Article Text

Abstract
Background Sarcomas exhibit low expression of factors related to immune response, which could explain the modest activity of PD-1 inhibitors. A potential strategy to convert a cold into an inflamed microenvironment lies on a combination therapy. As tumor angiogenesis promotes immunosuppression, we designed a phase Ib/II trial to test the double inhibition of angiogenesis (sunitinib) and PD-1/PD-L1 axis (nivolumab).
Methods This single-arm, phase Ib/II trial enrolled adult patients with selected subtypes of sarcoma. Phase Ib established two dose levels: level 0 with sunitinib 37.5 mg daily from day 1, plus nivolumab 3 mg/kg intravenously on day 15, and then every 2 weeks; and level −1 with sunitinib 37.5 mg on the first 14 days (induction) and then 25 mg per day plus nivolumab on the same schedule. The primary endpoint was to determine the recommended dose for phase II (phase I) and the 6-month progression-free survival rate, according to Response Evaluation Criteria in Solid Tumors 1.1 (phase II).
Results From May 2017 to April 2019, 68 patients were enrolled: 16 in phase Ib and 52 in phase II. The recommended dose of sunitinib for phase II was 37.5 mg as induction and then 25 mg in combination with nivolumab. After a median follow-up of 17 months (4–26), the 6-month progression-free survival rate was 48% (95% CI 41% to 55%). The most common grade 3–4 adverse events included transaminitis (17.3%) and neutropenia (11.5%).
Conclusions Sunitinib plus nivolumab is an active scheme with manageable toxicity in the treatment of selected patients with advanced soft tissue sarcoma, with almost half of patients free of progression at 6 months.
Trial registration number NCT03277924.
- sarcoma
- immunotherapy
- clinical trials as topic
- translational medical research
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Systemic treatment for advanced soft tissue sarcomas (STS) is an unmet need since no new drugs or combination schemes have offered significant overall survival (OS) benefit over doxorubicin alone in the last 40 years.1–3 Immunomodulation has become a topical area of interest in many tumors, being sarcoma not an exception. Nevertheless, while in paradigmatic immune-sensitive tumors such as melanoma the expression levels of tumor mutational burden (TMB), CD8+ lymphocytes and Programmed death-ligand 1 (PD-L1) were in the high range, with values of 14 mutations per DNA megabase (Mb), 42%±23% and 35%, respectively,4 5 sarcomas appear at the low range of these expression values. Indeed, the median TMB reported was 2 mutations per Mb,6 the percentage of CD8+ lymphocytes was 23%±13%7 and the tumorous PD-L1 expression was 6.6% in the largest series.8
Specific immune-checkpoint protein expression, such as PD-1/PD-L1 axis, has not demonstrated convincing prognostic or predictive value in sarcomas.9 10 Apart from the inherent heterogeneity of STS, other factors that introduce bias across these studies were different staging, diverse antibody clones used for PD-L1,11 12 and intrastudy variability between tissue microarray or complete tumor block section.13 A genomic approach that can circumvent some constraints of PD-L1 immunohistochemistry has confirmed an independent negative prognostic value for PD-L1 overexpression in a large study of patients with STS.14 The elapsed time between tumor sampling and the time frame considered for the analysis is a common limitation of immunostaining and genomics, which is only eluded if biopsies are mandatory at baseline.
Using transcriptomic analysis of the microenvironment cell population, which measures the expression of eight immune and two stromal cell populations,15 it was possible to classify sarcoma into five different sarcoma immune classes (SIC).16 Patients grouped as SIC E were considered the most immune-responsive subset, but only represented 17.8% of cases. Therefore, the immune cold microenvironment found in most sarcomas could explain the low probability of response or tumor control with anti-PD-1 compounds in monotherapy. In fact, two reported experiences with nivolumab showed a median progression-free survival (mPFS) of 1.7 months in 43 patients with STS17 and 1.8 months in 12 patients with STS,18 respectively. A pioneer study trialing pembrolizumab in the most frequent STS and bone sarcomas reported better outcomes with an mPFS of 4.2 months, 18% of responses and a 6-month progression-free survival rate (PFSR) of 32%.19
Immune barrier promoted by tumor angiogenesis is well established and there is an ever growing list of immune cells exhibiting the dual capacity of promoting immunosuppression and angiogenesis.20 This impelled us to design a double inhibition study with antiangiogenic and anti-PD-1 compounds, which could potentially synergize for the treatment of some advanced soft tissue and bone sarcomas. We present herein the phase Ib, common to both cohorts, and phase II part of the STS cohort of the IMMUNOSARC trial, which combined sunitinib and nivolumab. The bone sarcoma cohort will be reported separately.
