Article Text

Abstract
Background In the KEYNOTE-022 study, pembrolizumab with dabrafenib and trametinib (triplet) improved progression-free survival (PFS) versus placebo with dabrafenib and trametinib (doublet) without reaching statistical significance. Mature results on PFS, duration of response (DOR), and overall survival (OS) are reported.
Methods The double-blind, phase 2 part of KEYNOTE-022 enrolled patients with previously untreated BRAFV600E/K-mutated advanced melanoma from 22 sites in seven countries. Patients were randomly assigned 1:1 to intravenous pembrolizumab (200 mg every 3 weeks) or placebo plus dabrafenib (150 mg orally two times per day) and trametinib (2 mg orally one time a day). Primary endpoint was PFS. Secondary endpoints were objective response rate, DOR, and OS. Efficacy was assessed in the intention-to-treat population, and safety was assessed in all patients who received at least one dose of study drug. This analysis was not specified in the protocol.
Results Between November 30, 2015 and April 24, 2017, 120 patients were randomly assigned to triplet (n=60) or doublet (n=60) therapy. With 36.6 months of follow-up, median PFS was 16.9 months (95% CI 11.3 to 27.9) with triplet and 10.7 months (95% CI 7.2 to 16.8) with doublet (HR 0.53; 95% CI 0.34 to 0.83). With triplet and doublet, respectively, PFS at 24 months was 41.0% (95% CI 27.4% to 54.2%) and 16.3% (95% CI 8.1% to 27.1%); median DOR was 25.1 months (95% CI 14.1 to not reached) and 12.1 months (95% CI 6.0 to 15.7), respectively. Median OS was not reached with triplet and was 26.3 months with doublet (HR 0.64; 95% CI 0.38 to 1.06). With triplet and doublet, respectively, OS at 24 months was 63.0% (95% CI 49.4% to 73.9%) and 51.7% (95% CI 38.4% to 63.4%). Grade 3–5 treatment-related adverse events (TRAEs) occurred in 35 patients (58%, including one death) receiving triplet and 15 patients (25%) receiving doublet.
Conclusion In BRAFV600E/K-mutant advanced melanoma, pembrolizumab plus dabrafenib and trametinib substantially improved PFS, DOR, and OS with a higher incidence of TRAEs. Interpretation of these results is limited by the post hoc nature of the analysis.
- drug therapy
- combination
- immunotherapy
- melanoma
- programmed cell death 1 receptor
Data availability statement
Data are available upon reasonable request. Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Kenilworth, New Jersey, USA (MSD) is committed to providing qualified scientific researchers access to anonymized patient-level data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. The company is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The process includes submission of data requests to the MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php). Data will be made available for request after product approval in the US and EU or after product development is discontinued. There are circumstances that may prevent MSD from sharing the requested data.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immunotherapies with antiprogrammed death 1 (PD-1) monoclonal antibodies, either alone or in combination with anticytotoxic T-lymphocyte-associated antigen 4 monoclonal antibodies, and targeted therapies with BRAF plus MEK inhibitors have significantly improved the outcomes of patients with advanced melanoma.1 In particular, use of BRAF and MEK inhibitors generally results in higher objective response rates for patients with BRAFV600-mutant disease, whereas immune checkpoint inhibitors typically result in more durable responses. Importantly, these treatments have distinct mechanisms of action; thus, it has long been hypothesized that combining immune checkpoint inhibitors with BRAF and MEK inhibitors may result in a higher frequency of durable responses.2 3 Evidence from previous preclinical modeling and analysis of patient biopsy specimens suggest that inhibition of the BRAFV600 driver oncogene facilitates an antitumor immune response.4–7
Results of early clinical trials in patients with previously untreated metastatic BRAFV600-mutant melanoma highlight the promise of this strategy. For example, 15 patients were treated with the anti-PD-1 antibody pembrolizumab plus the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib in the phase 1 portion of the KEYNOTE-022 study,8 and 39 patients received the BRAF inhibitor vemurafenib and the MEK inhibitor cobimetinib in combination with the anti-PD ligand 1 (PD-L1) antibody atezolizumab in another phase 1 trial.9 Efficacy data obtained from these two studies were remarkably consistent, with the combination of BRAF, MEK, and immune checkpoint inhibitors producing objective response rates of 73% and 72%, respectively; median progression-free survival was 15.4 months and 12.9 months, respectively. Interestingly, tumor biopsy specimens showed increased tumor infiltration of CD8+ T cells and expression of PD-L1 and a T cell-inflamed gene signature after treatment with the combination of pembrolizumab, dabrafenib, and trametinib.8
Other studies investigating combinations used the anti-PD1-L1 antibody durvalumab10 or the anti-PD-1 antibody spartalizumab11 combined with the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib. In the latter study, the objective response rate was 75% (n=27), the complete response rate was 33% (n=12), and the 12 month progression-free survival rate was 65.3%.11
In addition, the randomized, double-blind, phase 2 part of KEYNOTE-022 compared the efficacy of the triplet combination of pembrolizumab, dabrafenib, and trametinib with the efficacy of the doublet combination of placebo with dabrafenib and trametinib.12 At a median follow-up of 9.6 months, although the primary endpoint of progression-free survival did not show statistically significant improvement in the triplet arm compared with the doublet arm, numerically higher values were observed (16.0 months vs 10.3 months; HR, 0.66; p=0.043). After additional follow-up of a median 36.6 months, we report mature results showing clinically substantial improvement in duration of response and survival.
