Article Text

Abstract
Background The therapeutic effect of immune checkpoint blockers, especially the neutralizing antibodies of programmed cell death (PD-1) and its ligand programmed death ligand 1 (PD-L1), has been well verified in melanoma. Nevertheless, the dissatisfactory response rate and the occurrence of resistance significantly hinder the treatment effect. Inflammation-related molecules like A20 are greatly implicated in cancer immune response, but the role of tumorous A20 in antitumor immunity and immunotherapy efficacy remains elusive.
Methods The association between tumorous A20 expression and the effect of anti-PD-1 immunotherapy was determined by immunoblotting, immunofluorescence staining and flow cytometry analysis of primary tumor specimens from melanoma patients. Preclinical mouse model, in vitro coculture system, immunohistochemical staining and flow cytometry analysis were employed to investigate the role of A20 in regulating the effect of anti-PD-1 immunotherapy. Bioinformatics, mass spectrum analysis and a set of biochemical analyzes were used to figure out the underlying mechanism.
Results We first discovered that upregulated A20 was associated with impaired antitumor capacity of CD8+T cells and poor response to anti-PD-1 immunotherapy in melanoma patients. Subsequent functional studies in preclinical mouse model and in vitro coculture system proved that targeting tumorous A20 prominently improved the effect of immunotherapy through the invigoration of infiltrating CD8+T cells via the regulation of PD-L1. Mechanistically, A20 facilitated the ubiquitination and degradation of prohibitin to potentiate STAT3 activation and PD-L1 expression. Moreover, tumorous A20 expression was highly associated with the ratio of Ki-67 percentage in circulating PD-1+CD8+T cells to tumor burden.
Conclusions Together, our findings uncover a novel crosstalk between inflammatory molecules and antitumor immunity in melanoma, and highlight that A20 can be exploited as a promising target to bring clinical benefit to melanomas refractory to immune checkpoint blockade.
- melanoma
- immunotherapy
- inflammation
- programmed cell death 1 receptor
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Eradication of immune destruction is a hallmark of cancer, which is primarily due to the abnormal activation of immune checkpoints and the termination of antitumor immune response. Programmed cell death (PD-1)/programmed death ligand 1 (PD-L1) have been demonstrated as a pair of major immune checkpoint molecules and valuable therapeutic targets for melanoma treatment.1 For instance, the binding of membrane PD-L1 on tumor cells to PD-1 on T cells evokes an immunosuppressive signal that results in the dysfunction and even the apoptosis of cytotoxic T cells, thereby impairing antitumor immunity.2 Therefore, PD-1/PD-L1 blockade is in a position to disrupt the interaction between tumor cells and T cells, which restores tumor-specific immune response.3 For melanoma patients receiving anti-PD-1 antibody monotherapy, the median overall survival could be up to 32.7 months after almost 5 years of follow-up.4 However, a proportion of 40%–60% of patients does not achieve any significant therapeutic response, and some patients even show complete resistance to PD-1/PD-L1 blockade.5 Accumulative evidence has revealed multiple primary and acquired alterations leading to treatment resistance of immunotherapy, including low mutational burden, defective antigenicity and antigen presentation of tumor cell and genomic dysregulation of interferon-γ (IFN-γ) signaling pathway.6–9 These reports have prompted potential molecular profile for predicting the treatment response and effective synergized therapeutic strategy for cancer immunotherapy. Notably, tumor cells can adaptively increase the expression of PD-L1 or other immune checkpoints under the control of IFN-γ after the treatment with anti-PD-1 antibody, rendering melanomas refractory to immunotherapy.10–12 Hence, the molecular mechanism underlying aberrant tumorous PD-L1 expression associated with the resistance to immunotherapy in melanoma is in urgent need to be clarified to improve the therapeutic efficacy.