Methods
Study design and subjects
In this phase Ib/II, single-arm trial, STS cohort, patients aged over 18 years with advanced STS were considered for eligibility and enrolled in eight centers in Spain and Italy with expertise in sarcoma care. Central pathology review was mandatory before accrual. Patients had to be in progression in the previous 6 months according to Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. Eligible subtypes of sarcoma were chosen based on several reasons: histologies reported in transplant recipients (undifferentiated pleomorphic sarcoma (UPS), angiosarcoma, and synovial sarcoma), higher tumor infiltrating lymphocytes (UPS and epithelioid sarcoma), or sensitivity to antiangiogenic compounds (solitary fibrous sarcoma, extraskeletal myxoid chondrosarcoma, clear cell sarcoma, alveolar soft part sarcoma (ASPS), epithelioid hemangioendothelioma and angiosarcoma). Other relevant inclusion criteria are described in online supplemental methods.
Supplemental material
Patients provided written informed consent before participating in the study.
Procedures
In phase I, two dose levels were defined: the starting dose (dose level 0) used sunitinib 37.5 mg orally on a daily basis from day 1. The de-escalation level (dose level −1) used sunitinib 37.5 mg on the first 14 days (induction) and from then on 25 mg per day. Nivolumab was given at 3 mg/kg intravenously on day 15 and every 2 weeks from then on at both dose levels. Phase I was completed when 10 dose-limiting toxicity (DLT) evaluable patients had been treated, associated with a DLT rate of less than 0.33. There was a de-escalation level if three or more DLTs occurred at the initial dose level.
Radiological assessment was performed at the local sites as well as by a mandatory independent central review. Tumor assessment was done every 8 weeks by CT scan or by MRI in accordance with RECIST 1.1. Other relevant procedures are described in online supplemental methods.
For translational purposes, tumor biopsies were required at baseline and at week 13, and blood samples were collected at baseline, day 1 of week 3, day 1 of week 13, and after each radiological evaluation.
The HTG EdgeSeq, ran in the VERIP service lab of HTG in Tucson (HTG Molecular Diagnostics, Tucson, Arizona, USA), and ProcartaPlex immunoassay (Invitrogen, Carlsbad, California, USA) protocols were applied in the exploratory translational study, and complete bioinformatic analyses are described in online supplemental methods.
Outcomes
The main endpoint for phase Ib was to determine the recommended dose for phase II. The main endpoint for phase II was 6-month PFSR according to RECIST. The secondary endpoints were median OS (mOS), overall response rate according to RECIST 1.1 and Choi criteria, toxicity profile according to Common Terminology Criteria for Adverse Events (CTCAE) V.4.0, and contribution to translational studies.
Sample size has been obtained for a one-arm, one-stage survival design based on Brookmeyer-Crowley-like test.21 The statistical test for survival probability was based on non-parametric estimation of survival distribution. For STS second-line cohort sample size has been obtained for the primary endpoint: 6 month PFSR. The estimated accrual time was 24 months. In this population, a 5% PFSR was considered not promising, whereas a 15% PFSR was considered promising. With a 0.05 type I error α and a power of 0.80, 48 patients were needed in this cohort.
Statistical analysis
The intention-to-treat population includes patients who had provided written informed consent, with central pathology confirmation, and fulfilling all the inclusion criteria and none of the exclusion criteria. The per-protocol population (efficacy) includes patients fulfilling the intention-to-treat population criteria, and additionally patients who had received sunitinib in the induction phase and at least one dose of nivolumab. The safety population includes all patients of the intention-to-treat population who had received at least one dose of sunitinib. The radiological evaluable population includes all patients of the intention-to-treat population who underwent at least one radiological assessment.
Time-to-event variables were measured from the date of enrollment and were estimated according to the Kaplan-Meier method. Comparisons between the variables of interest were performed by the log-rank test. For variables not available at baseline (ie, RECIST or Choi response), a landmark analysis was performed. False discovery rate was applied to regulate multiple comparisons. Other statistical methods are described in online supplemental methods.
