Article Text

Abstract
Background Concomitant tumor necrosis factor (TNF) neutralization in combination with immune checkpoint inhibitors (ICIs) reduces clinical immune-related adverse events (irAEs) and appears to improve antitumor efficacy in preclinical tumor models. Agonistic antibodies targeting costimulatory receptors such as CD40 represent an additional strategy to boost antitumor immune response and potentiate the activity of ICIs. However, the dose-limiting toxicities observed in anti-CD40-treated cancer patients have hindered its clinical development.
Methods We previously described a mouse model to assess both antitumor activity and irAEs induced by various effective combination immunotherapies. Using the BALB/c and C57BL/6 strains of FoxP3-GFP-DTR (FoxP3DTR) mice, transient depletion of T regulatory cells (Tregs) prior to immunotherapy with additional immunomodulatory antibodies, lowered immune self-tolerance, resulting in the development of a spectrum of physical and biochemical irAEs similar to that reported clinically. In MC38 and 4T1.2 tumor models, following transient Treg depletion, we evaluated the impact of anti-CD40 on antitumor efficacy and the development of irAEs and the impact of concomitant or delayed TNF blockade on both these parameters. Physical irAEs were scored and biochemical irAEs were measured in the serum (ALT and cytokine levels). Histopathological liver and colon tissue analysis were performed to assess immune cell infiltration and tissue damage.
Results Similar to early clinical trials of CD40 agonists, in our tumor models we observed liver toxicities and rapid release of proinflammatory cytokines (TNF, interleukin 6, interferon-γ). In the BALB/c strain, anti-CD40 induced severe physical and biochemical irAEs. Concomitant anti-TNF treatment abrogated weight loss, liver damage and colitis, which consequently resulted in an improved clinical score. However, concomitant anti-TNF impaired antitumor response in a proportion of anti-CD40-treated C57BL/6 FoxP3DTR mice. Delaying TNF blockade in these mice reduced biochemical but not physical irAEs while preserving antitumor efficacy.
Conclusions Our results suggest concomitant rather than delayed anti-TNF is most effective in reducing biochemical and physical irAEs induced by anti-CD40, although it had the potential to negatively impact antitumor efficacy. Furthermore, our findings highlight the utility of our mouse model to assess the severity of irAEs induced by novel immunotherapeutic agents and evaluate whether their toxicity and antitumor efficacy can be uncoupled.
- immunotherapy
- immunomodulation
- costimulatory and inhibitory T-cell receptors
- cytokines
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Immune checkpoint inhibitors (ICIs) targeting CTLA4 and/or PD1/PDL1 have considerably changed the landscape of cancer treatment, underlining the importance of the immune system as a major player to control and eradicate cancers. While the use of anti-PD1, alone or in combination with anti-CTLA4, has been significantly beneficial for some patients across a wide range of malignancies,1 2 there remains a significant proportion of cancer patients who do not respond to these ICIs.3 4
Agonistic monoclonal antibodies (mAb) against costimulatory receptors represent an additional strategy to boost antitumor immune response and potentiate the activity of ICIs. Based on its ability to bridge innate and adaptive immunity by activating antigen-presenting cells (APC), CD40 mAb was among the first agonistic treatment developed for cancer immunotherapy.5 6 Preclinical studies demonstrated that CD40 mAb therapy enhanced APC’s ability to prime CD8+ cytotoxic T lymphocytes, even in the absence of CD4+ T cell helper signals, resulting in a strong and durable antitumor immune response against B cell lymphomas and various solid tumors.7–9 CD40 agonists also modulated tumor-infiltrating myeloid cells from protumor to antitumor and could activate macrophage-dependent tumor matrix degradation.10–12 However, these promising preclinical studies failed to predict the dose-limiting toxicities observed in cancer patients and this has hindered the clinical development of CD40 mAbs.13 In the initial clinical trial from Vonderheide et al, grade 3 headaches and venous thromboembolism were observed in some patients, with the majority of the patients experiencing cytokine release syndrome (CRS) and transient liver toxicity 24–48 hours after infusion.14
Immune-related adverse events (irAEs) are a frequent complication observed after immunotherapy particularly following coblockade of CTLA4 and PD1. The reinvigoration of antitumor immunity can lead to breaches in immune tolerance, autoantigen reactivity and abnormal T cell proliferation and infiltration into various organs.15 16 The occurrence and management of irAEs are a major concern in immunotherapy development, with more than 50% of ICI-treated cancer patients experiencing some form of irAEs, which sometimes necessitates treatment discontinuation and occasionally can result in fatalities.17 18 Given the need to improve clinical benefits, there is a strong rationale to assess combination immunotherapies, although this may also increase the frequency and severity of irAEs. Hence, there is a need to understand the immune pathways that contribute to the development of severe irAEs following different immunotherapies and whether they can be specifically targeted without impacting on antitumor efficacy.
We previously described a mouse model to assess both antitumor activity and irAEs induced by various effective combination immunotherapies. Using the BALB/c and C57BL/6 strains of FoxP3-GFP-DTR (FoxP3DTR) mice, transient depletion of T regulatory cells (Tregs) prior to immunotherapy with additional immunomodulatory antibodies, lowered immune self-tolerance resulting in the development of a spectrum of physical and biochemical irAEs similar to that reported clinically. Generally, irAEs were more severe in the BALB/c strain compared with C57BL/6 strain of FoxP3DTR mice.19 In particular, tumor-bearing BALB/c FoxP3DTR mice that received transient Treg depletion followed by anti-CD137 developed irAEs similar to that observed with urelumab in humans.19 Furthermore, in this model, we demonstrated that administration of antitumor necrosis factor (TNF) alleviated severe irAEs induced by anti-CD137 therapy. Clinically, TNF blockade with infliximab is a second-line treatment for ICI-treated patients suffering from colitis who do not respond to corticosteroids.20 Recently, a preclinical study reported that concomitant TNF blockade in combination with anti-PD1 and anti-CTLA4 treatment not only reduced irAEs but also improved antitumor efficacy.21 In another study, addition of anti-TNF overcame resistance to anti-PD1 in an experimental melanoma mouse model.22 Given the dose-limiting toxicity profile of CD40 mAb, in this study, we used two strains of FoxP3DTR mice and two different tumor models to study how concomitant or delayed TNF neutralization impacted the balance between antitumor efficacy and irAEs induced after anti-CD40 treatment.
Methods
Mice
Inbred C57BL/6 FoxP3DTR were kindly provided by Geoffrey Hill,23 while BALB/c FoxP3DTR mice were generated by backcrossing C57BL/6 FoxP3DTR mice for 10 generations to BALB/c WT mice. All mice were bred and maintained at the QIMR Berghofer Medical Research Institute. Mice aged 6–12 weeks were used in all experiments and performed in accordance to QIMR Berghofer Medical Research Institute animal experimental ethics committee guidelines. Additionally, mice were scored for clinical symptoms of illness taking into account posture, activity, fur texture, blepharitis and weight change; as previously described.19 Mice were monitored daily and euthanized when clinical symptoms reached the cumulative limit outlined by animal ethics.
Cell lines
BALB/c-derived 4T1.2 mammary carcinoma and C57BL/6-derived MC38 colon adenocarcinoma cell lines were maintained in RPMI or DMEM supplemented with 10% FCS, penicillin/streptomycin, and additional L-glutamine (Gibco, Waltham, USA) as previously described.19 All cell lines were routinely tested as negative for mycoplasma. Cell line authentication was not routinely performed.