Methods
Study design
KEYNOTE-022 was a five-part, multicenter, phase 1/2 dose-finding and preliminary efficacy study of pembrolizumab plus dabrafenib and trametinib in patients with unresectable or metastatic melanoma or solid tumors (parts 4 and 5 only) (online supplemental table S1). Part 3 was the randomized, double-blind, phase 2 portion in which patients with previously untreated advanced melanoma were randomly assigned 1:1 to receive pembrolizumab or placebo combined with dabrafenib and trametinib. Patients were enrolled from 22 medical centers in seven countries.12
Supplemental material
Patients
Eligible patients were aged ≥18 years and had unresectable stage III or stage IV cutaneous melanoma, with at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumours version 1.1 (RECIST V.1.1)13 on imaging (CT or MRI). Patients were required to have BRAF -mutation positive (V600E or V600K) disease should not have previously received systemic therapy for advanced melanoma, and should not present with active central nervous system metastases and/or carcinomatous meningitis.
Procedures
Patients were randomly assigned to receive pembrolizumab plus dabrafenib and trametinib or placebo plus dabrafenib and trametinib using an interactive voice response system/integrated web response system. Randomization was stratified by Eastern Cooperative Oncology Group performance status (ECOG PS; 0 vs 1) and lactate dehydrogenase level (>1.1× upper limit of normal (ULN) vs ≤1.1× ULN). Because of the low number of patients, the ECOG PS 1 and lactate dehydrogenase level >1.1× ULN strata were combined.
Patients received pembrolizumab 2 mg/kg or placebo intravenously every 3 weeks combined with dabrafenib 150 mg orally two times per day and trametinib 2 mg orally one time a day. Treatment was continued for up to 24 months or until disease progression, unacceptable toxicity, or withdrawal from the study. Trametinib and/or dabrafenib treatment could be continued beyond 24 months per standard of care until disease progression. Dose modification or interruption of pembrolizumab, placebo, trametinib, or dabrafenib was allowed to manage toxicity per protocol-specified management guidelines.12
Response and disease progression were assessed radiologically per RECIST V.1.1 by investigator review. Tumor imaging was performed at baseline, at 12 weeks after the first study treatment, every 6 weeks following this up to 18 months, and every 12 weeks thereafter while the patient remained on study therapy. Patients were monitored throughout the study for adverse events, which were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (V.4.0). Immune-mediated adverse events are defined in the online supplemental. PD-L1 expression in tumor samples was determined using the PD-L1 IHC 22C3 pharmDx assay (Agilent Technologies).
Outcomes
The primary endpoint was progression-free survival, defined as the time from randomization to the first documented disease progression or death from any cause, whichever occurred first. Secondary endpoints were objective response rate, duration of response, and overall survival.