Results
From May 31, 2017 to April 18, 2019, 91 patients with advanced and progressing STS were assessed for eligibility in phase Ib and in the STS cohort of phase II. Twenty-three patients were excluded (considering both phases) after screening. Sixty-eight patients were enrolled and treated with the experimental compounds: 16 in phase Ib and 52 in the STS cohort of phase II. The intention-to-treat population and safety population for phase II consisted of 52 eligible patients, while the per-protocol population consisted of 49 patients, since 3 patients had not ever received nivolumab. Radiological evaluable patients in phase II according to RECIST and Choi criteria were 46 and 40, respectively (figure 1). Patients’ demographics are depicted in table 1.

CONSORT diagram. CONSORT, Consolidated Standards of Reporting Trials; ECOG, Eastern Cooperative Oncology Group; RECIST, Response Evaluation Criteria in Solid Tumors.
Baseline characteristics
The initial dose level (sunitinib 37.5 mg/day) in phase Ib was ceased after observing three DLTs in six accrued patients: grade 3 fatigue (n=2) and grade 4 septic shock (n=1). In the subsequent de-escalation (−1) level, only one DLT (febrile neutropenia) was reported in 10 accrued patients. Thus, the recommended dose of sunitinib for phase II was 37.5 mg/day as induction on the first 14 days, followed by sunitinib 25 mg/day plus nivolumab 3 mg/kg every 2 weeks from day 15 on. One hundred and sixty-eight 2-week cycles were administered in phase Ib. Phase Ib toxicities by patients are depicted in table 2.
Toxicity profile in phase Ib
According to RECIST there were seven partial responses among 14 evaluable patients enrolled in this part (figure 2): two patients with clear cell sarcoma, two with ASPS, and one occurring in each of the following histologies: synovial sarcoma, chondrosarcoma and angiosarcoma. In the remaining non-responder patients, three had stable disease and four had progressive disease.
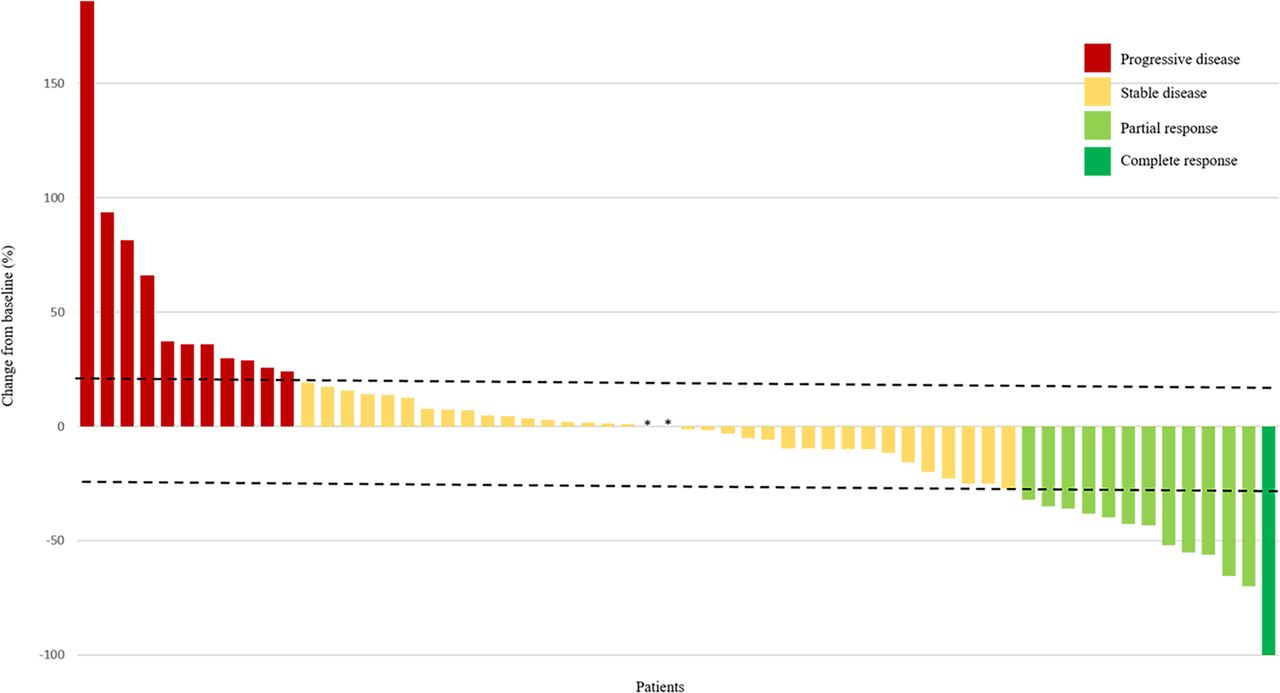
Response to treatment by patient according to RECIST. All evaluable patients (N=60) from phase Ib (n=14) and phase II (n=46) are shown. Tumor diameter was measured in millimeters. The dashed lines represent 20% increase in diameter and 30% decrease in diameter (RECIST progression and response cut-offs, respectively). *No size variation. RECIST, Response Evaluation Criteria in Solid Tumors.