Experimental tumor models
BALB/c or C57BL/6 FoxP3DTR mice were injected in the mammary fatpad or subcutaneously (s.c.) with either 4T1.2 (1 x 105) or MC38 cells (1×106), respectively. Tumor growth was measured regularly by caliper square measurements and treatment started when the mean tumor size was over 40 mm2. Mice were euthanized when tumor size reached 150 mm2. To deplete Tregs and uncover antibody treatment toxicity, mice were injected intraperitoneally with 250 ng of diphtheria toxin (DT) (Sigma-Aldrich, St Louis, USA), diluted in PBS. Some mice additionally received treatment at the indicated dose and schedule with 200 µg rat control IgG2a (1-1; Leinco Technologies, St. Louis, USA), 100 µg anti-CD40 (FGK4.5, BioXCell, West Lebanon, USA), 200 µg anti-TNFα (TN3-19.12; Leinco Technologies) and 500 µg anti-interleukin (IL)-6R (MR16-1; Chugai Pharma, Tokyo, Japan).
Serum analysis
Mice were bled from the retro-orbital sinus, allowed to clot, and centrifuged at 10 000 rpm for 10 min to separate sera. Cytokine levels (interferon (IFN)-γ, TNFα, IL-6) were determined using mouse cytometric bead array flex set as per manufacturer’s instructions (BD Biosciences, San Jose, USA). Serum alanine aminotransferase (ALT) levels were measured by a liquid ALT detection kit as per manufacturer’s instructions (Pointe Scientific, Canton Township, USA).
Histology
Mouse tissues were fixed in 10% neutral buffered formalin before being embedded in paraffin. Four µm sections were cut and stained with H&E. H&E-stained tissue sections were imaged using an Aperio AT slide scanner (Leica, Wetzlar, Germany) and analyzed by Aperio ImageScope. The pathology of mouse liver and colon tissues were scored blindly and independently by two persons, referring to Erben’s or Thoolen’s standards, respectively.24 25 Representative images of our scoring system are presented in online supplemental figure S1.
Supplemental material
Statistical analysis
Statistical analysis was performed using GraphPad Prism software V.8 (La Jolla, USA). Differences in tumor growth were determined by Mann-Whitney U test. Differences between measurements in groups were determined by a one-way analysis of variance (Kruskal-Wallis) test with Dunn’s post hoc analysis as indicated. Differences between survival curves were determined using a log-rank analysis. P values were considered significant with p<0.05 indicated with (*), p<0.01 with (**), p<0.001 with (***) and p<0.0001 with (****).
Results
Treg depletion and anti-CD40 treatment display different therapeutic index against established tumors
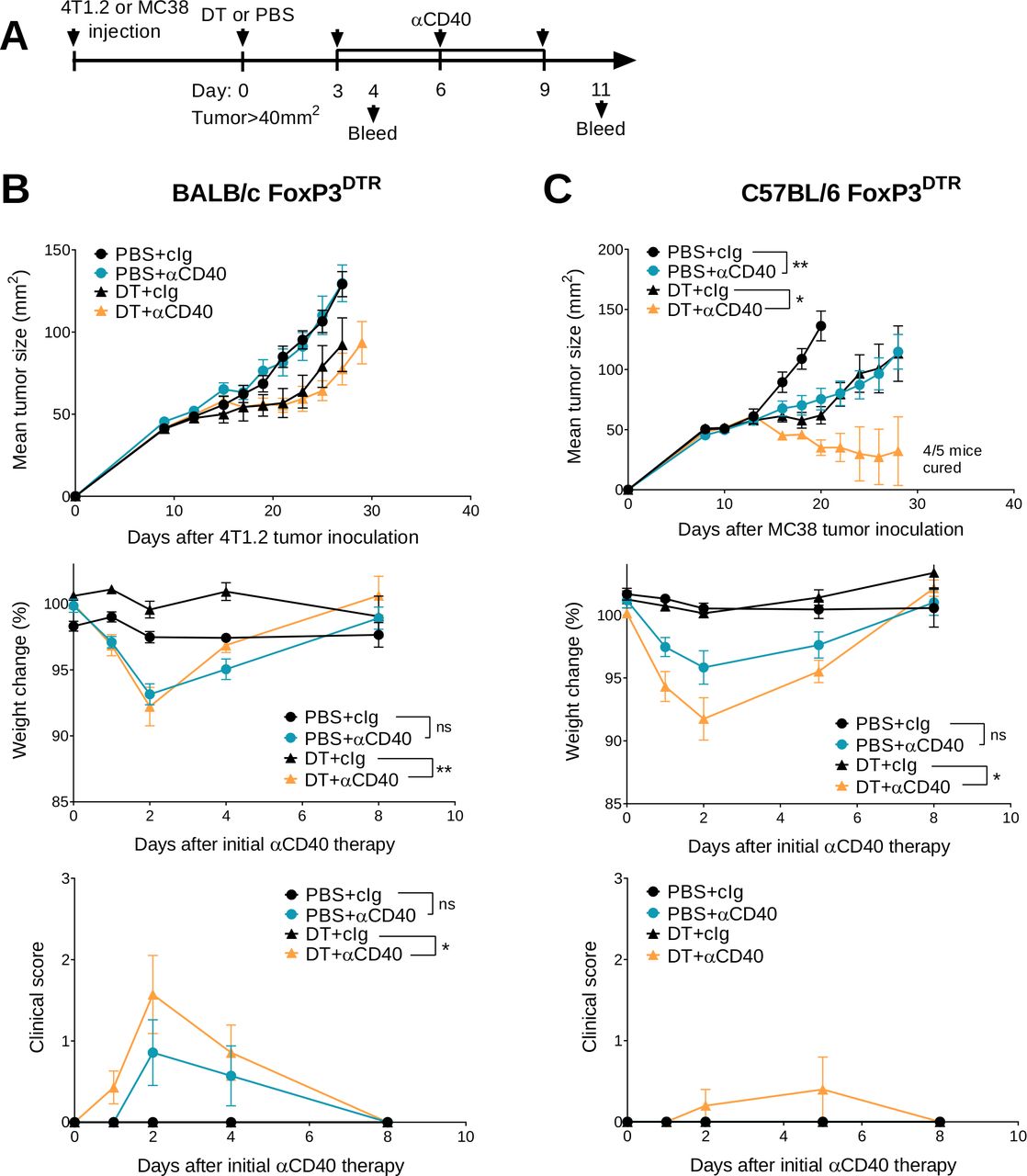
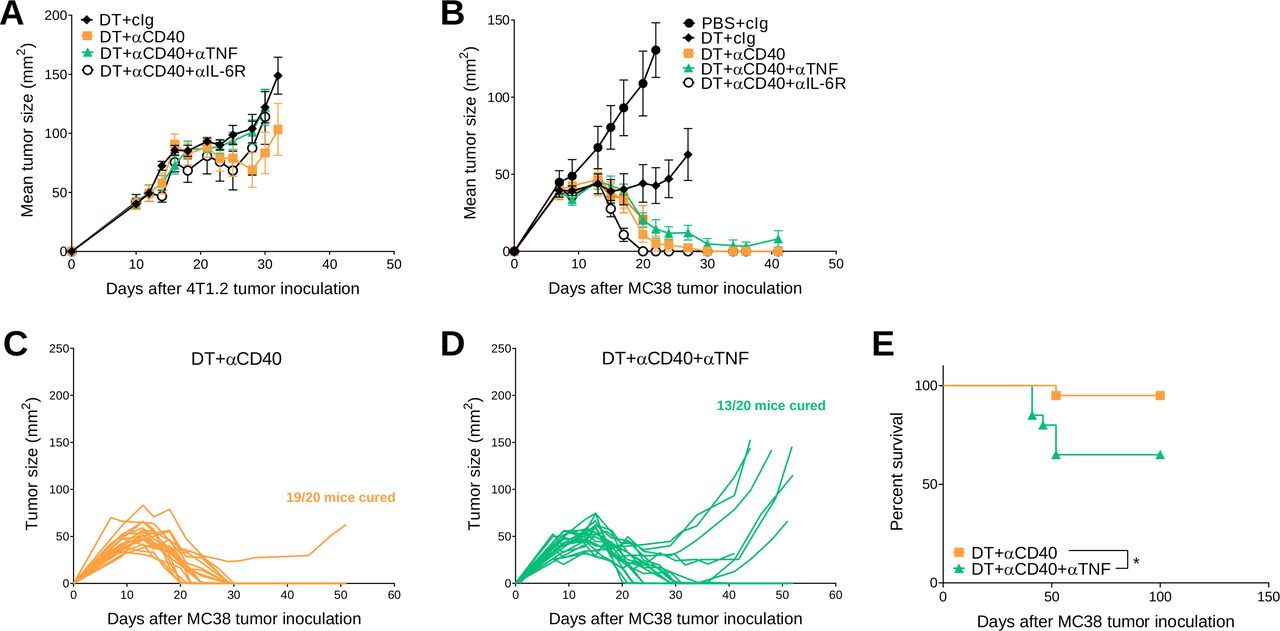