Inflammation is closely related to the development of various cancers including melanoma, with its pivotal role in the regulation of cancer immune response demonstrated recently.13–15 To be specific, tumor necrosis factor-α (TNF-α) that is a common proinflammatory cytokine and its downstream pathway could induce tumor immune escape by elevating the expressions of immune checkpoint molecules.13 16 On the other, TNF-α is also one of the major cytokines secreted by activated CD8+ T cells in tumor microenvironment that potentiates antitumor immune response.17 18 These reports highlight the vital impact of inflammatory signaling on antitumor immunity, though the actual function remains controversial. A20 is a primary responsive gene of TNF-α and is documented as a crucial negative mediator of inflammation as well as immune response in multiple immune cells.19 As a ubiquitin-editing enzyme, it is composed of an N-terminal OTU domain with deubiquitinase activity and seven Cys2-Cys2 zinc finger C-terminal domains functioning as a ubiquitin ligase.20 The cooperative activity of these two ubiquitin-editing domains mediates the negative regulatory role of A20 in NF-κB signaling,20 so that the genetic deficiency of TNFAIP3 that encodes A20 protein can result in the onset and progression of multiple autoimmune diseases by amplifying the pro-inflammatory NF-κB signaling.21 For cancer pathogenesis, previous investigations emphasized on A20 expressed in tumor-infiltrating immune cells to clarify its effect on antitumor immunity. In B16 mouse melanoma tumor model, silencing of A20 in dendritic cells (DCs) enhanced NF-κB activity followed by elevated expression of interleukin 6 (IL-6), TNF-α and IL-12, leading to potentiated antitumor immune responses.22 In addition, tumor-infiltrating CD8+ T with the deletion of A20 had stronger antitumor capacity by relieving the brake on NF-κB signaling pathway.23 Of note, A20 is also abundantly expressed in tumor cells and can directly influence their biological behaviors.24 25 Therefore, A20 expressed in immune cells and tumor cells in tumor microenvironment are both greatly implicated in the pathogenesis of cancer. Previous studies regarding antitumor immunity mainly focused on the role of A20 that is expressed in immune cells. However, whether tumorous A20 can affect the function of tumor-infiltrating immune cells and the therapeutic effect of immunotherapy remains unknown.
In the present study, A20 was initially identified to be highly related with the effect of anti-PD-1 immunotherapy among melanoma patients. Preclinical melanoma mouse model and in vitro coculture system were then established to evaluate the therapeutic effect of anti-PD-1 immunotherapy with synergized suppression of tumorous A20 expression. Subsequently, the potential mechanism of A20-mediated resistance to immunotherapy was further investigated by bioinformatics, mass spectrum analysis and a set of biochemical analyzes, with a particular emphasis on the regulation of PD-L1 expression. Furthermore, the relationship between tumorous A20 expression and the invigoration of circulating exhausted-phenotype CD8+ T cells after anti-PD-1 antibody treatment was analyzed in melanoma patients.
Methods
A detailed description of the methods used in this study is available in online supplemental methods.
Supplemental material
Statistical analysis
All experiments were repeated at least three times unless otherwise indicated. Error bars represent SE of the mean. Student’s t-test was used to compare two groups of independent samples. Liner regression was used to confirm the correlation between two groups of dependent samples. P<0.05 was considered statistically significant. Kaplan-Meier analysis and log-rank (Mantel-Cox) test were used to evaluate the statistical significance for comparison of survival curves. All statistical analyzes were performed with GraphPad Prism (GraphPad software V.6.0, La Jolla, USA).