Statistical analysis
Progression-free survival, objective response rate, and overall survival were assessed in the intention-to-treat population.12 Safety was assessed in patients who received at least one dose of study drug. Clinical data were collected using InForm V.4.6.5 software (Oracle Health Sciences, Redwood City, California, USA), and SAS V.9.4 software (SAS Institute, Cary, North Carolina, USA) was used for all statistical analyses. Progression-free survival was estimated using the Kaplan-Meier method with patients censored at the last disease assessment date or at the last disease assessment before initiation of a new anticancer treatment. A stratified log-rank test was used to assess between-group differences in progression-free survival. A Cox proportional hazards model with Efron’s method of tie-handling was used to estimate HRs and 95% CIs. The Kaplan-Meier method was used for summary statistics for duration of response and overall survival (see protocol for censoring rules). The treatment comparison for objective response rate was based on Miettinen and Nurminen’s method.14 Assuming an HR of 0.5, the study needed approximately 74 progression-free survival events to have 80% power to reject the null hypothesis at a one-sided 0.025 type I error. The observed HR had to be approximately 0.62 or lower for the treatment effect to be statistically significant. The primary analysis was conducted using all available alphas; therefore, this subsequent post hoc analysis cannot be considered to carry any statistical significance. The study was overseen by a data monitoring committee and is registered at ClinicalTrials.gov (NCT02130466).
Results
Between November 30, 2015 and April 24, 2017, 180 patients were screened for eligibility and 120 were randomly assigned to receive pembrolizumab plus dabrafenib and trametinib (triplet therapy; n=60 (50%)) or placebo plus dabrafenib and trametinib (doublet therapy; n=60 (50%))12 (online supplemental figure S1). All patients received at least one dose of study treatment. The median age was 56 years (range, 18–83), and 58% (n=69) of patients were men. Baseline characteristics were generally well balanced between arms,12 apart from a higher proportion of patients with M1c disease in the triplet arm (82% (n=49) triplet arm, 63% (n=38) doublet arm) (online supplemental table S2). The median exposure duration for each study drug is provided in online supplemental table S3.
At data cut-off (June 26, 2019), the median time from randomization to data cut-off was 36.6 months (range, 26.1–42.9) in the triplet arm and 36.4 months (range, 27.2–41.6) in the doublet arm. Fifteen patients (13%) were receiving treatment: 11 (18%) in the triplet arm and 4 (7%) in the doublet arm. Forty-three patients (72%) had discontinued treatment in the triplet arm and 56 (93%) in the doublet arm, mainly because of adverse events (18 (30%) triplet arm, 10 (17%) doublet arm) and progressive disease (17 (28%) triplet arm, 44 (73%) doublet arm).
Thirty-four patients (57%) in the triplet arm and 53 (88%) in the doublet arm had a progression event. Median progression-free survival was 16.9 months (95% CI 11.3 to 27.9) in the triplet arm and 10.7 months (95% CI 7.2 to 16.8) in the doublet arm (HR 0.53; 95% CI 0.34 to 0.83) (figure 1A). The 12-month progression-free survival rates were 62% (95% CI 48.1% to 73.5%) in the triplet arm and 47% (95% CI 33.4% to 58.7%) in the doublet arm, and the 24-month progression-free survival rates were 41% (95% CI 27.4% to 54.2%) and 16% (95% CI 8.1% to 27.1%), respectively.

Kaplan-Meier estimates. (A) Progression-free survival, defined as time from randomization to disease progression or death, whichever came first. Median progression-free survival was based on Kaplan-Meier estimate per investigator assessment. (B) Overall survival, defined as time from randomization to death. *Based on Kaplan-Meier estimates of progression-free survival (PFS) (investigator assessment by Response Evaluation Criteria in Solid Tumors, V.1.1) or overall survival (OS). †The HRs and 95% CIs were calculated using a Cox regression model with treatment as a covariate stratified by Eastern Cooperative Oncology Group performance status (ECOG PS) (0 vs 1) and lactate dehydrogenase (LDH) (>1.1× upper limit of normal (ULN) vs ≤1.1× ULN); because of the low number of patients enrolled in the ECOG PS 1 and LDH ≤1.1× ULN strata, these strata were combined. Doublet, placebo plus dabrafenib plus trametinib; mo, month; NR, not reached; triplet, pembrolizumab plus dabrafenib plus trametinib.
Exploratory subgroup analysis of progression-free survival showed that the HRs for the subgroups favored the triplet over the doublet arm in patients who were aged ≤65 years, men, had ECOG PS 0, or had elevated lactate dehydrogenase levels (online supplemental figure S2).
