Article Text

Abstract
T cells that recognize self-antigens and mutated neoantigens are thought to mediate antitumor activity of immune checkpoint blockade (ICB) in melanoma. Few studies have analyzed self and neoantigen-specific T cell responses in patients responding to ICB. Here, we report a patient with metastatic melanoma who had a durable clinical response after treatment with the programmed cell death protein 1 inhibitor, nivolumab, combined with the first-in-class CD122-preferential interleukin-2 pathway agonist, bempegaldesleukin (BEMPEG, NKTR-214). We used a combination of antigen-specific T cell expansion and measurement of interferon-γ secretion to identify multiple CD4+ and CD8+ T cell clones specific for neoantigens, lineage-specific antigens and cancer testis antigens in blood and tumor from this patient prior to and after therapy. Polyclonal CD4+ and CD8+ T cells specific to multiple neoantigens but not self-antigens were highly enriched in pretreatment tumor compared with peripheral blood. Neoantigen, but not self-antigen-specific T cell clones expanded in frequency in the blood during successful treatment. There was evidence of dramatic immune infiltration into the tumor on treatment, and a modest increase in the relative frequency of intratumoral neoantigen-specific T cells. These observations suggest that diverse CD8+ and CD4+ T cell clones specific for neoantigens present in tumor before treatment had a greater role in immune tumor rejection as compared with self-antigen-specific T cells in this patient. Trial registration number: NCT02983045.
- melanoma
- antigens
- neoplasm
- T-lymphocytes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
T cells that recognize neoantigens resulting from cancer-specific mutations are often detected in patients with melanoma and other cancers and are thought to contribute to effective antitumor immunity.1–3 In melanoma, T cells can also recognize antigens from melanocyte lineage specific proteins and re-expressed cancer-testis antigens.4
T cell receptor (TCR) sequencing allows for precise quantitation of individual T cell clonotypes within clinical samples, but identifying which T cell clones are specific for individual tumor antigens is challenging because such cells are usually present in low frequency in the blood5–7 and in tumors.8 Rare T cells can be expanded from the blood with antigen stimulation,9 allowing the use of TCR sequencing to detect clonotypes that increase in frequency following stimulation with peptide antigen to identify specific T cells.5 10 Here, we use peptide stimulation and TCR sequencing to identify clonally diverse CD8+ and CD4+ T cell responses to multiple neoantigens and self-antigens and study their dynamics and localization during successful treatment of a patient with metastatic melanoma.
Methods
Research samples
Research blood samples were collected at day −13, 42, 63 and 455 on Fred Hutchinson Cancer Research Center (FHCRC) protocol 1765 and tumor biopsies were obtained on the clinical trial protocol 6 days before and 17 days after initiation of treatment.
Whole exome sequencing, RNA sequencing, neoantigen prediction
Whole exome sequencing (WES) of pretreatment tumor biopsy and peripheral blood mononuclear cell (PBMC) was performed and single nucleotide variants called by both Mutect11 and Strelka12 were filtered for a variant allele frequency (VAF) >0.2 (online supplemental figure S1). The 45 mutations with the highest level of mRNA expression (TPM >12) were selected for screening (online supplemental table S1). RNA sequencing was performed as described previously and in the online supplemental methods.13
Supplemental material
Supplemental material
Supplemental material
T cell culture and TCRVβ sequencing
PBMC were cultured for 13 days in the presence of 2 µg/mL peptide encompassing each neoantigen (27-mer with mutated amino acid at position 14, 80% pure, Elim Biopharma) or self-antigen (Peptivator, Milltenyi) in the presence of interleukin (IL)-21, IL-15, and IL-7 (Peprotech) as described previously.13 14 Reactivity of cultures with individual peptides was assayed by interferon (IFN)-γ ELISpot assay (10 µg/mL peptide, ELISpot Pro human IFN-γ, Mabtech).13 14 IFN-γ secretion assay used the IFN-γ secretion kit (Milltenyi). Sorted IFN-γ-positive cells were expanded in 96-well plates with allogeneic irradiated PBMC, phytohemagglutinin and IL-2 for 14–21 days as described.14
TCRVβ sequencing was performed on DNA samples using the human TCRB kit from Adaptive Biotechnologies, and data were analyzed using Adaptive software. Frequencies of TCR clonotypes were determined by amino acid sequence (online supplemental table S2). Clonotypes identified in the T cell cultures were presumed antigen specific if they met the following criteria: (1) expansion to >5 templates with specific stimulation and at least 10-fold enrichment in frequency over any other stimulation and (2) greater frequency of antigen-dependent IFN-γ secreting cells than either prestimulation sample or irrelevant antigen control culture, with at least five templates observed in the IFN-γ-enriched sample. CDR3 consensus sequences were generated using weblogo15 (weblogo.berkeley.edu).
