Article Text

Abstract
The synapses between immune cells and their targets are 150 Å wide. They regulate immune cell responses (IRs) to cognate antigens. Here, I outline a potential mechanism for self-nonself discrimination based on the C3d and iC3b proteolytic fragments of complement protein C3. The proposed C3 checkpoint works through complement receptor 3 (CR3), which binds both C3d and iC3b. The CR3 conformations involved differ; the bent, cis-acting CR3 engages C3d, activating the immune cell expressing CR3; the extended, transacting CR3 conformer binds iC3b on another cell, inhibiting IRs. The CR3 complexes formed with iC3b and C3d vary greatly in size. Only bound C3d is small enough to fit within the synapse. It stimulates IRs by countering the inhibitory signals that iC3b generates at the synapse edge. The competition between C3d and iC3b dynamically determines whether or not an immune cell activates. Host cells use regulators of complement activation (RCA) to coat themselves with iC3b, silencing IRs against self by preventing synapse formation. Tumors exploit this process by overexpressing C3 and RCA to masquerade as ‘super-self’, with iC3b masking neoantigens. Enhancing synapse formation by specifically labeling cancer cells as nonself with targeted C3d therapeutics offers a new strategy for boosting tumor-specific immunity.
- immunity
- innate
- immune tolerance
- immunotherapy
- inflammation
- tumor escape
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
The paradox of C3
The complement system is an ancient mechanism for self–nonself discrimination. Its role in cancer is paradoxical.1 Both complement activation and complement deficiency can promote tumor growth. For example, in genetic and syngeneic models of epithelial ovarian carcinoma, deletion of the complement C3 gene inhibits tumor growth.1 Similarly, in mouse models of melanoma, complement C3 deficiency delays progression.1 In contrast, C3 deficiency in the Her2/neu autochthonous mouse mammary carcinoma model leads to an earlier onset and accelerated tumor spread. In this opinion piece, I explore the complement paradox and integrate existing data to advance its resolution. I will describe a complement C3 checkpoint used by tumors to label themselves as ‘super-self’. By doing so, cancer cells silence immune responses (IRs) directed at neoantigens. In this scenario, iC3b leads to tolerance by labeling cells as ‘self’ while C3d initiates immune activation by signaling ‘non-self’ (figure 1).

The role of complement C3 in self–nonself discrimination. (A) The alternative (AP), classical (CP), lectin (LP) and other16 pathways drive the complement amplification loop driven by complement factors B and D (FB, FD) to produce the C3b convertase that activates additional C3 and C5 proteins. C3b is inactivated by compactor factor I (FI) to form either iC3b or C3d fragments. Complement factor H (FH), CD46 and CD55 favor iC3b production while Complement Receptor 1 (CR1) favors C3d formation by releasing the large C3c fragment. Tumors use the iC3b fragment to label themselves as ‘super-self’ to silence immune responses against the abnormal proteins they produce. CD59 prevents complement-mediated lysis of tumors by Complement C6, C7, C8 and C9 that together form a membrane pore. It is proposed here that C3d tags cells as nonself and favors antitumor responses. (B) Linear representation of C3b domains with cleavage sites indicated by ∧ and a white line. The order of cleavage is indicated by the numbers. Cleavages 1 and 2 produce iC3b by releasing C3f. The third cleavage at the other end of the thioester domain (TED) results in the production of C3dg which is trimmed by tissue proteases to give C3d. Cleavage releases C3c which consists of the β-chain connected to the α'1 and α'2 domains by disulfide bonds (indicated by lines above the domains). The C345C domain combines with C3d to create the CR3 binding site. (C) The four conformations of complement receptor 3 (CR3) with active states colored red. The bent (E−) or extended (E+) conformations and the low (H−) or high affinity (H+) affinity states of each play different roles in immune regulation as described in the text. The open state is associated with outside-in signaling. CR3 can transition from the bent inactive state to the fully extended high affinity state by path 1→2→ or 1→3→4.7 The E-H+ state is visualized in the crystal structure of the αXβ2 integrin ectodomain.17 (D) The domains of the CR3 α (CD11b) and β (CD18) chains with the I domain and I-like domain shown in blue and the flexible knee that bends in red.