We previously demonstrated that transient Treg depletion in tumor-bearing FoxP3DTR mice (one dose DT) lowered immune tolerance and allowed biochemical and physical irAEs induced by any subsequent immunotherapy treatment to be more easily detected.19 Using this model, we assessed the efficacy and safety of anti-CD40 therapy in BALB/c or C57BL/6 FoxP3DTR mice bearing established 4T1.2 or MC38 tumors, respectively (figure 1A). Anti-CD40 treatment in combination with Treg depletion (DT+anti-CD40) displayed modest suppression of 4T1.2 tumor growth compared with the anti-CD40 alone or PBS+cIg-treated group (figure 1B and online supplemental figure S2A). In contrast, DT+anti-CD40 was effective against MC38 tumors, resulting in 80% cures (4/5) of mice (figure 1C and online supplemental figure S2B). Although PBS+anti-CD40- or DT+ cIg-treated groups suppressed MC38 tumor growth compared with the control treated group, this did not lead to cures. In both models, the majority of mice treated with DT+anti-CD40 developed visible signs of illness as measured by weight loss and increase in clinical score, as previously defined.19 This includes the presence of blepharitis, weight loss, changes in posture, activity and fur texture (figure 1B,C). As expected, clinical scores were higher in the BALB/c compared with the C57BL/6 strain where irAEs was observed even in the PBS+anti-CD40-treated group.
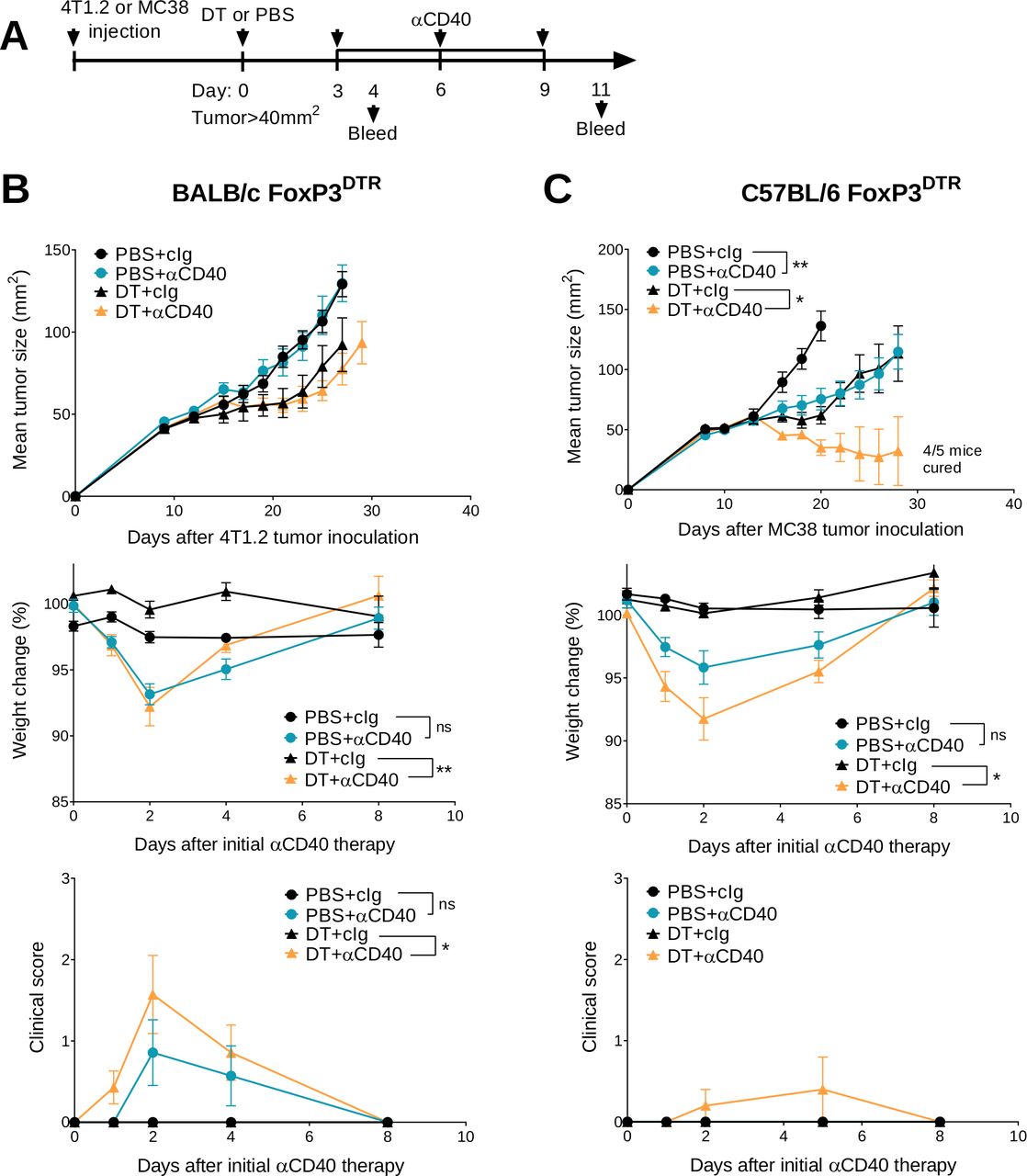
Transient Treg depletion and anti-CD40 treatment display different therapeutic index against established MC38 and 4T1.2 tumors. (A) Schematic representation of the treatment protocol. (B) BALB/c or (C) C57BL/6 FoxP3DTR mice were injected with 1×105 4T1.2 or 1×106 MC38 tumor cells, respectively. When tumors reached a mean size of 40 mm2, mice were treated intraperitoneally (i.p.) with PBS or DT and 3 days later treated i.p. with control IgG (cIg) or anti-CD40. Antibody treatment continued for two more doses given 3 days apart. Mean tumor size (mm2) with their corresponding weight change and clinical score are shown. Data representative of two experiments (n=5–7/group; mean±SEM). Mice were monitored for tumor growth and development of irAEs and euthanized when tumor size or clinical signs of illness reached cumulative ethical limits. Statistical comparisons between tumor sizes were performed with Mann-Whitney U test analysis at the final measurement of the PBS+ CIg-treated group, while a Kruskal-Wallis test with Dunn’s post hoc analysis was used to compare weight change and clinical score between the indicated groups (at day 2), *p<0.05, **p<0.01. DT, diphtheria toxin; irAEs, immune-related adverse events; ns, not significant; Treg, T regulatory cells.