Results
A20 expression is correlated with the clinical response to anti-PD-1 antibody treatment in melanoma patients
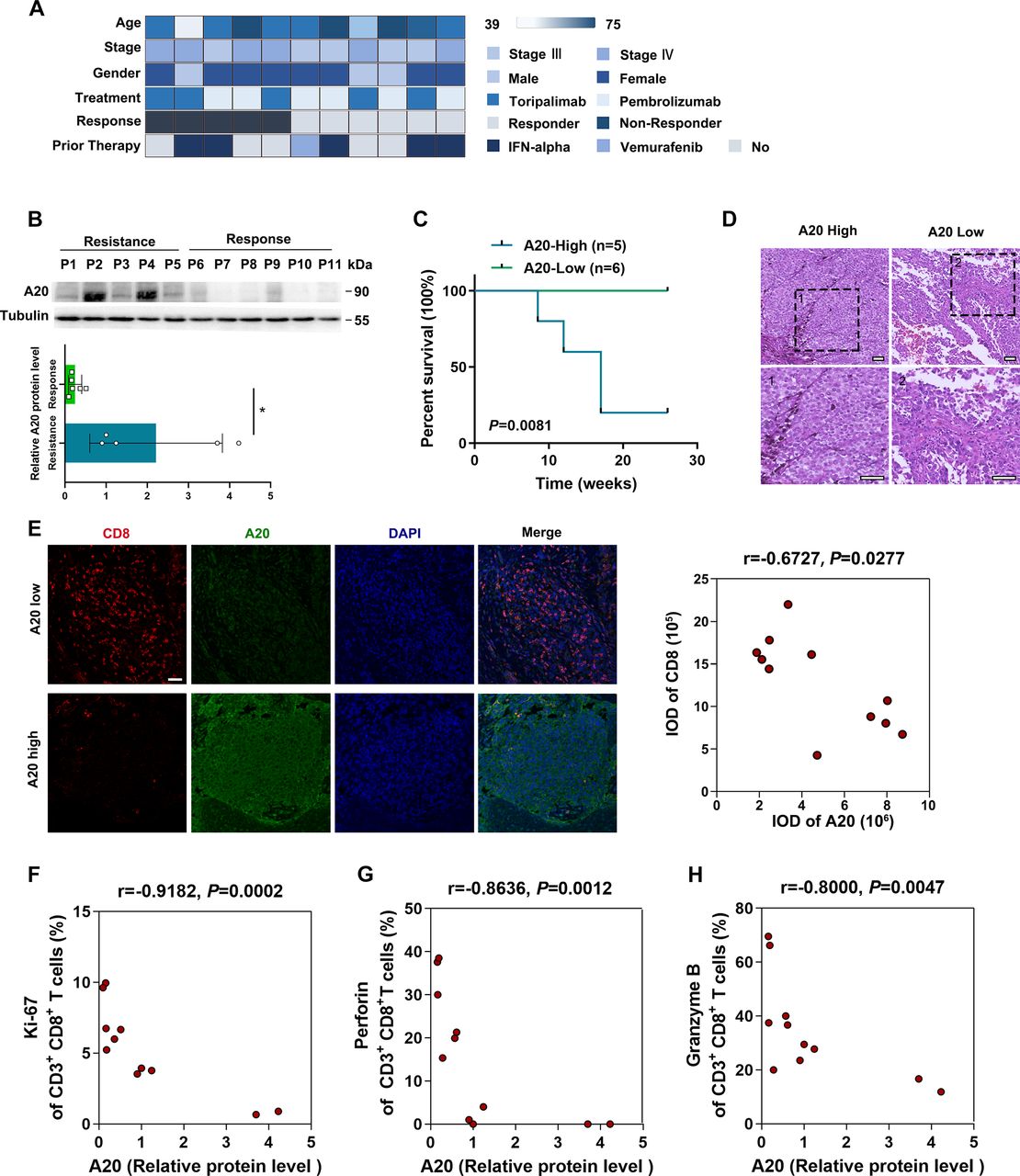
Previously, several transcriptomic and proteomic researches have demonstrated that inflammation-related signals and molecules were highly correlated with the therapeutic effect of anti-PD-1 immunotherapy in melanoma.26–29 Of note, in a cohort of 54 melanoma patients receiving anti-PD-1 antibody monotherapy, we noticed that a high fraction of non-responding patients had abundant expressions of immune gene markers like TNFAIP3 (encoding A20) and TLR3 in tumor, which was associated with dampened immune response.29 A20 is a primary responsive gene of TNF-α and is documented as a negative mediator of inflammation. For cancer pathogenesis, previous investigations emphasized on A20 expressed in tumor-infiltrating immune cells and its effect on antitumor immunity.22 23 However, our immunofluorescence staining assay revealed that A20 was mainly distributed in tumor cells (Melan A-positive) rather than lymphocytes (CD45-positive) in melanoma specimens (online supplemental figure 1A). We have recently demonstrated that upregulated A20 contributed to cell proliferation, metastasis and the treatment resistance to vemurafenib in melanoma (data not shown), whereas the role of tumorous A20 in the regulation of antitumor immunity and immunotherapy effect remains elusive. To this end, we collected baseline primary tumors from 11 patients who subsequently treated with anti-PD-1 antibody. According to the response status, these patients were separated into response group (responder, n=6, partial response) and resistance group (non-responder, n=5, progressive disease) after 6 months of follow-up visit (figure 1A; online supplemental table 1). The age of patients showed no prominent difference between both groups (online supplemental table 1). We digested and fractionated melanoma specimens with trypsin to obtain protein lysates and performed immunoblotting analysis. As was shown, the protein level of tumorous A20 was remarkably increased in resistance group compared with that in response group (figure 1B). Moreover, the survival analysis of the patients revealed that high tumorous A20 expression was associated with poor prognosis after receiving anti-PD-1 antibody treatment (figure 1C).
Supplemental material
Supplemental material

A20 expression is correlated with the efficacy of anti-PD-1 antibody treatment in melanoma patients. (A) The study cohort includes 11 patients receiving anti-PD-1 antibody treatment. Clinical parameters are indicated in the heat map. (B) Immunoblotting analysis of A20 expression in melanomas derived from 11 patients related to (A). The densitometric analysis was displayed below. (C) Kaplan-Meier survival analysis of melanoma patients with high A20 expression (n=5, resistance group) or low A20 expression (n=6, response group). (D) Representative H&E staining images of melanomas with relative high expression of A20 and low expression of A20 from 11 patients indicated in (A). Scale bars=50 µm. (E) Representative immunofluorescence staining images and analysis of CD8 expression in melanomas with relative high expression of A20 and low expression of A20 from 11 patients indicated in (A). The correlation analysis of A20 and CD8 expression was displayed on the right. Scale bars=50 µm. (F–H) The scatter diagram shows the linear correlation between relative tumorous A20 protein expression and the percentage of Ki-67 (F), Perforin (G) and Granzyme B (H) in infiltrated CD3+CD8+T cells in melanomas from 11 patients indicated in (A). P values were calculated by Spearman’s rank correlation coefficient analysis. *P< 0.05. IFN, interferon; PD-1, programmed cell death.