The clinical cut-off for the final data analyses for phase II was February 27, 2020. At that time, 5 (10%) of the 52 patients were still under treatment and 47 (90%) have discontinued the scheme. Of the 52 patients, 40 (77%) discontinued due to progression, 4 (7%) following patient decision and 3 (6%) due to toxicity. The latter included grade 2 cystitis (n=1), grade 4 psoriatic arthritis (n=1) and grade 3 peripheral ischemia (n=1). The median elapsed time between previous progression and study enrollment was 1.17 months (0.03–5.17).
Seven hundred and sixty-three 2-week cycles were administered in 49 patients of the per-protocol population, with a median of 6 (1–23) and 12 (2–46) cycles of sunitinib and nivolumab per patient, respectively. The median dose intensity for sunitinib and nivolumab was 97% (20–142) and 100% (33–100), respectively. One (2%) patient had dose reductions and 29 (59%) had dose interruptions of sunitinib, whereas 21 (43%) patients had dose interruptions of nivolumab.
The most frequent treatment-related toxicities per subject in phase II were fatigue in 33 of 52 patients (63.5%) and increased aspartate aminotransferase (AST) in 25 out of 52 patients (48.0%). The most common reported grade 3 or 4 side effects were transaminitis in 9 out of 52 patients (17.3%) and neutropenia in 6 out of 52 patients (11.5%) (online supplemental table 1).
At a median follow-up of 17 months (4–26), 37 of 49 (76%) per-protocol evaluable patients experienced progression according to central assessment and 18 of 49 (37%) patients died due to disease progression.
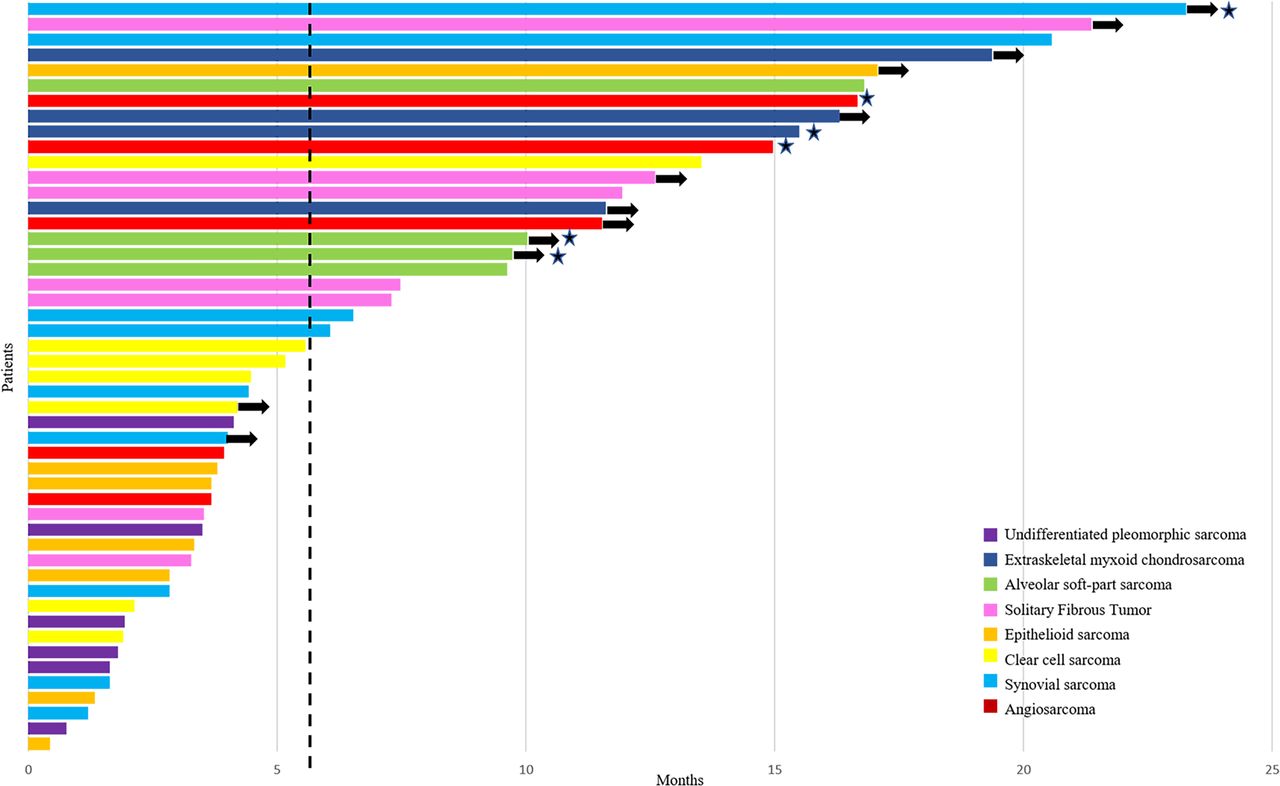
The 6-month PFSR according to central and local assessments was 48% (95% CI 41 to 55) and 51% (95% CI 44 to 58), respectively (figure 3). The mPFS for central and local assessments was 5.6 months (3.0–8.1) and 6.0 months (3.1–9.0), respectively. Remarkably, the proportion of patients alive at 12 and 18 months was 75% (95% CI 68 to 81) and 67% (95% CI 59 to 74), respectively, and the mOS was 24 months (95% CI NA). Central radiological assessment according to RECIST (figure 2) reported 1 complete response in 46 evaluable patients (2%), 5 partial responses (11%), 33 stabilizations (72%) and 7 progressions (15%). Complete response was observed in one patient with angiosarcoma and partial response in patients diagnosed with ASPS (n=2), angiosarcoma (n=1), extraskeletal myxoid chondrosarcoma (n=1) and synovial sarcoma (n=1). Central assessment according to Choi criteria showed 25 patients with partial response (63%), 10 with stable disease (25%) and 5 with progressive disease (12%). Response assessment according to RECIST showed significant prognostic difference for PFS and OS; by contrast, Choi assessment only had prognostic relevance for PFS. Adding the 12 evaluable STS cases of phase I to the 46 evaluable patients with STS in phase II, the RECIST Overall Response Rate (ORR) was 21% (12 out of 58). The 18-month OS proportion was 100%, 75% and 44% for those with response, stable disease and progressive disease, according to RECIST, respectively (p=0.01) (table 3).
{kind=link}
{kind=link}
{kind=link}
Progression-free survival to treatment by patient, based on RECIST central radiological assessment. Each patient in the efficacy population in phase II is represented as bars (n=49). The vertical dashed line represents the median progression-free survival (5.6 months). The stars represent patients achieving RECIST objective responses. The arrows represent patients non-progressing in the last central radiological assessment. RECIST, Response Evaluation Criteria in Solid Tumors.
Univariate analysis
Ad hoc analyses showed a growth modulation index (GMI) in phase II of 0.7 (0.1–11.6), while 31% of patients exhibited GMI >1.33. On the other hand, the GMI in phase II in subtypes with known sensitivity to antiangiogenic compounds was 1.01 (0.04–26.82).
In the translational study, among 732 genes related to tumor immune response, 50 and 102 genes showed to have potential prognostic value for PFS and OS, respectively. Hierarchical clustering, considering the 102 genes with impact on OS, distributed samples into two groups with a distinct molecular pattern (online supplemental figure 1). The different outcome of these two groups was confirmed (table 3). Functional enrichment analysis showed that the genes upregulated in group 2 (molecular group with better survival) were highly associated with Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (online supplemental figure 2) cytokine-cytokine receptor interaction (hsa04060) (adjusted p=3.69×10−53) or chemokine signaling pathway (hsa04062) (adjusted p=2.13×10−22). On the other hand, genes upregulated in group 1 (molecular group with worse outcome) were greatly associated with metabolic and energetic processes (online supplemental figure 3).