Complement basics
Complement was discovered by Buchner and Bordet in the 1890s and has since been intensely studied by many distinguished scientists to give a detailed view of the proteins and the molecular interactions involved. The C3 protein plays a key role in the complement cascade.2 3 Once activated to form C3b, a positive feedback loop accelerates the conversion of additional C3 to C3b (figure 1A). The cycle is broken by the regulators of complement activity (RCA). RCAs act on C3b to coat host cells with iC3b, producing non-inflammatory outcomes. Pathogens lacking RCAs are tagged instead with the proteolytic fragment C3d, which promotes immunity.4
The Tag team
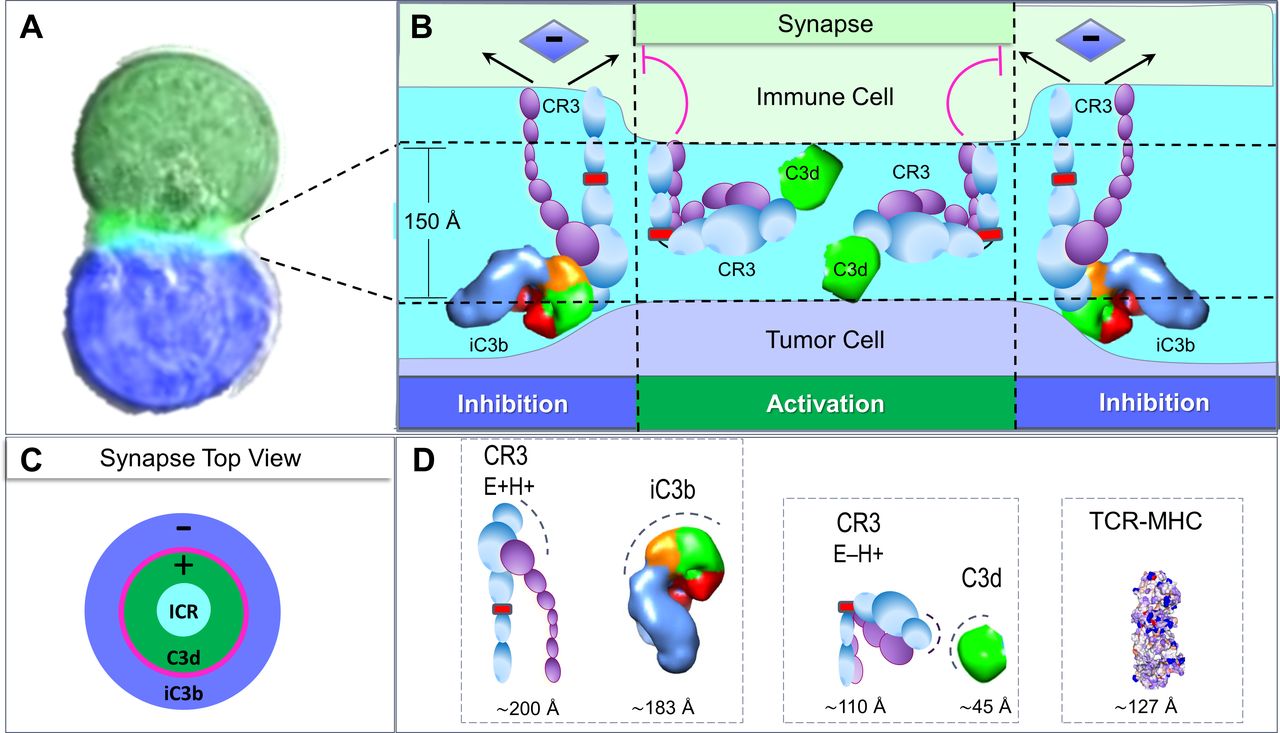
The biology of iC3b and C3d is fundamentally different. The receptors they engage and their size likely determines the outcomes they produce (figure 1C,D.). Size matters. At ~45 Å (PDB: 4M76), C3d is likely small enough to fit within the immune synapse (IS), which is 150 Å wide (figure 2).5 There it potentially modulates the signals that induce immune cell (IC) responses. In contrast, iC3b with a maximal dimension of ~183 Å6 is likely too large to fit within the IS.