Supplemental material
Statistics
Data were analyzed using GraphPad Prism software. ELISpot data were analyzed using unpaired t-tests without correction for multiple comparisons. Relative enrichment of TCRVβ templates was performed using Fisher’s exact test.
Results
Clinical presentation
A man aged 21 years presented with a lung nodule that was determined by core needle biopsy to be metastatic melanoma, BRAF V600E mutated. Staging showed multiple lung nodules and mediastinal lymphadenopathy, without a detectable primary skin lesion. The patient was enrolled in a phase I clinical trial combining nivolumab 360 mg and bempegaldesleukin 0.06 mg/kg16 intravenously every 3 weeks. A CT scan at 4.5 months showed a partial response by RECIST V.1.1 (figure 1A,B) and positron emission tomography showed no Fluorodeoxyglucose (FDG) avid disease after 13 months of therapy. The patient developed vitiligo 1 year following initiation of treatment and bempegaldesleukin was discontinued after 15 months due to recurrent grade 1 fatigue and myalgias. Nivolumab was discontinued after 2 years and the patient has not progressed after >3 years (figure 1A).
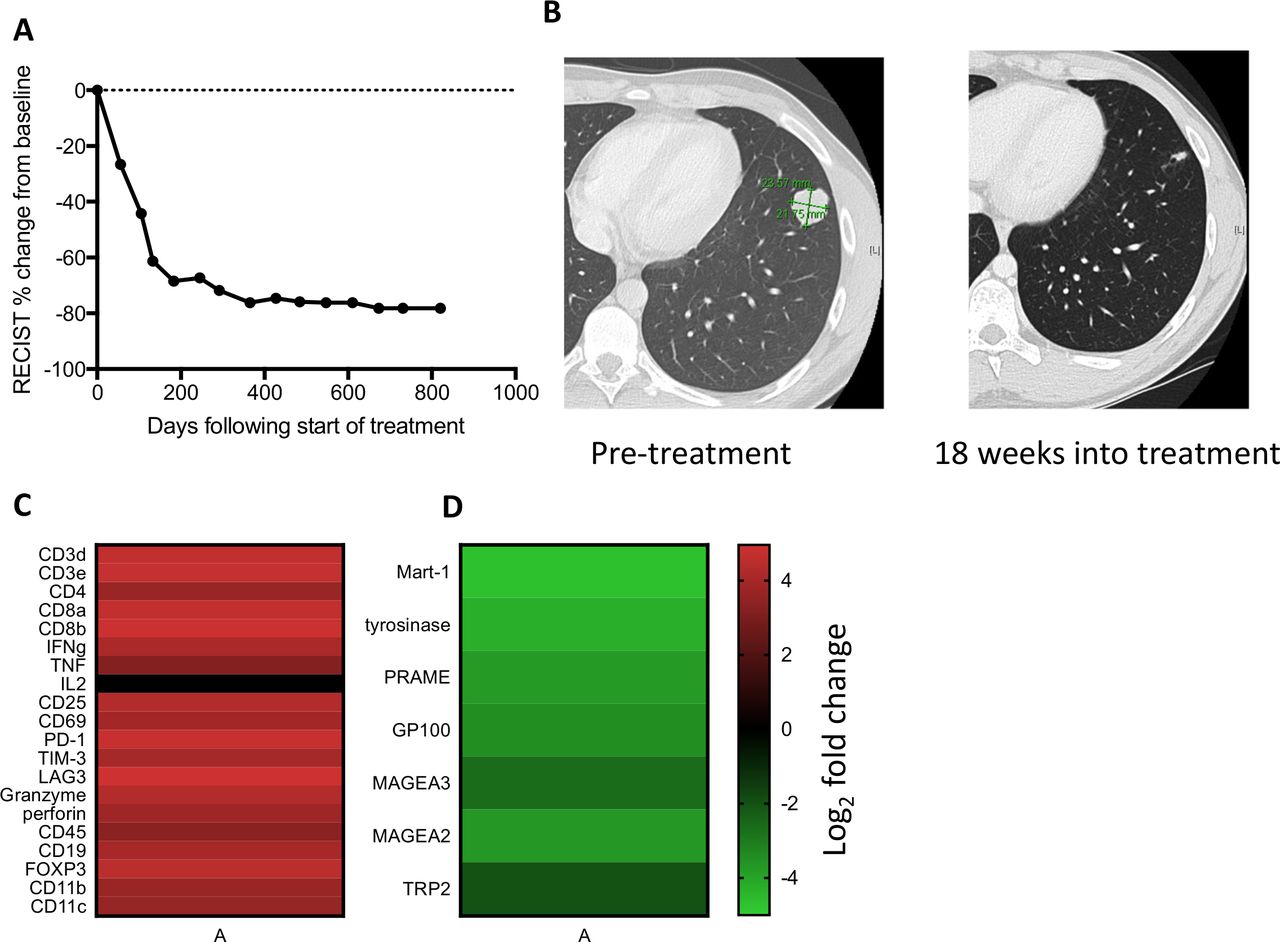
Treatment response and transcriptome analysis. (A) RECIST clinical disease burden following initiation of treatment. (B) Lung nodule on CT scan that underwent needle biopsy at day 19 of treatment. (C) Log2 fold change of immune-related genes between on-treatment and pretreatment biopsy by whole transcriptome sequencing. (D) Log2 fold change of melanocytic lineage-specific and cancer-testis antigens between on-treatment and pre-treatment tumor biopsy.
Tumor regression is accompanied by an immune cell transcriptional signature