Twenty-six patients (43%) in the triplet arm and 36 (60%) in the doublet arm died. Median overall survival was not reached in the triplet arm and was 26.3 months in the doublet arm (HR, 0.64; 95% CI 0.38 to 1.06) (figure 1B). The 12-month overall survival rates were 80% (95% CI 67.5% to 88.1%) in the triplet arm and 73% (95% CI 60.2% to 82.7%) in the doublet arm, and the 24-month overall survival rates were 63% (95% CI 49.4% to 73.9%) and 52% (95% CI 38.4% to 63.4%), respectively. Exploratory subgroup analysis of overall survival showed no difference between the triplet and doublet arms in any subgroup (online supplemental figure S3).
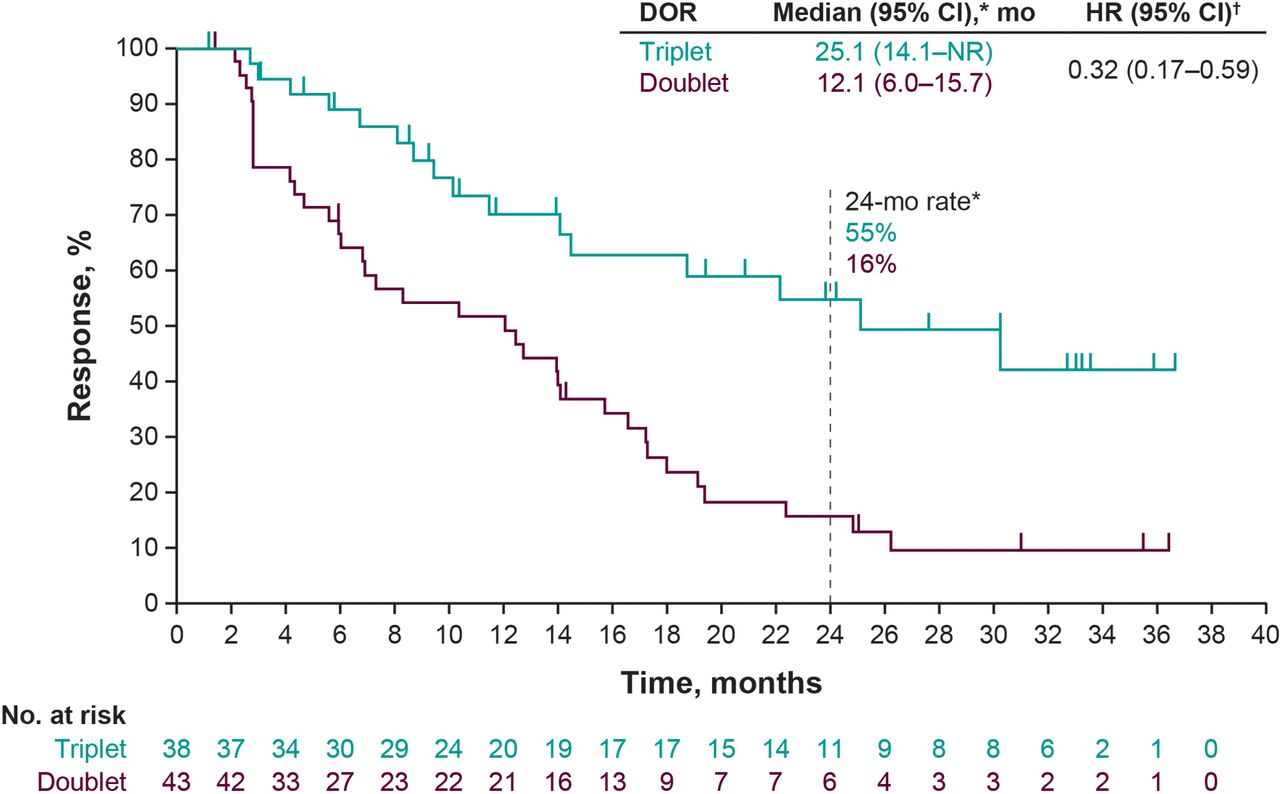
The objective response rate was 63% in the triplet arm and 72% in the doublet arm (difference in rate −8.5%; 95% CI −24.8 to 8.3). A complete response was reported in 12 patients (20%) in the triplet arm and 9 patients (15%) in the doublet arm (table 1). The median duration of response was 25.1 months in the triplet arm and 12.1 months in the doublet arm (HR, 0.32; 95% CI 0.17 to 0.59) (figure 2). At the data cut-off, responses were ongoing in 11 of 38 patients (29%) in the triplet arm and 4 of 43 patients (9%) in the doublet arm (online supplemental figure S4). A reduction in target lesion size of at least 50% was observed in 44 patients (73%) in the triplet arm and 38 patients (63%) in the doublet arm (online supplemental figure S5).