Other clinical and translational results are described in online supplemental results.
Discussion
In this phase Ib/II trial, we found that the 6-month PFSR was 48% according to RECIST and independent central review. This outcome widely exceeds the 15% considered promising in the statistical assumption of this trial. This threshold was based on the European Organisation for Research and Treatment of Cancer (EORTC) recommendation cut-off for activity, in terms of 6-month PFSR, in second line drugs of advanced STS.22 This indicator was chosen considering that a valuable immunotherapy should induce durable disease control; accordingly, 3-month PFSR would not be useful enough in detecting the potential added value of immunomodulation in sarcoma. The scheme was in general manageable even when it required transient dose interruptions in up to 59% of patients. Neutropenia, which could be related to FLT3 inhibition by sunitinib,23 was found at a higher proportion than sarcoma trials with anti-PD-1 alone19 or sunitinib alone.24 The difference with the latter could be explained by the shorter use of sunitinib due to earlier progression in the monotherapy trial.
The mOS of 24 months is worth mentioning; phase III trials showing significant benefits of some active drugs in STS second line reported mOS ranging from 12.5 to 13.5 months.25–27 It will be important to analyze postprotocol therapies and microenvironment transformation after the immunomodulatory treatment since the clinical impact of the sunitinib and nivolumab scheme could also have an influence on subsequent postprotocol therapies. We have analyzed the potential bias of selecting some more indolent histologies or those histologies more sensitive to antiangiogenics as the reason for this prolonged survival, but we did not find significant differences in survival between groups.
The 6-month PFSR of 48% and the mOS of 24 months showed in our trial favorably compare with the activity reported with anti-PD-1 or sunitinib in monotherapy in previous STS trials. The SARC028 study reported 32% and 11.4 months for 6-month PFSR and mOS, respectively, in 42 patients with STS treated with pembrolizumab.19 Nevertheless, the SARC028 study selected the four more frequent histotypes for the STS cohort, making comparison with our study difficult since only synovial sarcoma and UPS were common histologies in both trials. The Alliance-091401 trial reported 15% and 10.7 months, respectively, for the same indicators in 43 patients with STS treated in the monotherapy arm with nivolumab.17 Another study was prematurely closed, with 12 patients with uterine leiomyosarcoma treated with nivolumab, after observing an mPFS of 1.8 months at the interim analysis and with no patient free of progression at 6 months.18 Sunitinib was also trialed as monotherapy in a phase II study of 50 patients with STS, reporting a 6-month PFSR of 22% with an mPFS of 1.8 months, while OS data were not provided.24 Of note, the GMI in our study is comparable with those reported with active chemotherapy in second line,28 and the median elapsed time of 1.17 months between previous progression and enrollment reinforces the outcome in terms of PFS. Furthermore, the GMI in the subset of subtypes with higher potential sensitivity to antiangiogenics was even better. A limitation of this study is inherent to design assumption: it is not possible to distinguish separately the efficacy of antiangiogenic or anti-PD-1. The differences detected between immunomodulation with combination approach and monotherapy underline the feature that STS exhibits, in general, a cold immune microenvironment. In line with this, a combined immunotherapy approach tried to convert a cold into inflamed microenvironment in sarcomas. Thus, the combination arm of Alliance-091401 addressed a double immune-checkpoint inhibition with nivolumab plus ipilimumab. Patients in the combination arm had a 6-month PFSR of 28% and a mOS of 14.3 months, all the efficacy indicators being superior to monotherapy arm. The lower 6-month PFSR and the shorter OS in the combination arm in comparison with our trial could be explained, at least partially, by the restriction in the number of ipilimumab cycles (four at maximum).17 Conceptually similar to our study, the administration of antiangiogenic plus anti-PD-1 agents was explored in a single-center phase II trial with axitinib plus pembrolizumab . The 6-month PFSR was 46.9% and the mOS was 18.7 months, which were both closely similar to our study. A substantially different aspect was based on the proportion of ASPS; while in the axitinib-pembrolizumab 36% of the patients had this subtype, in