Whole transcriptome sequencing performed on pretreatment and on-treatment tumor tissue from a lung lesion that subsequently completely regressed showed a >10-fold increase in expression of multiple genes annotated as immune response genes (3.47-fold enrichment, p=1.3e-81), including those reflecting T cell infiltration (CD3, CD4 and CD8) and activation (IFN-γ, CD69, CD25, programmed cell death protein 1, Lag-3, Tim-3, granzyme, perforin and a decrease in lineage-specific melanocyte proteins tyrosinase, GP100, Mart1 and TRP2 and cancer testis antigens Mage A3, Mage A2 and PRAME (figure 1C,D, online supplemental table S2), consistent with nivolumab and bempegaldesleukin resulting in rapid immune infiltration and tumor regression.16 17
Identification of immunogenic neoantigens
The oncogenic BRAF V600E mutation and 279 additional single nucleotide variants were identified as candidate neoantigens by WES of pretreatment tumor and germline DNA (online supplemental table S1). T cell responses were evaluated to 45 candidates with VAF >0.2 and expression >12 TPM. PBMCs were stimulated with a single pool of two 20 amino acid peptides encompassing each of the mutations and reactivity evaluated by IFN-γ ELISpot (figure 2A). Following stimulation and prior to ELISpot assay, CD8+ T cells were enriched by positive immunomagnetic selection and analyzed separately from CD8+ depleted PBMC. Candidate mutations that showed qualitative responses over baseline with crude mutated peptides (online supplemental figure S2A–C) were tested for IFN-γ production in response to 80% purified 27-mer peptides corresponding to the mutant and wild-type sequences in a confirmatory assay. We identified CD8+ T cell responses to mutations in SLC39A14 (p<0.0001 vs negative control, p=0.0096 vs wild-type) and to NACC1 (p=0.0075 vs negative control, p=0.09 vs wild-type) (figure 2B). We also identified CD4+ T cell responses to the same mutation in SLC39A14 (p<0.0001 vs negative control and p=0.0004 vs wild-type) and a mutation in CAPG (p<0.0001 vs negative control and p=0.052 vs the wild-type peptide, figure 2C).