{kind=link}
{kind=link}
Kaplan-Meier estimates of duration of response. Duration of response (DOR) was defined as the time from the first response to disease progression or death, whichever occurred first. *Median DOR was based on Kaplan-Meier estimate per investigator assessment. †The HRs and 95% CIs were calculated using a Cox regression model with treatment as a covariate stratified by Eastern Cooperative Oncology Group performance status (ECOG PS) (0 vs 1) and lactate dehydrogenase (LDH) (LDH>1.1× upper limit of normal (ULN) vs ≤1.1× ULN); because of the low number of patients enrolled in the ECOG PS 1 and LDH ≤1.1× ULN strata, these strata were combined. Doublet, placebo plus dabrafenib plus trametinib; mo, month; NR, not reached; Triplet, pembrolizumab plus dabrafenib plus trametinib.
Best overall response (intention-to-treat population). Responses are based on investigator best assessment across time points per Response Evaluation Criteria in Solid Tumors v1.1 with confirmation
Grade 3–5 adverse events occurred in 42 patients (70%) in the triplet arm and 27 patients (45%) in the doublet arm; grade 3–5 treatment-related adverse events were reported in 35 patients (58%) and 15 patients (25%), respectively. Adverse events led to dose reductions in 16 patients (27%) and 9 patients (15%). Adverse events leading to dose interruption occurred in 50 patients (83%) and 41 patients (68%), respectively; the most common were fever (55%; n=33) and diarrhea (15%; n=9) in the triplet arm and fever (45%; n=27) and neutropenia (10%; n=6) in the doublet arm. Adverse events led to treatment discontinuation in 28 patients (47%) in the triplet arm and 12 patients (20%) in the doublet arm (online supplemental table S4). Treatment-related adverse events occurred in 57 patients (95%) in the triplet arm and 56 patients (93%) in the doublet arm (table 2); the most common were fever (72%; n=43), rash (37%; n=22), and chills (35%; n=21) in the triplet arm and fever (68%; n=41), chills (38%; n=23), and fatigue (38%; n=23) in the doublet arm. Immune-mediated adverse events occurred in 31 patients (52%) in the triplet arm and 9 patients (15%) in the doublet arm (table 3); the most frequently occurring in the triplet arm were pneumonitis (17%; n=10) and hypothyroidism (8%; n=5). Serious treatment-related adverse events occurred in 24 patients (40%) and 14 patients (23%), respectively. One patient (1.7%) in the triplet arm died of treatment-related pneumonitis.
Treatment-related adverse events occurring in ≥15% of patients in either treatment arm (all-subjects-as-treated population)
Immune-mediated adverse events* occurring in at least one patient in either treatment arm (all-subjects-as-treated population)
Discussion
In our previous report on part 3 of the KEYNOTE-022 trial,12 despite a median progression-free survival of 16.0 months in the triplet arm versus 10.3 months in the doublet arm and an HR of 0.66, the study did not meet the prespecified primary endpoint of progression-free survival. Analysis was performed after 9.5 months of follow-up at the occurrence of 72 progression-free survival events; the study plan required 74 events to have 80% power to reject the null hypothesis. The short follow-up time could have influenced the results. Although the current analysis was not protocol-specified, as data matured with a longer median follow-up of 36.6 months, improvements in progression-free survival (16.9 months and 10.7 months, respectively), differences in duration of response (25.1 months and 12.1 months, respectively), and overall survival (not reached and 26.3 months, respectively) became more pronounced between the two arms.