our trial only 7% had this diagnosis. Thus, for the non-ASPS cases of that trial, the mPFS was 3 months, the 6-month PFSR was 38.1%, the mOS was 13.1 months, while no patient survived beyond 2 years. All these parameters seemed to be worse than ours, although limited by indirect comparisons. The indolence and special sensitivity of this subtype to both antiangiogenic and immunomodulation could have influenced the survival results. In our series, four of seven accrued patients with ASPS (considering both phases) had partial response (57%), whereas in a randomized phase II trial exploring cediranib versus placebo in ASPS the authors reported responses in 6 of 31 (19%) patients.29 In fact, excluding ASPS from the analysis of PFS due to its inherent indolence, we were not able to find statistical differences between potential antiangiogenic sensitive subtypes and others. In other words, ASPS cases could represent bias for the main endpoint of 6-month PFSR. Intriguingly, ASPS does not exhibit some features of the classic immune sensitive microenvironment; thus, the proportion of tumor infiltrating lymphocytes, or specifically CD8+ or FOXP3+ subpopulations, per cubic millimeter is lower in ASPS than in non-translocation-related sarcomas.30 Additionally, TMB is lower in ASPS compared with other sarcomas, such as synovial sarcoma or Ewing sarcoma. The activity of anti-PD-1 in ASPS has been related with molecular mismatch-repair deficiency signature, even in the absence of high TMB, but a broader number of cases to confirm this point will be required.31
The proportion of responding patients, 13% in phase II or 21% joining STS evaluable patients from phase Ib and II of our trial, is similar to the 18% reported with pembrolizumab in the SARC028 study. This could be explained by STS heterogeneity, even within specific subtype; for instance, from seven evaluable patients with UPS in our phase II, only two had stabilization and five had progressive disease, with an mPFS of 1.8 months. By contrast, in the SARC028 study, from nine patients with UPS, four responded, three were stable and two progressed, with an mPFS of 7 months. This emphasizes the concept of the relevance of the microenvironment in the immunomodulation results in sarcoma. A potential molecular prognostic signature with impact on survival has been proposed in our exploratory translational research. Of note, inflammatory processes were associated with better survival, whereas metabolic processes were linked to worse outcome. The role of metabolism in tumor immune evasion has been previously described.32 33 Also, the significant prognostic value of PDGFD or IL16 overexpression in longer survival, shown in online supplemental results, is a finding that deserves to be further explored.
Based on the findings of this study, six subtypes of sarcoma were selected for a multicohort phase II trial, which has been recently activated, with the same regimen. In the translational research, only 28 samples were used for transcriptomics, which is a limitation of this study. However, a broad comprehensive analysis of the immune populations within the tumor and their association with gene signatures and clinical outcomes is currently ongoing and expected to be published as a separate correlative study. Sunitinib inhibits several tyrosine kinase receptors (TKR), including angiogenic (eg, Vascular Endothelial Growth Factor Receptor (VEGFR) types 1 and 2, and PDGFR-α and PDGFR-β), pro-oncogenic (eg, fms like tyrosine kinase 3 (FLT3), rearranged during transfection (RET), and KIT) or immune-related pathways (eg, Colony stimulating factor 1 receptor (CSF1R)). Although the main hypothesis of this study is to convert the sarcoma cold microenvironment into an inflamed one, by inhibiting immunosuppression driven by angiogenesis, we cannot discard the potential added value of targeting other TKRs. In fact, it seems that CSF1 expression has been described as an adaptive mechanism of resistance to anti-PD-1 inhibition in melanoma, while the combination of anti-CSF1R and anti-PD-1 inhibitions was demonstrated to be active in BRAFV600E-driven, transplant mouse melanomas. The efficacy of this combination was dependent on the elimination of tumor-associated macrophages (TAMs).34 Likewise, it has been reported that the combination of CSF1R inhibitor with anti-PD-L1/PD-1 axis blockage is also highly active in hepatocellular carcinoma mouse models, increasing intratumoral CD8+ T cells infiltrate and reducing TAMs.35 Translational and preclinical studies will also address the potential role of targeting these TKRs for anti-PD-1/PD-L1 axis inhibition efficacy in STS.