Identification of neoantigen-specific T cell clones. (A) Schematic of approach for identifying T cell responses to neoantigens. (B–C) Peripheral blood mononuclear cells (PBMCs) were stimulated with pools of crude peptides and then magnetically enriched into CD8+ or CD4+ T cell subsets. CD8+ (B) and CD4+ (C) T cells were restimulated with single purified wild-type or mutant peptides and interferon (IFN)-γ secretion was quantitated by ELISpot. (D) PBMCs were stimulated with purified peptides containing mutations and TCRVβ clonotypes were quantitated by sequencing (left panel). IFN-γ secreting cells were identified following stimulation with peptide or no peptide (control) and sorted by fluorescence activated cell sorting (FACS) and subjected to TCRVβ sequencing. (E) Multiple CD4+ and CD8+ clonotypes were enriched after IFN-γ secretion after stimulation with the peptide containing the mutation in SLC39A14, CAPG or NACC1.
Identification of neoantigen-specific TCR clonotypes
We then used TCRVβ deep sequencing of PBMC that had been stimulated with individual peptides to identify putative antigen-specific T cell clonotypes that expand to one antigen and not others.10 TCR seq is robust in this assay to identify TCR clones present at high enough frequencies to be evenly distributed across parallel cultures. A limitation with this method is that clones present in only one culture could expand non-specifically. Indeed, it was common for some T cell clones to expand without antigen-specific stimulation in one but not in multiple other parallel cultures (online supplemental figure S3). As a second method of establishing antigen specificity, we tested whether restimulating the cultures with peptide and selecting IFN-γ secreting T cells by fluorescence activated cell sorting would further enrich antigen-specific cells (figure 2D) but not non-specifically expanded cells. Many T cells in these cultures secreted IFN-γ even without antigen restimulation (figure 2D), therefore CD4+ and CD8+ IFN-γ secreting cells were also sorted following control stimulations without peptide. With this combination of assays, we assigned the TCRVβ of putative antigen-specific cells to be those that 1) expanded with one antigen and no other antigens and 2) were enriched in IFN-γ secreting cells with specific antigen stimulation to a greater degree than with control stimulation without antigen.
We then applied these methods to identifying putative neoantigen-specific TCRVβ clones in stimulations of post-treatment PBMC in separate cultures with purified peptides containing the mutations in CAPG, SLC39A14 and NACC1. We identified 16 CD8+ clonotypes specific for SLC39A14, 8 CD4+ clonotypes specific for SLC39A14, 16 CD8+ clonotypes specific for NACC1 and a single CD4+ clonotype specific for CAPG (figure 2E, online supplemental table S3). Many of these TCR clonotypes were expanded in only one out of two independent stimulations of 5 million PBMC (seven CD8+ clones and all CD4+ clones specific for SLC39A14, and nine CD8+ clones specific for NACC1, figure 2E) indicating the low frequency of many of these clonotypes capable of expanding with peptide stimulation.
Supplemental material
Isolation of T cell lines and confirmation of neoantigen specificity
To confirm specificity, neoantigen-reactive T cell lines were obtained by expanding small numbers (3–30) of IFN-γ secreting T cells after stimulation with mutated NACC1 and SLC39A14 peptides. T cell lines were then restimulated with peptide to identify those containing >90% antigen-specific cells (figure 3A). TCRVβ deep sequencing of these lines was performed and demonstrated that many were highly oligoclonal (figure 3B–D). TCRVβ clonotypes present at a level of >10% in these cultures were presumed to be antigen specific, which identified four CD4+ T cell clones specific for SLC39A14, seven CD8+ T cell clones specific for SLC39A14 and four CD8+ T cell clones specific for NACC1 (figure 3B–D, online supplemental table S3). Five of these 15 TCR clonotypes from this analysis overlapped with those previously identified, but multiple additional clonotypes were identified in cultures for each antigen (online supplemental table S3). To assess neoantigen specificity, oligoclonal CD8+ T cell lines derived by stimulation with SLC39A14 and NACC1 mutant peptides and CD4+ T cell lines derived by stimulation with the SLC39A14 mutant peptide were tested for reactivity with the mutant and wild-type peptides at various concentrations. Two different CD8+ T cell lines showed preferential activation with a 9-mer peptide containing the SLC39A14 mutation predicted to bind HLA-A:02, relative to the corresponding wild-type peptide (figure 3E), and two different CD8+ T cell lines reacted preferentially with a 10-mer mutated NACC1 mutant peptide predicted to bind HLA B15 (figure 3F). Similarly, two different CD4+ T cell lines were reactive with a 27-mer peptide containing the mutation in SLC39A14 and not the wild-type peptide (figure 3G). There was sequence similarity between TCRVβ hypervariable CDR3 sequences between independent clonotypes, such as nine different NACC1 mutation-specific CDR3 sequences of length 12 (figure 3H). These data show that the expanded T cells were high avidity and neoantigen-specific.