Given that the main adverse events seen in humans following anti-CD40 therapy are rapid CRS and transient liver toxicity,14 we measured serum concentrations of pro-inflammatory cytokines and liver ALT in these two mouse models, 24 hours after the first anti-CD40 injection (figure 2). DT +anti-CD40 treatment induced liver damage in BALB/c FoxP3DTR mice (figure 2A) and to a lesser extent in C57BL/6 FoxP3DTR mice (figure 2B), as evidenced by higher levels of ALT in the former compared with the latter. A significant increase in ALT in PBS+anti-CD40-treated groups was observed for both strains, although the levels were again higher in the BALB/c mice compared with the C57BL/6 mice. TNF and IL-6 production were also increased and in both models the levels were higher in the DT+ anti-CD40 group. The production of these inflammatory cytokines is likely to correlate with the clinical score observed (figure 1). IFN-γ levels also increased although their levels were similar between the DT+anti-CD40- and PBS+anti-CD40-treated groups. Consistent with the transient symptoms observed in humans, the increase in ALT and inflammatory cytokines in our mouse models also occurred early with levels dropping 8 days after treatment initiation (online supplemental figure S3). By assessing both antitumor efficacy and irAEs, these results demonstrate the better therapeutic index of anti-CD40 therapy in MC38 compared with 4T1.2 tumor-bearing mice.
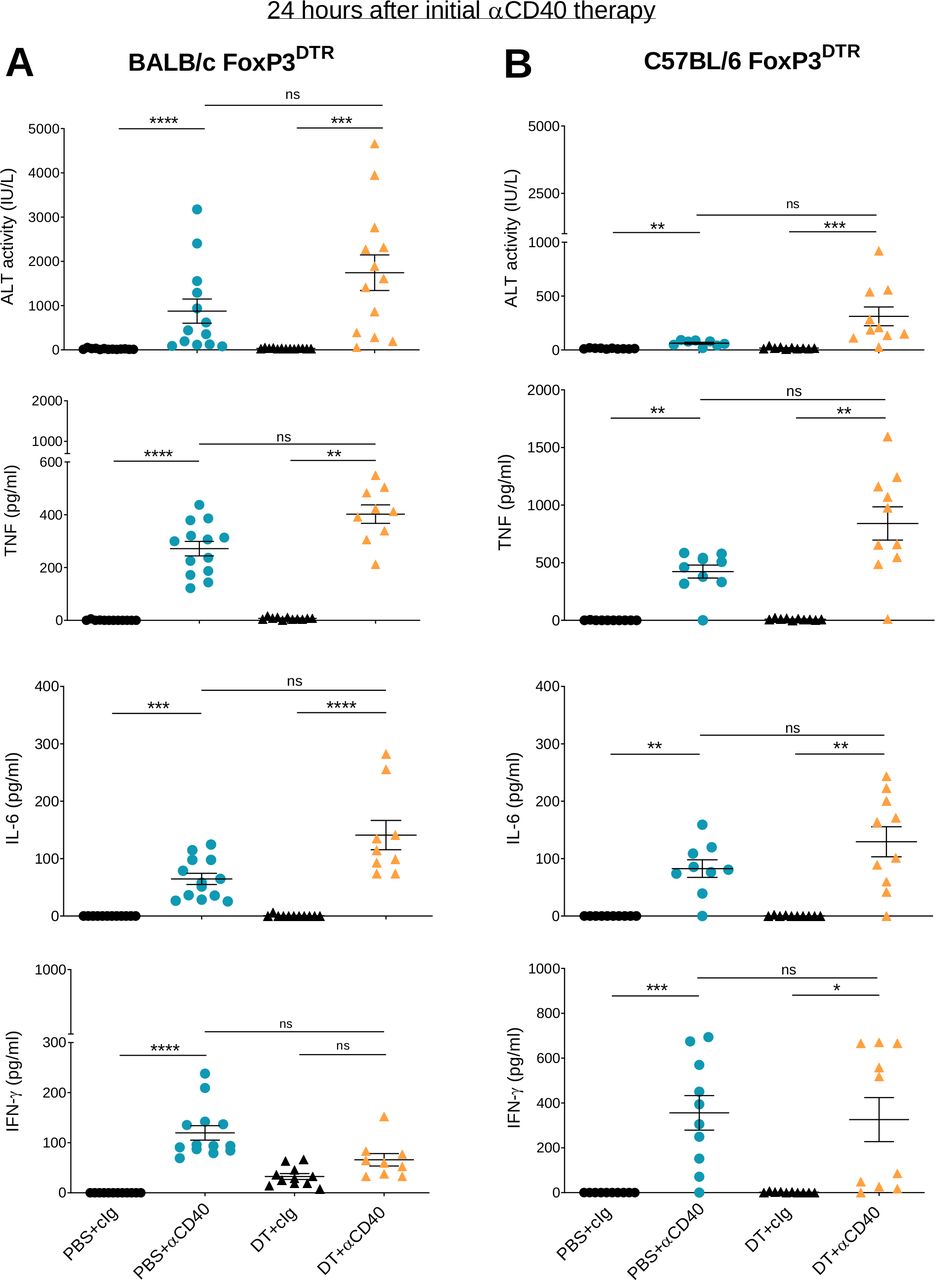
Anti-CD40 induces liver damage and release of inflammatory cytokines within 24 hours of administration. From the same experiments as described in figure 1, sera from (A) 4T1.2 and (B) MC38 tumor-bearing mice were collected 24 hours after the start of anti-CD40 treatment. ALT activity, TNF, IL-6 and IFN-γ levels were measured. Data pooled from two independent experiments (n=4–7/group/experiment; mean±SEM) with significant differences determined by Kruskal-Wallis test with Dunn’s post hoc analysis, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ALT, alanine aminotransferase; IFN-γ, interferon-γ; IL-6, interleukin 6; ns, not significant; TNF, tumor necrosis factor.