Patients in the doublet arm had a higher initial objective response rate than patients in the triplet arm at the time of the primary analysis, which was considered to have been likely due to an imbalance in the baseline prognostic factors between the treatment arms (more patients in the triplet arm had M1c disease).12 This result persisted in the mature analysis, even though the survival benefit became more pronounced in the triplet arm. Although it is unclear whether other factors may have contributed to this dichotomy, the additional follow-up in the patients in the triplet arm showed deeper and more durable responses than in the doublet arm and a higher rate of complete responses, a lower rate of relapses, and progressive separation in the curves of duration of response. Improved depth of response was associated with improved survival in pooled analyses of trials in melanoma; therefore, it is likely that these response characteristics resulted in the further separation of the progression-free survival and overall survival curves.15 16 These data confirm the rationale of the study and are also consistent with the notion that adding anti-PD-1 blocking antibodies to BRAF plus MEK inhibitor therapy may not improve the initial objective response rates but instead prevent or delay the onset of acquired resistance, as demonstrated by the progressive separation of the time-to-event curves.17 18 Exploratory analysis of progression-free survival showed a substantial advantage of triplet versus doublet therapy in all patient subgroups, particularly in patients younger than 65 years of age, male patients, and patients with elevated lactate dehydrogenase levels at baseline. Patients with elevated lactate dehydrogenase levels have adverse features for durable response with dabrafenib and trametinib or anti-PD-1 therapy alone, have worse prognosis, and are at higher risk for early progression with immune or targeted therapies.19–21 Our findings support the idea that these patients can be offered triplet combination therapy to circumvent the development of resistance.
Although the use of BRAF and MEK inhibitors is recommended for patients with BRAF-mutant disease,22 immune checkpoint inhibitor combinations have also been shown to be effective in this population. In the CheckMate 067 trial, nivolumab plus ipilimumab (5-year overall survival rate, 60%) or nivolumab alone (46%) improved long-term survival compared with ipilimumab (30%) in patients with previously untreated BRAF-mutant melanoma.23 The 5-year progression-free survival rate in CheckMate 067 was 38% with nivolumab plus ipilimumab, 22% with nivolumab alone, and 11% with ipilimumab in patients with previously untreated BRAF-mutant melanoma. Notably, 24-month overall survival and progression-free survival rates with nivolumab plus ipilimumab are similar to those with triplet therapy in the current analysis (63% and 41%, respectively). Robust antitumor activity has also been observed with pembrolizumab plus reduced-dose ipilimumab in the phase 1b KEYNOTE-029 study of patients with BRAF-mutant melanoma in which the majority (87%) of patients had not received prior systemic therapy. Patients with BRAF-mutant melanoma who had not previously received BRAF or MEK inhibitor therapy had a response rate of 60%, whereas those who had previously received BRAF or MEK inhibitor therapy had a response rate of 38%.24 While the results of these studies suggest that patients with BRAF-mutant melanoma may benefit from immune checkpoint inhibitor combinations, it remains unknown how such regimens compare with targeted therapies, or indeed with the triplet therapy investigated in the current analysis, as cross-trial comparisons are hindered by differences in study design and patient population. Trials directly comparing these therapeutic combinations are required to definitively ascertain which regimen is more effective in patients with BRAF-mutant melanoma. Meanwhile, several clinical trials currently underway are investigating the optimal sequencing of targeted and immune checkpoint inhibitor therapy, including COWBOY (NCT02968303), ImmunoCobiVem (NCT02902029), and EBIN (NCT03235245); the results of these should provide insight into which regimen is more effective as front-line therapy.
In the current analysis, the triplet combination was associated with greater toxicity compared with the doublet regimen. Patients who received pembrolizumab plus dabrafenib and trametinib experienced a higher rate of grades 3–5 treatment-related adverse events than patients who received placebo plus dabrafenib and trametinib (58% vs 25%), leading to a higher discontinuation rate because of toxicity. Both immune-mediated adverse events (52% vs 15%) and serious treatment-related adverse events (40% vs 23%) were also more common in the triplet arm compared with the doublet arm. Interestingly, discontinuation resulted primarily from disease progression (44 patients (73%)) in the doublet arm but from adverse events (18 patients (30%)) in the triplet arm. Studies are ongoing to address whether different intermittent or dose-sequencing regimens, combining immunotherapies with targeted therapies for melanoma, may be able to maintain the benefit of the concomitant triple therapy while reducing toxicity (NCT02858921, NCT02224781, and NCT02631447 (Sequential COMBo Immuno and Targeted therapy)).