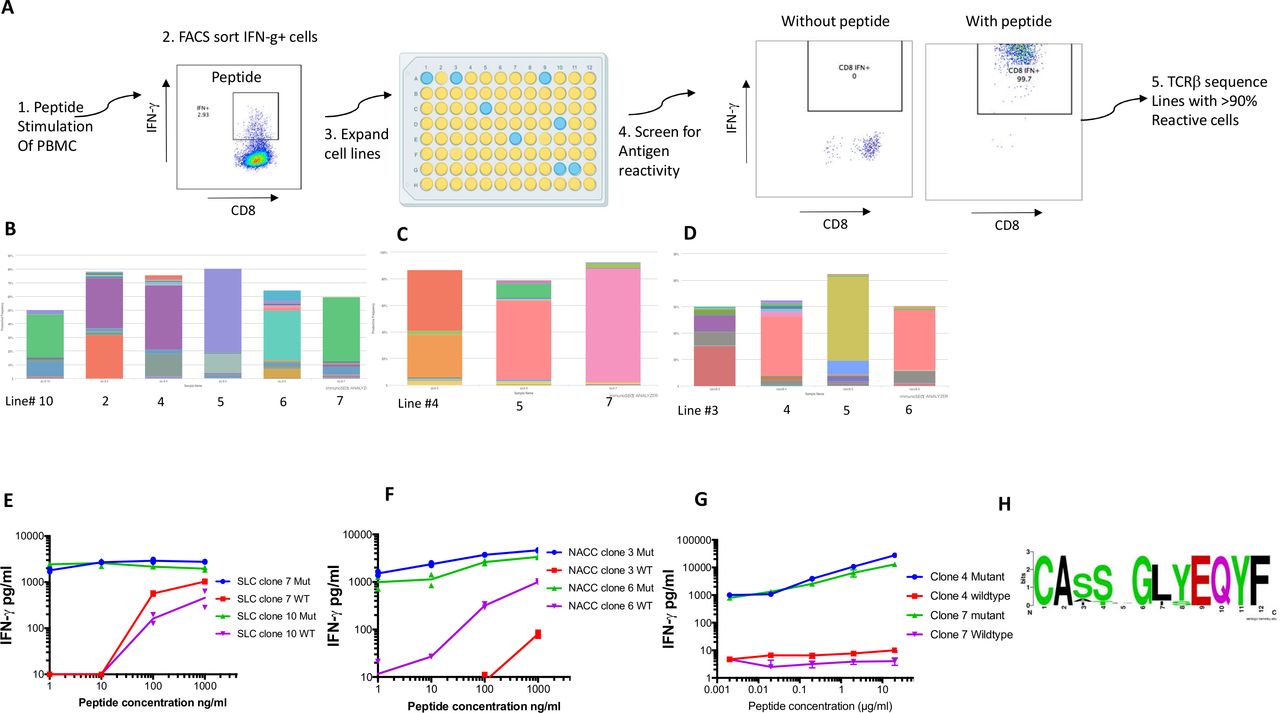
Isolation of patient-derived neoantigen-specific CD8 and CD4 T cells. (A) Schematic of strategy for isolating neoantigen-specific T cells: stimulation of peripheral blood mononuclear cell (PBMC) with peptides followed by isolation of interferon (IFN)-γ secreting cells, expansion of T cell lines and screening of T cell lines for antigen specificity and TCRVβ sequencing of reactive T cell lines. (B–D) Clonotype analysis of six reactive CD8+ T cell lines with each color representing an individual clone (B) and three CD4+ T cell lines (C) specific for mutation in SLC39A14 and four CD8+ T cell lines specific for a mutation in NACC1 (D). (E–G) Expanded CD8+ T cell lines were incubated with different concentrations of mutant and wild-type peptides containing mutations in SLC39A14 (E) and NACC1 (F) and CD4+ T cell lines were incubated with peptides containing mutant and wild-type SLC39A14. (G) Sequences and IFN-γ secretion was measured by ELISA assay. (H) Consensus sequence of nine NACC1-specific TCRVβ CDR3 amino acid sequences of length 12 were generated using weblogo.