TNF blockade reduces physical and biochemical irAEs induced by anti-CD40 treatment
Cytokine blockade is a recommended second-line therapy to treat irAEs induced by different immunotherapies. Infliximab, a TNF neutralizing antibody, is used to treat severe colitis induced by ICI that is refractory to steroids,26 27 and recently, preclinical studies have suggested its use in a concomitant manner.21 22 Targeting the IL-6/IL-6R signaling pathway is also another approach, with the IL-6R blocking antibody tocilizumab already used for the management of severe CRS that can occur following CAR-T cell therapy.28 Therefore, we asked whether concomitant injection of TNF or IL-6R blocking antibodies improved the therapeutic window of 4T1.2 or MC38 tumor-bearing mice treated with DT+ anti-CD40 displaying high and low grade irAEs, respectively (figure 3A). TNF neutralization significantly reduced clinical score (figure 3B,E and online supplemental video 1), diminished weight loss (figure 3C,F) and reduced ALT levels in DT+ anti-CD40-treated mice (figure 3D,G). As expected TNF levels were significantly reduced in the DT+anti-CD40-treated groups that received anti-TNF compared with those that did not, while IL-6 levels were similar between these two groups (figure 3H,I). Interestingly, in the BALB/c mice treated with DT+anti-CD40+anti TNF, the levels of IFN-γ significantly increased compared with the DT+anti-CD40-treated group, although this effect was not observed in the C57BL/6 strain (figure 3H,I). While anti-TNF ameliorated both physical and biochemical irAEs induced by DT+anti-CD40, we did not observe this effect in mice treated with anti-IL-6R (figure 3B–G). Furthermore, in DT+anti-CD40-treated mice, TNF levels significantly increased in the group that received IL-6R blockade (figure 3H,I), although by day 8 the levels had decreased (online supplemental figure S4). This may explain why the clinical symptoms of these mice were not improved and ALT levels still significantly increased by day 8 (online supplemental figure S4). An increase in IL-6 was also observed (figure 3H,I) which is in line with clinical observations following tocilizumab injection.29 Overall, these observations suggest that neutralizing TNF rather than blocking IL-6R is more efficacious in managing irAEs and reducing liver injury induced by agonistic anti-CD40 treatment.
Supplementary video

Anti-TNF but not anti-IL-6R reduces physical and biochemical irAEs induced by anti-CD40. (A) Schematic representation of the treatment protocol. (B–D, H) BALB/c or (C–E, I) C57BL/6 FoxP3DTR mice were injected with 1×105 4T1.2 or 1×106 MC38 tumor cells, respectively. When tumors reached a mean size of 40 mm2, mice were treated i.p. with DT and 3 days later treated i.p. with cIg or anti-CD40 and continued for two more doses given 3 days apart. In, some groups, mice were additionally treated with anti-TNF or anti-IL-6R at the same time as anti-CD40. (B, E) Clinical score and (C, F) weight change are shown. (D, G) 24 hours after the start of anti-CD40 treatment, sera were collected and ALT activity was measured. (H–I) From the same sera samples, IL-6, TNF and IFN-γ levels were measured. Data pooled from two experiments for BALB/c and performed once for C57BL/6 (n=5–7/group/experiment; mean±SEM). Kruskal-Wallis test with Dunn’s post hoc analysis was used to compare the indicated groups (clinical score and weight was compared at day 5), *p<0.05, **p<0.01, ***p<0.001, ***p<0.0001. ALT, alanine aminotransferase; DT, diptheria toxin; IFN-γ, interferon-γ; IL-6, interleukin 6; i.p., intraperitoneally; irAEs, immune-related adverse events; ns, not significant; TNF, tumor necrosis factor.
Concomitant TNF neutralization may negatively impact on antitumor efficacy of DT plus anti-CD40 treatment
Next, we assessed whether cytokine neutralization or blockade affected the antitumor efficacy of DT +anti-CD40-treated tumor-bearing BALB/c or C57BL/6 FoxP3DTR mice (figure 4 and online supplemental figure S5). As shown previously (figure 1), DT+anti-CD40 was generally ineffective against 4T1.2 tumors and cotreatment with anti-TNF or anti-IL-6R did not positively or negatively affect tumor growth (figure 4A). In C57BL/6 FoxP3DTR mice treated with DT+anti-CD40, all mice rejected their tumors (7/7), but interestingly in the DT+anti-CD40+anti-TNF-treated group, not all mice were cured (5/7)(figure 4B), suggesting that concomitant TNF neutralization may affect the antitumor efficacy of anti-CD40 in a proportion of these mice. Therefore, we repeated this experiment where a larger cohort of MC38 tumor-bearing mice were treated with DT+ anti-CD40 or DT+anti-CD40+anti TNF (figure 4C,D) and we confirmed that the rate of tumor rejection (19/20 vs 13/20 cures, respectively) was negatively affected in the group that received concomitant TNF neutralization. This translated to a significant loss in survival in the anti-TNF treated group (figure 4E). These data contradicted a previous report from Perez-Ruiz et al, who showed that concomitant TNF neutralization in combination with ICIs (anti-PD1/anti-CTLA4) ameliorated irAEs and improved the survival of MC38-tumor-bearing mice.21 Given that the TNF antibody clone used in our study was different to that used in the Perez-Ruiz study, we set up an experiment to replicate their findings and confirmed that TNF blockade did not negatively impact on the antitumor efficacy of anti-PD1/anti-CTLA4 (online supplemental figure S6).

Concomitant anti-TNF negatively impacts antitumor efficacy of DT+ anti-CD40 treated MC38 tumor-bearing mice. (A, B) From the same experiments as described in figure 3 (A) 4T1.2 or (B–E) MC38 tumor growth curves. (A) Mean tumor size represented as mean±SEM (n=6/group). Data representative of two experiments. (B) Mean tumor size represented as mean±SEM (n=5–7/group). Experiment performed once. (C, D) In a similar treatment protocol as figure 4B, individual tumor growth curves of MC38 tumor-bearing mice treated with (C) DT+ anti-CD40 or (D) DT+ anti-CD40+ anti-TNF are shown (n=20/group). (E) Corresponding survival of pooled experiments from B to D. Statistical comparisons between the indicated groups were performed with a log-rank analysis, respectively, *p<0.05. DT, diphtheria toxin; IL-6R, interleukin 6 receptor; TNF, tumor necrosis factor.