{kind=link}
{kind=link}
Complement and the immune synapse. It is proposed that complement dependent immune outcomes depend on latent binding sites on both CR3 and C3 as well as the size of the C3 proteolytic Fragments. (A) An immunological synapse formed between two cells is shown in light green (source https://en.wikipedia.org/wiki/Immunological_synapse). (B) The complex of iC3b with the high affinity, extended form of CR3(E+H+) (see legend figure 1) is likely too large to fit within the synapse width. Instead, the complex acts on the cytoskeleton to oppose synapse formation and inhibit immune cell activation. The small size of C3d and the bent CR3(E−H+) receptor likely allows their accommodation within a synapse. The activated bent CR3(E−H+) opposes the cytoskeletal forces generated by iC3b that would otherwise collapse the synapse. Instead those forces, now unbalanced, promote synapse expansion by pulling outwards on the synapse edge. (C) A top overview of the synapse illustrating the different zones of activation and inhibition surrounding the central zone of antigen recognition by the immune cell receptor (ICR). The magenta line indicates the ring of competition between bent CR3(E−H+) and extended CR3(E+H+). (D) The size of iC3b (PDB: 2A73) and C3d (PDB: 4M76) fragments are compared with those of the extended and bent form of CR3 with latent binding sites indicated by a dashed line (adapted from6). The knee at which CR3 flexes is shown in red. The C3d domain is colored green and the C345C domain orange as in figure 1. The size of the T-cell receptor, major histocompatibility complex, peptide is shown for comparison (PDB: 3RGV). CR3, complement receptor 3; E+H+, extended and high affinity.
The many faces of complement receptor 3
C3d and iC3b interact with the complement receptor 3 (CR3, Mac-1, CD18/CD11b, αM/β2 encoded by ITGAM/ITG2B2 respectively), but they bind different CR3 conformations.7 CR3 traditionally is considered to adopt three states: extended and high affinity (E+H+), bent and low affinity (E–H–), extended and low affinity (E+H–).7 Recent electron-microscopy evidence supports a fourth CR3 state that is bent and of high affinity (E–H+) (figure 1C) that binds extracellular ligands through the αM I-domain (figure 1D).8 The bent state projects only 110 Å from the cell surface (figure 2D) and is small enough to fit within an IS. FRET studies of neutrophils reveal that bent CR3(E-H+) binds the intercellular adhesion molecule 1 (ICAM1) on the same cell (ie, in cis) and inhibits binding of extended CR3(E+H+) to ICAM1 on another surface (ie, in trans).7 Super-resolution microscopy further enables quantitation of the how the four CR3 states cluster together and how these interactions affect cellular responses.7
The bent CR3 (E–H+) can only bind complement fragments through its αM I-domain as other binding surfaces are buried (figure 2D).6 The αM I-domain binds C3d with high nanomolar affinity (KD ∼0.4 µM).2 The lower overall affinity of bent CR3(E–H+) for iC3b suggests that its C3d domain is often occluded and not available for binding.2 6
The interaction of bent CR3(E-H+) with C3d likely occurs in cis (ie, on the same cell surface that CR3 is on) (figure 2B), similar to the interactions of bent CR3(E-H+) with ICAM1.7 The complex formed is likely small enough to fit within an IS (figure 2A,B). The affinity of extended CR3(E+H+) for iC3b in trans is high due to binding sites newly exposed that engage iC3b by bringing its C3d and C345C domains together (figure 2D).6 By analogy with the neutrophil findings, the binding of bent CR3(E−H+) to C3d in cis likely competes with the interactions of extended CR3(E+H+) with iC3b in trans. Whether cis or trans outcomes predominate depends on the density of C3d and iC3b on each cell partner. The outcomes are binary as each ligand inhibits the binding of CR3 to the other.