Neoantigen-specific T cell clones are present in the pretreatment tumor and expand in the blood during treatment
In total, there were 54 TCRVβ clonotypes identified to be neoantigen-specific (online supplemental table S3). To determine the localization of neoantigen-specific T cell clones during treatment, we performed TCRVβ sequencing of blood and tumor samples and found that 32 of 54 neoantigen-specific clones were detectable in the pretreatment tumor, which in total made up 0.71% of the TCRVβ templates in the pretreatment tumor (figure 4A, online supplemental table S2). In contrast, only four of these clones were detected in unstimulated pretreatment PBMC, making up 0.005% of TCRVβ sequences in the PBMC (150-fold enrichment in tumor, p<0.0001). Furthermore, only six of these clones could be detected in pretreatment PBMC after peptide stimulation of 5 million PBMC (online supplemental table S3).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neoantigen, but not self-antigen-specific T cell clones localize to tumor and expand in the blood following treatment. (A–B) Cumulative frequency (left) or number of distinct neoantigen-specific TCRVβ clones (right) in tumor and blood samples at the indicated timepoints following treatment for total clones (A) and separated into CD4+ and CD8+ clones (B). (C) Peripheral blood mononuclear cell (PBMC) was stimulated with purified peptides containing mutations or peptide pools of self-antigens and TCRVβ clonotypes were quantitated by sequencing in two independent replicates. These cultures were restimulated with peptides or no peptide controls, interferon (IFN)-γ secreting cells were sorted by fluorescence activated cell sorting (FACS) and TCRVβ clonotypes were quantitated by sequencing. The frequency of putative antigen-specific clones is shown for CD4+ and CD8+ T cells for the indicated antigens. (D) Cumulative frequency (top panel) and number (bottom panel) of distinct neoantigen and self-antigen-specific TCRVβ clones in tumor and blood samples following treatment.
Neoantigen-specific T cell clones expanded in frequency in the peripheral blood in the first 50 days following treatment (fivefold, p<0.0001) and contracted in the blood 13 months after treatment (fivefold, p<0.0001, figure 4A). RNA sequencing of a tumor sample after treatment demonstrated a prominent increase T cell transcripts (figure 1C), and the cumulative frequency of intratumoral neoantigen-specific TCRVβ increased modestly (30%, p<0.0001, figure 4A), although the overall clonal diversity was similar prior to and on treatment (online supplemental figure S4). The findings that neoantigen-specific T cell clones were enriched in the tumor and expanded in the blood held true for neoantigen-specific CD8+ T cells (p<0.0001 for enrichment in tumor, expansion in the blood following treatment and contraction following tumor regression) and for neoantigen-specific CD4+ T cells (p<0.0001 for enrichment in the tumor, p=0.1 for expansion in the blood post-treatment and p=0.01 for contraction in the blood post-treatment, figure 4B). Taken together, these data support the methodology for identifying CD4+ and CD8+ T cells that recognize tumor neoantigens, and demonstrate such clones are present in pretreatment tumor, rare in the peripheral blood before treatment and transiently expand in the blood during treatment.