Delaying anti-TNF uncouples toxicity and antitumor efficacy of anti-CD40 treatment
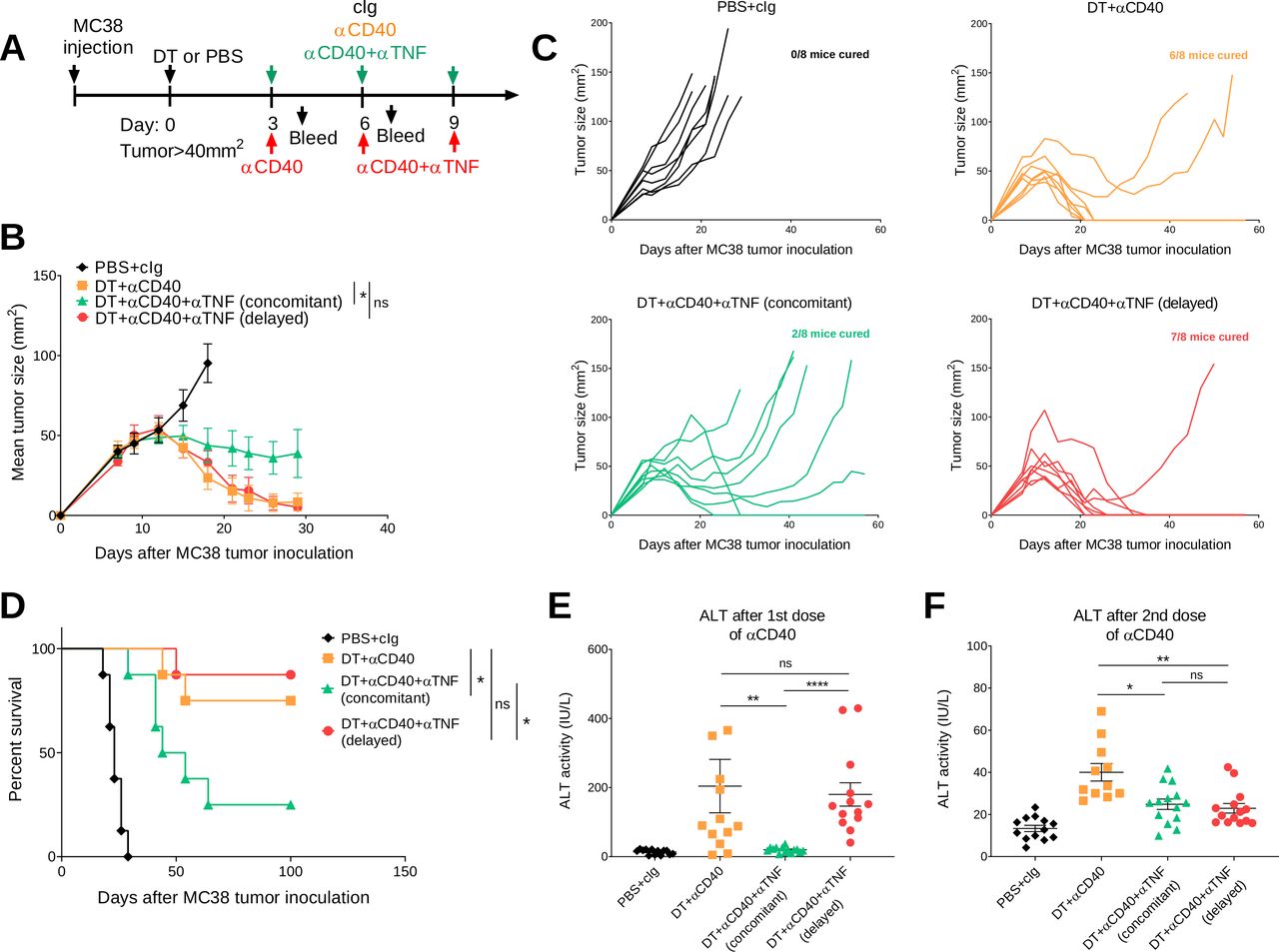
Clinically, irAEs are generally treated when they arise rather than concomitantly. In our study, anti-TNF was given concomitantly, but we wanted to examine how delaying anti-TNF impacted on the antitumor efficacy and irAEs induced by anti-CD40 therapy in both the C57BL/6 and BALB/c FoxP3DTR mouse strains (figures 5 and 6). In one group of DT+anti-CD40-treated mice bearing MC38 tumors, anti-TNF was given 3 days after mice received their first anti-CD40 injection (figure 5A). Delaying the injection of anti-TNF rescued the antitumor efficacy of anti-CD40 therapy in comparison with the group receiving concomitant injections from the start (7/8 vs 2/8 cures, respectively, figure 5B–D). In the C57BL/6 strain, which presents with lower grade irAEs compared with the BALB/c strain, delayed TNF blockade in DT+anti-CD40-treated mice could still decrease ALT and inflammatory cytokines to levels comparable to mice that received concomitant anti-TNF (figure 5E,F) (online supplemental figure 7). However, delayed anti-TNF did not improve the physical irAEs of these mice as measured by their clinical score and weight loss (online supplemental figure 7). Overall, in the MC38 tumor model, delaying anti-TNF treatment improved the therapeutic window of anti-CD40 treatment in contrast to concomitant treatment with anti-TNF.
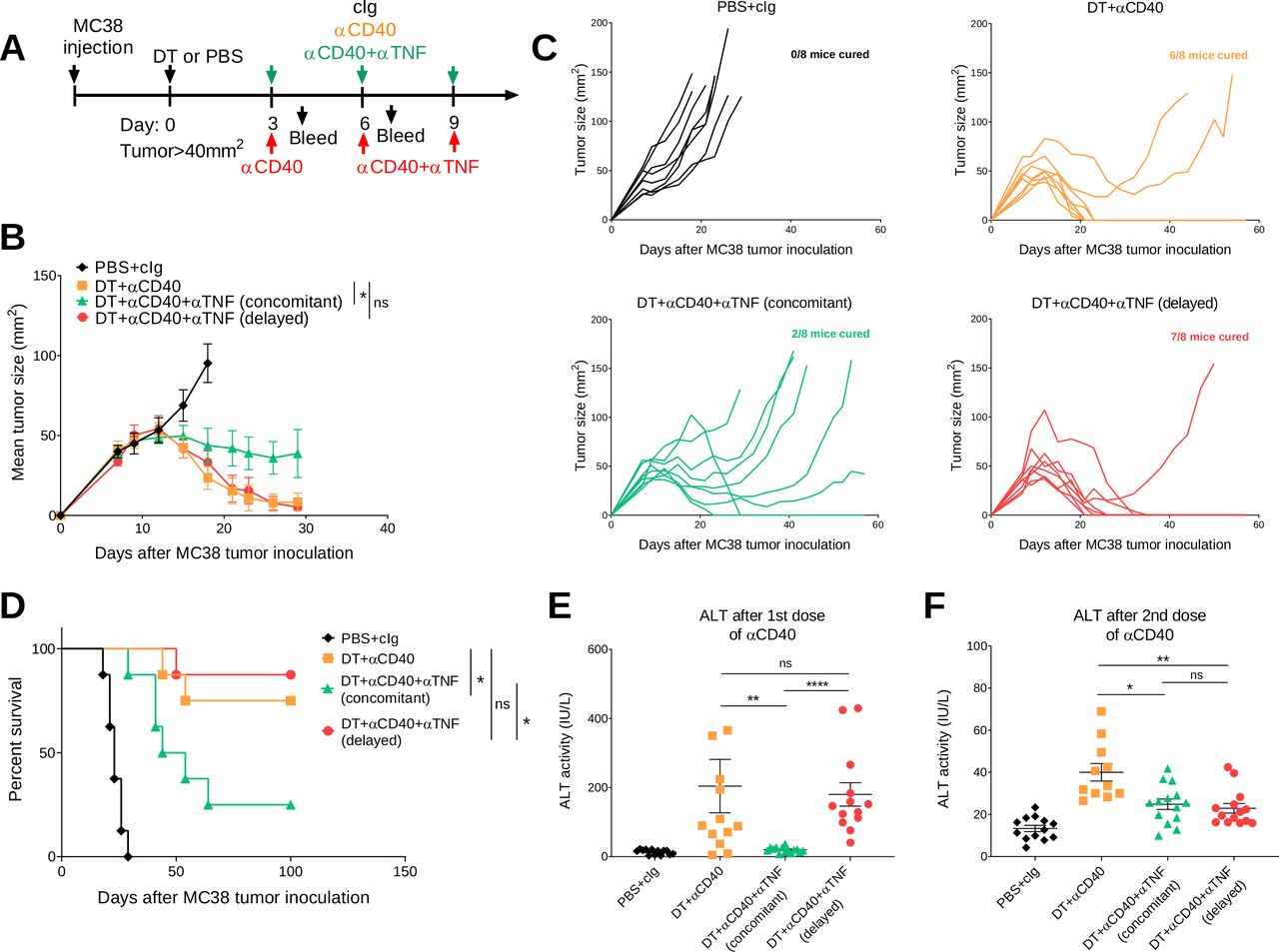
Delayed anti-TNF reduces irAEs while maintaining antitumor efficacy in DT+ anti-CD40 treated MC38 tumor-bearing mice. (A) Schematic representation of the treatment protocol. C57BL/6 FoxP3DTR mice were injected s.c. with 1×106 MC38 tumor cells. When tumors reached a mean size of 40 mm2, mice were treated i.p. with PBS or DT and 3 days later treated i.p. with cIg, anti-CD40 or anti-CD40+anti TNF (concomitant), for three doses given 3 days apart. In one group, anti-TNF treatment (delayed) was given at the time of the second dose of anti-CD40 and then given for one more dose. (B) Mean tumor size represented as mean±SEM (n=8/group). (C) Individual tumor growth curves of the indicated groups. Data representative of two experiments. (D) Corresponding survival curves. Statistical comparisons between tumor sizes and survival curves were performed with Mann-Whitney analysis at the final measurement of the PBS+ CIg-treated group or log-rank analysis, respectively, *p<0.05. From the same mice, sera was collected (E) 24 hours after the start of anti-CD40 treatment or (F) 24 hours after the second anti-CD40 treatment and ALT activity was measured. Data pooled from two independent experiments (n=5–8/group/experiment; mean±SEM) with significant differences determined by Kruskal-Wallis test with Dunn’s post hoc analysis, *p<0.05, **p<0.01, ****p<0.0001. ALT, alanine aminotransferase; DT, diphtheria toxin; i.p., intraperitoneally; irAEs, immune-related adverse events; ns, not significant; TNF, tumor necrosis factor.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Concomitant compared with delayed anti-TNF is more effective at reducing both physical and biochemical irAEs in DT+anti-CD40 treated 4T1.2 tumor-bearing mice. (A) Schematic representation of the treatment protocol. BALB/c FoxP3DTR mice were injected with 1×105 4T1.2 tumor cells. When tumors reached a mean size of 40 mm2, mice were treated i.p. with PBS or DT and 3 days later treated i.p. with cIg, anti-CD40 or anti-CD40+ anti-TNF (concomitant). One group was treated with anti-TNF 24 hours later (delayed). (B) Weight change and (C) clinical score are shown. (D–F) Indicated organs were taken 96 hours after the start of anti-CD40 treatment. The organs were fixed in paraformaldehyde for 24 hours (colon) or 72 hours (liver), embedded, H&E stained and sliced. (D) Representative images from H&E-stained sections of colon (scale bar 100 µm), liver (scale bar 300 µm) and necropsy pictures of the liver. Histological (E) colon and (F) liver scores. Data representative of two experiments (n=6–8/group). Kruskal-Wallis test with Dunn’s post hoc analysis (clinical score and weight was compared at day 4), colon and liver scores of indicated groups were compared with the corresponding DT+ cIg treated group, *p<0.05, **p<0.01, ***p<0.001. i.p., DT, diphtheria toxin; intraperitoneally; irAEs, immune-related adverse events; ns, not significant; TNF, tumor necrosis factor.
Concomitant anti-TNF is more effective than delayed TNF neutralization