Going big
The CR3(E+H+) extended conformation is the default conformation on macrophages.9 The interaction with iC3b inhibits T-Cell activation independently of any process related to antigen presentation.9 The clusters formed promote phagocytosis.10 In the case of tumors, the outcome is non-inflammatory.4
Antibody-mediated phagocytosis is more complex. It involves both extended CR3(E+H+) and bent CR3(E−H+). Here antibodies bridge small antigens (less than 150 Å) on one cell to a Fc receptor (FcR) on the other (figure 2E).10 The FcR interacts with bent CR3(E−H+) in cis to form a synapse that leads to phagocyte activation. The structure formed is encircled by extended CR3(E+H+)/iC3b complexes that signal in trans, excluding inhibitory phosphatases like CD45 and CD148 from the synapse while engaging the cytoskeleton to initiate phagocytosis.9 10 The process is dynamic, with competition between bent and extended CR3 determining the threshold for phagocytosis.7 9 10
Staying small
In contrast to the inhibition of T-Cells by fully extended CR3(E+H+), bent CR3(E-H+) can activate ICs. CR3 is expressed on T-Cells following antigen exposure and during primary and secondary antiviral responses in mice. Mice with CR3 deficiency show impaired T-Cell responses to the T-Cell super antigen staphylococcal enterotoxin A.9 The defect is in the T-cells themselves, not in the myeloid cells they bind. Stimulation of allogeneic IRs also depends on T-Cell expression of CR3.9 Human T-cells coated with covalently bound C3d show enhanced cytokine secretion after stimulation compared with C3d negative cells, both in patients with systemic lupus erythematosus and in normal subjects.11 Unmodified C3d expressed in tumors also improves their effectiveness as anti-tumor vaccines and enhances checkpoint inhibitor efficacy.12 Collectively these results are consistent with the notion that engagement of bent CR3(E-H+) with C3d activates ICs. This cis signaling event potentially involves C3d on both sides of the synapse as the two cell membranes are closely approximated (figure 2B). In this scenario, C3d initiates immune activation by signaling ‘non-self’ while iC3b produces tolerance by tagging cells as ‘self’.
Tumors own the IR
The competition between trans-inhibition and cis-stimulation of cell-mediated immunity offers new mechanistic insight into how tumors might evade IRs. Overproduction by tumors of both C3 (see https://www.proteinatlas.org/ENSG00000125730-C3/pathology for immunohistopathology performed with different antibodies) and RCA favors iC3b formation and the extended CR3(E+H+) interactions that lead to immune suppression. In the scenario proposed here, iC3b allows tumors to mask neoantigens by labeling themselves as ‘super-self’. By not generating C3d, cancer cells would then preclude the cis signaling needed for IC activation. Localizing iC3b and C3d fragments within tumors using appropriate antibodies would help experimentally validate this mechanism.
The battle between iC3b and C3d is likely mediated through engagement of the cytoskeleton by CR3, analogous to those events experimentally observed during antibody-mediated phagocytosis.10 The front line of the proposed clash is depicted by the ring in figure 2C. In this scenario, attachment of iC3b bound, extended CR3(E+H+) to actin generates forces that move proteins in the direction the fibers point. When unopposed, the strain compels the synapse to collapse. By inhibiting actin attachment within the synapse, C3d bound to bent CR3(E−H+) prevents synapse closure. Instead, the cytoskeletal forces become unbalanced and pull outwards to expand the synapse (figure 2B). Repair of the resulting membrane disruption by endosomal vesicles then allows the delivery of new antigen receptor complexes to the synapse outer edge, enhancing signaling.13
While the effects of iC3b and C3d described here are on the local IS, they also alter the makeup of cell membrane fragments released when tumor cells die. Whether the membranes bear C3d or iC3b affects the nature of the IR induced at distal sites, like lymph nodes. Killing tumor cells is not enough; they need to be tagged the right way to induce antitumor immunity.
Tumors often deliver a back-handed complement
Tumors can influence outcomes by taking over complement production. Those that are capable of inducing immune silencing have a survival advantage. They can produce complement components internally. Alternatively, tumor cells may import C3(H2O), which forms spontaneously in the extracellular environment when water hydrolyzes C3, along with complement factor H (CFH) and complement factor I protease. Internal cleavage of C3 then occurs,14 with the possibility that iC3b is returned to the cell surface, with CFH uptake promoting surface deposition of iC3b as it does during apoptosis.15 Intratumoral bacteria represent another possible source of complement activation, with iC3b deposition on the cancer cell membrane guaranteed by the RCAs present there.
Future complementary directions
Targeted delivery of C3d to the tumor membrane offers a new approach for inducing neoantigen-specific IRs against tumors. Amplification of the IC generated likely requires administration with other immunomodulators. Combining current agents with C3 checkpoint modulators should reduce immune adverse events, not enhance them, as immunomodulators will be more effective when dosed at levels below those that cause a breach of self-tolerance.
Acknowledgments
InsideOutBio acknowledges and thanks the many talented investigators whose work cannot be cited within this article format.
References
Footnotes
Twitter @insideoutbio
Contributors AH conceived, wrote, illustrated and edited the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Disclaimer No external funding was received for this work.
Competing interests The author is the founder the company InsideOutBio that is committed to open science and working across disciplines. The company is actively developing complement therapeutics for the immunotherapy of cancer. The information presented here is all derived from publicly available sources.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.