, robust cellular platform that reproduces the requirements for IL-2 production, the hallmark of clonal expansion that follows T cell activation.
Constitutive PD-1 expression in TPR-PD-1 cells did not alter CD28 costimulatory signals, but simultaneous triggering of the two receptors by use of CD86/PD-L1-bearing TCS cells markedly abrogated IL-2 production, which was reinstated by the anti-PD-1 blocking Ab nivolumab.59 This genuinely reproduces the PD-1 inhibitory effects on CD28-triggered costimulatory functions,60 and confirms the potential of this platform for use in trials to test combinatorial treatments, and in the search for inhibitors that target T cell response modulators such as DGKζ.
Dual stimulation of PD-1 and CD28 provides interesting conclusions regarding the effect of PD-1 triggering on CD28-dependent signals. Whereas PD-1 ligation reduced NFAT and AP-1 induction, we observed no repression of NFκB activation. These results correlate well with the CD28 ability to elicit NFκB activation in the absence of TCR triggering in human T cells.44 Interestingly, DGKζ-silenced cells showed resistance to PD-1-dependent inhibition of AP-1, which confirms the role of this enzyme as a negative regulator of the Ras/ERK pathway in anergic/exhausted T cells. This nonetheless does not preclude inhibition of IL-2 production, suggesting that diminishing DGKζ is not enough to counteract the potent limitation imposed by PD-1 triggering.
Although useful in the search for pharmacological inhibitors, the TPR model is an artificial system in which the effect of DGKζ silencing can only be explored in conditions of constitutive PD-1 expression. DGKζ deficiency in mouse and Jurkat T cells facilitates enhanced IL-2 expression27 that, as we demonstrate here, limits PD-1 expression, linking this DGK isoform to the control of this immune checkpoint. This explains the partial insensitivity to PD-1/PD-L1-derived immunosuppression observed in DGKζ-deficient T cells.61 It might also have clinical implications. The success of anti-PD-1 therapies is described to be dependent on PD-1 expression not surpassing a determined threshold.10 Targeting DGKζ could limit PD-1 expression, and thus favor the effectiveness of anti-PD-1 blocking therapies. Our observations also suggest the possibility of using DGKζ expression as a biomarker in the clinic, especially in those cases in which ICT directed to the PD-1/PD-L1 axis are used, either to monitor the success of these therapies or when resistances to them are developed.
The DGKζ-imposed negative control of IL-2 production translates to enhanced PD-1 expression, an observation that fully correlates with the identification of DGKζ as a gene upregulated during tolerance induction.62 The DGKζ contribution to CD8+ T cell fate decisions between tolerance and CTL differentiation provides a mechanistic explanation for the notably diminished growth of MC38 adenocarcinoma cells in DGKζ-deficient mice. The MC38 tumor model is highly dependent on PD-1/PD-L1-mediated immunosuppression.53 63 64 DGKζ-deficient TIL analyzes confirmed significant presence of functionally activated CTL, identified as CD25+ CD8+ T cells. Our combined assessment of CD25 and PD-1 expression shows reduced PD-1 expression within CD25+ CD8+ T cells, which confirms that DGKζ deletion facilitates strong T cell responses even in the context of immunosuppressive milieus. In spite of the marked differences in tumor growth, single analysis of PD-1 expression in TIL indicated similar percentages of PD-1+ CD8+ T cells in wt and DGKζ-deficient mice. Whereas this observation fully agrees with induction of PD-1 expression following T cell activation,50 our experiments cast doubt on the use of PD-1 single expression studies to monitor T cell responses.
In addition to DGKζ, TIL also upregulate DGKα. Studies in human and mouse CAR T cells demonstrate enhanced functions following individual silencing of each isoform.29 65 These data contrast with the predominant function of DGKζ in the control of ERK activation, also observed in CAR T cells.65 A role for DGKα as a negative regulator of CD8+ cytotoxic functions was nonetheless previously characterized in mouse models. DGKα-deficient CD8+ T cells show increased proliferation that correlates with increased production of cytotoxic molecules such as perforin.66 Individual silencing of DGKα or DGKζ in human CAR T cells fails to improve antitumor effects in vivo. Only dual DGK-silenced CAR T cells show enhanced tumor control and higher T cell infiltration. This observation correlates with the synergistic effect on cytokine production,65 and suggests safeguarding properties for the individual DGK. Dual targeting of DGKα and ζ could constitute a powerful strategy for improving CAR T cell efficiency in solid tumors, particularly in those cases refractory to immune checkpoint blockage.
Our experiments confirm data from mouse studies regarding DGKζ depletion-derived effects in a robust, reliable human T cell platform. The results also extend knowledge of the DGKζ role in limiting T cell responses, specifically of the DGKζ interrelation with an important immune checkpoint such as PD-1; these findings indicate a promising horizon to be explored for further success of cancer immunotherapies.
Acknowledgments
We thank all members of the Mérida Lab for discussion and suggestions and C. Mark for expert editorial assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conceptualization: JA-N, AA-F and IM. Generation of TPR and TCS cells: JL and PS. TPR cell experiments: JA-N and AA-F. Mouse experiments: JA-N. Jurkat cell experiments: JA-N and CR-R. MC38 tumor cell engraftment: MM-S and RL. TIL isolation experiments: RL and AA-F. Development and optimization of flow cytometry protocols for TIL studies: MCM-O and AA-F. Writing manuscript: JA-N, AA-F and IM. All authors read and gave input on the manuscript.
Funding This work was supported in part by grants from the MINECO (PID2019-108357RB-I00) to AA-F and IM and MINECO (BFU2016-77207-R), the Spanish Association Against Cancer (AECC-1518), the Madrid regional government (IMMUNOTHERCAM Consortium B2017/BMD-3733) and the Aplastic Anemia and MDS International Foundation (AAMDSIF OPE01644), to IM. CR-R is a predoctoral fellow of the Álvaro Entrecanales and Jerome Lejeune Foundations.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Protocols approved by the CNB-CSIC Ethics Committee on Animal Experimentation.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplemental information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.