We next assessed the impact of delaying TNF blockade in DT+anti-CD40-treated BALB/c FoxP3DTR mice, given they displayed more severe irAEs. Here, anti-TNF was given concomitantly or 24 hours after anti-CD40 treatment, when irAEs developed (figure 6A). In this model, delayed anti-TNF was not as effective as when it was given concomitantly, since weight loss was not prevented (figure 6B). While concomitant anti-TNF-treated mice never displayed increased clinical scores, delayed anti-TNF rapidly decreased clinical score compared with DT+anti-CD40-treated mice (figure 6C and online supplemental video 2). Although delaying TNF neutralization did not reduce ALT levels in sera obtained 24, 48 and 72 hours after anti-CD40 treatment, TNF levels decreased at 48 hours while IL-6 and IFN-γ levels were not affected (online supplemental figure S8A‒C). Interestingly, in the mice that received concomitant anti-TNF, TNF levels initially decreased at the 24 and 48 hours time points, but surprisingly increased at the 72 hours time point compared with the DT+anti-CD40-treated group, although this increase did not affect the clinical score.
Supplementary video
Clinically, agonist CD40 antibody treatment was reported to induce acute inflammation and hepatitis,14 30 and was also linked to the induction of colitis in preclinical models.31 In our study, mild diarrhea was also observed in DT+ anti-CD40-treated BALB/c and C57BL/6 FoxP3DTR mice (data not shown). Therefore, we performed a histological analysis of various organs 4 days after treatment with anti-CD40 to understand the impact of concomitant or delayed TNF neutralization in the most affected tissues. Strikingly, we observed a strong immune cell infiltration in the colon and liver of every mouse treated with DT plus anti-CD40, resulting in severe lobular necrosis, noticeable white spots in the liver tissue and colon inflammation (figure 6D–F). In contrast, concomitant anti-TNF completely abrogated immune-related hepatitis and colitis induced by DT+ anti-CD40, while delayed TNF blockade merely reduced colon and liver scores, even when the clinical scores of these mice were significantly improved (figure 6C–E). A mild increase in spleen, but not colon weights was detected in all anti-CD40-treated groups compared with cIg-treated groups (online supplemental figure 8D,E). However, a reduction in colon lengths was only observed in the DT+anti-CD40-treated group, suggestive of ongoing colitis (online supplemental figure S8F). Overall, these data suggest concomitant compared with delayed anti-TNF was more effective in reducing high-grade irAEs.
Discussion
Agonistic CD40 antibodies were among the first agents to demonstrate the therapeutic advantage of targeting immune costimulatory receptors in cancer.7 However, the dose-limiting toxicity of this treatment hampered its use in the clinic, stressing the need to better understand whether its toxicities could be uncoupled from its antitumor activity. In this study, we used transient Treg depletion in two strains of FoxP3DTR mice to lower immune tolerance and study the irAEs and antitumor activity induced by subsequent anti-CD40 therapy. In both strains, we observed transient liver toxicity and rapid release of proinflammatory cytokines similar to what was observed in early clinical trials of CD40 agonists. In particular, in the BALB/c strain, DT+anti-CD40 treatment induced severe physical and biochemical irAEs rapidly after treatment while having modest effects on tumor growth. This included significant weight loss and an increase in clinical score, ALT levels and the proinflammatory cytokines, IL-6 and TNF. Importantly, concomitant compared with delayed anti-TNF in DT+anti-CD40-treated BALB/c FoxP3DTR mice reduced weight loss, liver damage and colitis, which consequently resulted in an improved clinical score. Similarly, concomitant compared with delayed anti-TNF was more effective in reducing biochemical and physical irAEs in DT+anti-CD40-treated tumor-bearing C57BL/6 FoxP3DTR mice. However, this came at a cost, given that concomitant anti-TNF reduced antitumor efficacy in a proportion of these mice.
The irAEs displayed in our mouse models, particularly in the BALB/c strain, mimicked the symptoms and kinetics observed in humans treated with CD40 agonists. This is in contrast to previous studies of agonistic antibodies, which generally evaluated irAEs in naïve wild-type mice.32 33 In our study, we observed that the increase in ALT levels following DT+anti-CD40 treatment was five times lower when injected into naïve mice (data not shown). This finding suggested that tumor-bearing mice should be used, considering that inflamed tissues and the immune system might exacerbate the toxicity of an antibody treatment. Indeed, it was shown that tumor-bearing mice displayed higher levels of liver damage compared with tumor-free mice.30 While C57BL/6 mice are the most commonly used strain to study antitumor responses, they do not display the full spectrum of irAEs and severity seen in humans.34 Similarly in our study, the irAEs observed in DT+anti-CD40-treated C57BL/6 FoxP3DTR mice were less severe compared with their BALB/c FoxP3DTR counterparts.