Recently, positive data were reported from the phase 3 IMspire 150 trial comparing the frontline use of atezolizumab plus vemurafenib and cobimetinib with that of vemurafenib and cobimetinib alone in patients with BRAFV600-mutation-positive-advanced melanoma.25 This study reported a significant improvement in progression-free survival with the combination of atezolizumab, vemurafenib, and cobimetinib versus vemurafenib and cobimetinib alone (15.1 vs 10.6 months) and a 2-year overall survival rate of 60% vs 53%, respectively; progression-free survival and overall survival rates were similar to those observed in the current analysis. While the lead-in of targeted therapies before initiating atezolizumab in the IMspire150 trial could have affected the early efficacy results, it seems not to have influenced toxicity. In fact, the incidence of grade 3/4 treatment-related adverse events in IMspire 150 was substantially higher than in the current analysis (atezolizumab, vemurafenib, and cobimetinib arm, 79%; vemurafenib and cobimetinib arm, 73%). In contrast, a recent analysis of part 3 of the COMBI-I trial reported that spartalizumab plus dabrafenib and trametinib did not significantly improve progression-free survival compared with placebo plus dabrafenib and trametinib among patients with previously untreated BRAFV600-mutant melanoma.26 In the COMBI-I study, the median progression-free survival was 16.2 months for spartalizumab plus dabrafenib and trametinib versus 12.0 months for placebo plus dabrafenib and trametinib (HR, 0.82; 95% CI 0.655 to 1.027).26 Median overall survival was not reached in either arm, with analyses ongoing. Grade 3 or higher treatment-related adverse events were reported in 55% of patients receiving spartalizumab plus dabrafenib and trametinib compared with 33% of patients receiving placebo plus dabrafenib and trametinib. Although the results of these studies provide insight into the role of the combination of PD-1/PD-L1 inhibitors with BRAF and MEK inhibitors in patients with advanced, treatment-naive BRAFV600-mutant melanoma, differences in agents investigated, study design, and patient population preclude direct comparison. In conclusion, pembrolizumab plus dabrafenib and trametinib as first-line therapy in patients with advanced BRAFV600-mutant melanoma provided a clinically substantial improvement in progression-free survival, longer duration of response, and higher overall survival rate compared with placebo plus dabrafenib and trametinib. An important lesson from this analysis is the further improvement in outcomes observed with longer follow-up. However, the results should be interpreted with caution given the post hoc nature of the analysis. This finding should be considered in the design of future studies evaluating immunotherapy combinations, as duration of response may be a critical factor. Indeed, durable responses can allow the differences between treatments to become more pronounced over time, and an early data cut-off may not provide the full picture of the true clinical benefit.27
Data availability statement
Data are available upon reasonable request. Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Kenilworth, New Jersey, USA (MSD) is committed to providing qualified scientific researchers access to anonymized patient-level data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. The company is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The process includes submission of data requests to the MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php). Data will be made available for request after product approval in the US and EU or after product development is discontinued. There are circumstances that may prevent MSD from sharing the requested data.
Ethics statements
Patient consent for publication
Ethics approval
The study protocol and all amendments were approved by the relevant institutional review board or ethics committee at each study site. The study was conducted in accordance with Good Clinical Practice guidelines, as defined by the International Conference on Harmonization, and the Declaration of Helsinki. All patients provided written informed consent.
Acknowledgments
The authors thank the patients and their families and the investigators and site personnel. They also thank the following employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA: Scot Ebbinghaus, Nageatte Ibrahim, Blanca Homet Moreno, Melanie Leiby, Maureen Bucci, Jesse Siegel, Diana Yurewicz, Karen Favata, Nadine Cote, James Anderson, and Rahul Yadav Baddula. Editorial assistance in preparation of this report was provided by Jemimah Walker, PhD, and Doyel Mitra, PhD, CMPP, of ApotheCom, Yardley, PA, and was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Contributors named in this report gave their permission to be acknowledged for their contributions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @PAscierto
Presented at Presented at Society for Melanoma Research; November 20-23, 2019; Salt Lake City, UT, USA.
Correction notice This article has been corrected since it was first published. Dr. Queirolo’s affiliations have been updated.