Self-antigen-specific T cell clones do not localize to the tumor or expand during treatment
We also assessed localization and expansion of T cell clones specific for self-antigens using similar stimulation and IFN-γ secretion assays with overlapping long peptide pools encompassing lineage-specific (Tyrosinase, GP100, Mart1, TRP2) and cancer testis (MAGE A3, PRAME) self-antigens that were expressed in the tumor by RNA-seq. We identified CD8+ (n=2) and CD4+ (n=1) clonotypes specific for GP100; CD4+ (n=3) and CD8+ (n=2) clonotypes specific for MAGE A3; CD4+ (n=7) and CD8+ (n=1) clonotypes specific for PRAME; CD8+ (n=2) clonotypes specific for TRP2 and CD4+ (n=6) and CD8+ (n=12) clonotypes specific for tyrosinase (figure 4C, online supplemental table S3). We then compared self-antigen and neoantigen-specific clonotypes identified by the same method from the same blood sample and found, in contrast to neoantigen-specific T cell clones, self-antigen-specific T cell clones did not expand in the PBMC or localize to the tumor (figure 4D).
Discussion
In this case report of effective immunotherapy in melanoma, we determined that diverse polyclonal CD8+ and CD4+ responses to neoantigens were pre-existing in the patient, 150-fold enriched in tumor relative to blood and expanded 5-fold in the blood during the initial stages of treatment with nivolumab and bempegaldesleukin. In contrast, T cell responses to lineage-specific and cancer testis antigens did not localize to the tumor or expand with treatment. We cannot rule out contributions from T cell responses to self-antigens that were not identified in our assays, and the late clinical development of vitiligo suggested such responses were present at some point.18 19 However, our data are consistent with a prominent role for neoantigen relative to self-antigen-specific T cells in the clinical response.1 20 This could be due to central tolerance mechanisms that delete high avidity T cells recognizing self-antigens or due to tumor escape mechanisms that are selective for self-antigens.4 21 22
T cell receptor sequencing of PBMC stimulated with specific peptides has been used as a method for identifying neoantigen-specific T cell clones,10 and we introduced a second step in which antigen-specific IFN-γ secretion with antigen restimulation is used for further enrichment, followed by confirmatory analysis of recognition of mutant and wild-type peptides. We identified polyclonal CD4+ and CD8+ T cell responses to multiple different neoantigens as well as lineage specific and cancer-testis antigens.
Pre-existing T cell responses to neoantigens were present in tumor but rare in the blood, suggesting challenges to isolating such T cells from the blood if they are not mobilized by effective treatment or enriched for a more tumor reactive subset.9 Indeed, the modest overlap in clonotypes identified by relative expansion versus limiting dilution cloning implies that both methods undersample the full set of neoantigen-specific clones, possibly a consequence of how rare these clones are in the periphery. Our RNA sequencing data suggested robust immune infiltration of the tumor 3 weeks post-treatment and a modest increase in the relative frequency of intratumoral neoantigen-specific cells.
In summary, we have described a method for systematically identifying neoantigen and self-antigen-specific T cell clones from the peripheral blood of a patient undergoing a clinical response to immunotherapy in melanoma, and highlighted a likely role for a pre-existing, polyclonal CD4+ and CD8+ T cell response to tumor neoantigens, but not self-antigens, in the patient’s clinical response.
Acknowledgments
The authors would like to thank the patient and their family for participating in this research, and the Lembersky family for their generous support of this and other projects.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This paper has been updated since first published to correct author name 'Ernesto Iacucci'.
Contributors JRV, KGP, EI, JZ and SR designed the experiments; JRV, NS and BJ performed the experiments; JV and SR analyzed the data; JRV, ST and SR wrote the paper.
Funding This work was funded by the NIH K12 grant CA076930-16A1 for JRV and generous support from the Bezos and Lembersky Families.
Competing interests JRV, BJ and SR have equity interest in Lyell Immunopharma, and JZ and EE are employees of Nektar therapeutics, and this paper discusses use of an investigational drug owned by Nektar therapeutics.
Patient consent for publication Obtained.
Ethics approval The patient signed informed consent for all protocols and all studies were approved by the institutional review board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Exome sequencing and RNA sequencing from this case will be submitted to the DbGap limited access database.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.