The anti-TNF blocking antibody infliximab is currently used as a second-line treatment for the management of gastrointestinal toxicity induced by ICIs, but its use for immune-related hepatotoxicity is more controversial. The current ESMO clinical practice guidelines do not recommend using anti-TNF treatment for hepatitis, based on case reports correlating infliximab treatment with drug-induced liver injury.35 Although significant toxicities were observed in some patients, the incidence of these events was fairly low and liver injury was reversible after anti-TNF discontinuation.36 37 In the current study, we showed that concomitant TNF neutralization completely abrogated immune-related colitis and hepatitis in mice treated with anti-CD40, similar to that previously shown in a pre-clinical study assessing the use of concomitant anti-TNF to prevent irAEs induced by anti-PD1/anti-CTLA4.21 Anti-TNF has also previously been shown to reduce anti-CD40 induced liver toxicities.38 Interestingly in our study, IL-6R blockade did not attenuate physical and biochemical irAEs. This is despite previous reports suggesting that blockade of IL-6R might be used to manage irAEs after immunotherapy in solid tumors,29 39 and that IL-6R blockade had synergistic antitumor efficacy when combined with ICIs.40 These findings demonstrate the importance of understanding the hierarchy of immune pathways activated or released by the specific immunotherapy that is causing the irAEs. Going forward, further studies are necessary to evaluate and reconsider the clinical benefit of TNF neutralization in the management of hepatic irAEs.
TNF can exert positive and negative effects on cancer immunity.41 42 In our study, we showed that TNF was involved in the induction of irAEs but also seemed to have a role in the antitumor efficacy of anti-CD40 as seen by a loss of tumor growth suppression in a proportion of DT+ anti-CD40-treated MC38 tumor-bearing mice that received concomitant anti-TNF. By delaying anti-TNF, we prevented this loss of antitumor efficacy but the trade-off was a reduction in biochemical but not physical irAEs. Interestingly, in DT+anti-CD40-treated MC38-tumor-bearing mice given concomitant anti-TNF, early tumor growth suppression was observed in most mice but over time, the antitumor immune response failed in a proportion of these mice. Analysis of T cell numbers and their immune phenotype in these tumors may shed light on the reason(s) for their outgrowth. Future studies will need to investigate why concomitant anti-TNF negatively impacts anti-CD40 treatment while having a positive effect when combined with ICIs in the same tumor model.21 22 One possibility to explain the negative impact of concomitant anti-TNF is the possible role of this cytokine in the immune response triggered by CD40 agonist treatment. Several studies have shown that TNF is a potent antitumor factor,43 44 and that the expression of CD40 and TNF correlated with better survival of colorectal cancer patients.45 It was also shown that TNF neutralization decreased the expression of CD40 on intestinal microvascular cells in Crohn disease patients.46 One potential mechanism that may explain the detrimental effect of anti-TNF in combination with anti-CD40 might then be that anti-TNF decreased CD40 expression on APCs. Clinically, questions remains as to the role anti-TNF may play on promoting or suppressing the antitumor efficacy of ICIs.41 Currently, two trials addressing this question have obtained contradictory results with one study demonstrating infliximab abrogated the survival advantage given by ICI treatment47 while another study reported improved survival of patients with cancer.27 Considering the known dual role of TNF in tumor development, addressing this question is of the utmost importance before generalizing the use of infliximab in the clinic.
While anti-TNF is one strategy to attenuate irAEs, other approaches to reduce anti-CD40 dose-limiting toxicity are also being investigated.5 13 In a preclinical study, an engineered CD40 mAb conjugated with extracellular matrix-binding peptide improved antitumor efficacy by improving local delivery and reducing systemic treatment-related adverse events.48 Alternatively, in preclinical mouse models, local treatment of slow-release agonistic anti-CD40 antibody directly into tumors was shown to induce robust antitumor CD8+ T cell responses without systemic toxicity.49 In patients with metastatic melanoma, a phase I/II trial is currently assessing the safety of intratumor administration of CD40 agonist mAb APX005M in combination with systemic anti-PD1 (NCT02706353).50 Two clinical trials have also included arms where the CD40 agonist selicrelumab is injected s.c. to limit adverse events, in combination with anti-VEGF (vascular endothelial growth factor) (NCT02665416) or anti-PD-L1 (NCT02304393). Positive results from these clinical trials could inform on how best to administer CD40 agonists in the clinic to minimize its toxicity while preserving its antitumor efficacy. Finally, studies are also underway to design new CD40 mAb with enhanced agonistic function. Several studies demonstrated the importance of the inhibitory receptor FcγRIIb to crosslink CD40 agonistic antibodies,51 52 leading to the creation of human CD40 mAb Fc variants with selectively enhanced binding to FcγRIIB.53 These variants had a 30-fold to 90-fold increased binding ability and improved antitumor efficacy, but a higher dose-dependent toxicity that was abrogated by intratumor injection.33 Another study showed that a switch from IgG4 to IgG2 isotype transformed a human antagonist CD40 mAb into a super-agonist with improved antitumor efficacy, highlighting the importance of the appropriate isotype in the design of CD40 agonist antibodies.54 However, the mouse strain and experimental assays used in this study does not fully inform on the potential toxicity of the new variant. Additional studies are required to assess if improving agonistic function and modulating FcγR binding of CD40 mAb is the safest option to increase its therapeutic window.
Overall, our study demonstrated the strength of an antitumor response and irAEs severity induced by any particular immunotherapy can differ depending on the tumor type and its microenvironment and host genetics. In the 4T1.2 tumor model, anti-CD40 displayed a low therapeutic index given it had modest antitumor efficacy but induced severe irAEs. In contrast, the same immunotherapy had a higher therapeutic index against the MC38 tumor model given it eradicated tumors in a proportion of treated mice while inducing less severe irAEs. The challenge for any immunotherapies whether it is used alone or in combination involves finding a balance between inducing a potent antitumor response, while limiting the severity of any potential irAEs. In this study, we demonstrate the utility of our mouse models to evaluate both the antitumor efficacy and irAEs induced by different immunotherapies. Furthermore, our models offer the opportunity to understand and decipher which immune pathways are linked to development of irAEs and/or required for antitumor immunity.
Acknowledgments
We thank Liam Town, Brodie Quine, and Andreea Zaharia for breeding, genotyping, and maintenance and care of the mice used in this study. We thank Eric Thomas for help in editing the supplementary videos.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @michele_teng
Contributors Conception and design: CJ-F, SB and MT. Development of methodology: CJ-F, SB and JL. Acquisition of data: CJ-F, SB, JL, EM and HT. Analysis and interpretation of data: CJ-F, SB, JL, EM, KN and MT. Study supervision: MT. Writing, review and/or revision of the manuscript: all authors.
Funding MT was supported by a NH&MRC Career Development Fellowship (1159655). MS was supported by a NH&MRC Senior Principal Research Fellowship (1078671) and a NH&MRC Program, grant (1132519).
Competing interests MJS is funded by research agreements from Bristol Myers Squib and Tizona Therapeutics and has an advisory board role for Tizona Therapeutics and Compass Therapeutics.
Patient consent for publication Not required.
Ethics approval All animal experiments were performed in accordance with QIMR Berghofer Medical Research Institute animal experimental ethics committee guidelines.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data sharing not applicable as no datasets generated and/or analyzed for this study.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.