Collaborators Antoni Ribas, Sachin Agarwal, Charles Cowey, Wilson Miller, Teresa Petrella, Felix Couture, Paolo Ascierto, Michele Del Vecchio, Michele Maio, Francesco Spagnolo, Pier Francesco Ferrucci, Jacob Schachter, Michal Lotem, Karen Drumea, Eva Ellebaek, Inge Marie Svane, Lars Basrholt, Henrik Schmidt, Victoria Atkinson, Georgina V. Long, Matteo Carlino, Michael McCrystal
Contributors GVL, SJD, and AR were involved in the conception, design, or planning of the study. PFF, AMDG, MDV, VA, JS, PQ, GVL, RS, IMS, ML, ESC, MA-A, and AR acquired data for the study. PFF, MDV, VA, PQ, GVL, IMS, RG, ESC, AR, and PAA analyzed the data. PFF, AMDG, MDV, VA, HS, JS, PQ, GVL, EG, RG, SJD, ESC, AR, and PAA interpreted the results. PFF, MDV, VA, EG, AR, and PAA drafted the manuscript with contribution from all authors. PFF, AMDG, MDV, VA, HS, JS, PQ, GVL, RS, IMS, EG, RG, SJD, ESC, MA-A, AR, and PAA critically reviewed or revised the manuscript for important intellectual content. MDV also contributed to the provision of study materials and patient data. All authors reviewed the interim drafts and the final version of the manuscript and agree with its content and submission. All authors had access to all the relevant study data and related analyses and vouch for the completeness and accuracy of the presented data.
Funding Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Study drug was provided by Novartis and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Competing interests PFF has received personal fees from Novartis, Roche, Bristol-Myers Squibb, Pierre Fabre, and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Kenilworth, New Jersey, USA (MSD). AMDG reports other from MSD during the conduct of the study; reports other from Incyte, GlaxoSmithKline, Pierre Fabre, and Sanofi outside the submitted work; and has served as an advisor and consultant for MSD, Incyte, GlaxoSmithKline, Pierre Fabre, and Sanofi. MDV has received personal fees from Novartis, MSD, and BMS outside the submitted work and has served as an advisor and consultant for MSD, BMS, Novartis, Pierre Fabre and Sanofi. VA has received personal fees and travel support from Bristol-Myers Squibb; personal fees from MSD, Merck Serono, Novartis, Pierre Fabre, Roche and Nektar; and travel support from OncoSec Medical, during the conduct of the study. HS received a research grant and personal fees from MSD and has received personal fees from Bristol-Myers Squibb, Incyte, Novartis, and Pierre Fabre outside the submitted work. PQ has received personal fees from Roche, Novartis, Bristol-Myers Squibb, MSD, Sanofi, and Pierre Fabre. GVL is consultant advisor for Aduro Biotech Inc, Amgen Inc, Array Biopharma inc, Boehringer Ingelheim International GmbH, Bristol-Myers Squibb, Highlight Therapeutics S.L., MSD, Novartis Pharma AG, Pierre Fabre, QBiotics Group Limited, Regeneron Pharmaceuticals Inc, SkylineDx B.V. RS reports personal fees from MSD New Zealand, outside the submitted work. IMS reports other (trial expenses) from MSD. IMS has received other funds from MSD during the conduct of the study. EG is an employee and stockholder of Novartis. RG is an employee of MSD. SJD is an employee and stockholder of MSD. ESC was an employee of MSD at the time of this study. AR has received personal fees from Amgen, Chugai, Novartis, Genentech-Roche, Sanofi, and Merck and serves as an advisor for, holds stock in, and has received personal fees from Arcus, Bioncotech, Compugen, CytomX, Five Prime, RAPT Therapeutics, Merus, Rgenix, PACT Pharma, and Tango Therapeutics outside the submitted work. PAA has received grants and personal fees from Bristol-Myers Squib, Roche-Genentech, and Array BioPharma; personal fees and other from MSD; and personal fees from Novartis, Merck Serono, Pierre Fabre, Incyte, Genmab, NewLink Genetics, Medimmune, AstraZeneca, Syndax, Sun Pharma, Sanofi, Idera, Ultimovacs, Sandoz, Immunocore, 4SC, Alkermes, Italfarmaco, Nektar, and Boehringer Ingelheim. JS, ML, and MA-A report no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.