In conclusion, our study supports the idea that the PD-1/PD-L1 axis inhibition per se is not enough in the context of STS. Combination strategies promoting inflamed microenvironment resulted in a higher efficacy in this setting. Targeting new immune-checkpoint inhibitors and selecting histological subtypes could also be advantageous in future trials. Correlative studies searching predictive signatures will allow a better selection of patients for immunotherapy.
Acknowledgments
The authors would like to thank Patricio Ledesma Figueroa, Araceli Rodriguez Morales and Emanuela Marchesi for data management.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Presented at Poster discussion at ASCO Annual Meeting 2018, Chicago, Illinois, USA (doi: 10.1200/JCO.2018.36.15_suppl.11515, Journal of Clinical Oncology 36, no. 15_suppl (May 20, 2018) 11515–11515.); oral presentation at ESMO Annual Meeting 2019, Barcelona, Spain (Annals of Oncology (2019) 30 (suppl_5): v683-v709. 10.1093/annonc/mdz283); and oral presentation at CTOS Annual Meeting 2019, Tokyo, Japan.
Contributors Conception and design of the study: JM-B and JAL-M. Collection and assembly of clinical data: JM-B, NH, GG, JM-T, AR, CV, SS, AL-P, LD, IC-G and JAL-M. Collection and execution of correlative studies: JM-B and DSM. Data analysis and interpretation of results: JM-B, NH, AG, HP-V, VE-T, EdA, PC, MP-C, JD, ML-A, DSM and JAL-M. Writing and revision of the manuscript: all authors. Manuscript final approval: all authors.
Funding This work was supported by GEIS and ISG. BMS and Pfizer provided drug supply and partial funding for shipping. Translational studies were partially funded by Beca Buesa from the GEIS group.
Competing interests JM-B reports research grants from PharmaMar, Eisai, Immix BioPharma and Novartis, outside the submitted work; honoraria for advisory board participation and expert testimony from PharmaMar, Eli Lilly and Company, Bayer and Eisai; and research funding for clinical studies (institutional) from PharmaMar, Eli Lilly and Company, AROG, Bayer, Eisai, Lixte, Karyopharm, Deciphera, GSK, Novartis, Blueprint, Nektar, Forma, Amgen and Daiichi Sankyo. NH reports grants, personal fees and non-financial support from PharmaMar, personal fees from Lilly, and grants from Eisai and Novartis, outside the submitted work, and research funding for clinical studies (institutional) from PharmaMar, Eli Lilly and Company, AROG, Bayer, Eisai, Lixte, Karyopharm, Deciphera, GSK, Novartis, Blueprint, Nektar, Forma, Amgen and Daiichi Sankyo. GG reports grants and personal fees from PharmaMar, grants from Novartis, and personal fees from Lilly, Pfizer, Bayer and Eisai, outside the submitted work. AR reports grants and personal fees from PharmaMar, personal fees from Lilly, Novartis, Amgen, AstraZeneca and Tesaro, grants and personal fees from Roche, and grants from Eisai, outside the submitted work. SS reports grants and personal fees from Bayer, Lilly and PharmaMar, and grants from GlaxoSmithKline, Novartis and Pfizer, outside the submitted work. EdA reports personal fees and non-financial support from Roche, BMS and PharmaMar, and personal fees from Bayer, outside the submitted work. ML-A declares institutional research grants from PharmaMar, Eisai, Immix BioPharma and Novartis, outside the submitted work. DSM reports institutional research grants from PharmaMar, Eisai, Immix BioPharma and Novartis, outside the submitted work, and travel support from PharmaMar, Eisai, Celgene, Bayer and Pfizer. JAL-M reports honoraria for advisory board participation and travel support from PharmaMar, Eli Lilly and Company, Bayer, Eisai, Novartis, BMS, MSD, Roche, Celgene, Pierre Fabre, Pfizer, GSK, Daiichi Sankyo, Amgen, and Chobani. All other authors declare no relevant relationship to disclose.
Patient consent for publication Not required.
Ethics approval Study procedures were in accordance with the guidelines established by the ethics committee of each hospital and with the Declaration of Helsinki. Approval was obtained from the ethics committees.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Raw data from HTG Molecular OBP panel experiments are available in the Gene Expression Omnibus (GEO) from NCBI.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.