To further illustrate the relationship between the tumorous A20 expression and antitumor immunity, we employed H&E staining and found that the number of tumor-infiltrating lymphocytes was significantly reduced in tumors with high A20 expression (figure 1D). Subsequent immunofluorescence staining analysis forwardly revealed that the number of infiltrated CD8+ T cells was negatively correlated with tumorous A20 expression in these tumors (figure 1E). What’s more, we performed flow cytometry analysis on the cell suspensions derived from these tumors. It revealed that tumorous A20 expression was negatively correlated with the percentage of Ki-67-positive CD8+ T cells (figure 1F), so were the percentages of Perforin-positive and Granzyme B-positive CD8+ T cells (figure 1G,H). These results suggested that the loss of tumorous A20 might induce stronger antitumor immune response in melanoma, which was probably involved in rendering therapeutic resistance to anti-PD-1 immunotherapy.
A20 regulates the therapeutic response to anti-PD-1 antibody treatment in melanoma
Then, we established a preclinical xenograft mouse model to investigate whether tumorous A20 could regulate the treatment effect of anti-PD-1 immunotherapy in melanoma. C57BL/6 mice were subcutaneously injected with wild-type murine B16F10 cells, a cell line that is resistant to PD-1 antibody,30 or B16F10 cells with the knockout of A20 by CRISPR/Cas9 (online supplemental figure 1B), followed with or without the treatment with anti-PD-1 antibody. Compared with the control group, the implantation with A20-deficient tumors or the monotreatment with anti-PD-1 antibody slightly prolonged the survival of mice (figure 2A). However, mice implanted with A20-deficient B16F10 tumor that also received anti-PD-1 antibody treatment displayed superior survival rate (figure 2A). Moreover, we examined the alteration of tumor growth in another cohort of mice. The knockdown of A20 or the monotreatment with anti-PD-1 antibody significantly led to the reduction of tumor volume and tumor weight (figure 2B,C). More importantly, A20 knockout could sensitize melanoma to anti-PD-1 antibody treatment, leading to more prominent regression of xenograft tumors in mice (figure 2B,C). Together, tumorous A20 could impair the efficacy of anti-PD-1 immunotherapy in melanoma.

A20 regulates the therapeutic effect of anti-PD-1 antibody treatment in melanoma. (A) Kaplan-Meier survival analysis of preclinical xenograft mice implanted with melanomas with or without the knockout of A20 expression, followed by the treatment with anti-PD-1 treatment (n=7 per group). P values were calculated by Mantel-Cox test. (B, C) Tumor growth (B) and tumor weight (C) of xenograft tumors in mice implanted with melanomas with or without the knockout of A20 expression followed by the treatment with anti-PD-1 treatment (n=4 per group). (D) Representative H&E staining images of excised tumors in mice receiving the treatment with anti-PD-1 antibody related to (B, C). Scale bars=50 µm. (E, F) Flow cytometry analysis of percentage of the percentage of Ki-67 and Granzyme B in tumor infiltrated CD8+T cells of xenograft tumors related to (B, C) (n=4 per group). (G) The flow chart shows PBMC extraction from xenograft mice and subsequent coculture with B16F10 cells with or without the knockout of A20. The cocultured melanoma cells and PBMCs were then processed to flow cytometry analysis. (H, I) Flow cytometry analysis of the percentage of Ki-67 and Granzyme B in cocultured CD8+T cells related to (G) (n=4 per group). (J) The percentage of apoptotic melanoma cells in cocultured system related to (G). (K, L) Tumor growth (K) and tumor weight (L) of xenograft tumors in mice that were implanted with melanomas with or without the knockout of A20 and received the treatment with anti-PD-1 antibody, followed by the intraperitoneal injection of anti-CD8 antibody (n=4 per group). P value was calculated by two-tailed Student’s t-test. Data represent the mean±SEM of at least triplicates. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001. ns, not significant; PD, programmed cell death; SEM, SE of the mean.
We then performed immunohistochemical staining analysis of xenograft tumors after anti-PD-1 antibody treatment and found that A20 deficiency led to significant enrichment of infiltrated lymphocytes in tumor (figure 2D). Subsequent flow cytometry analysis of the cell suspensions derived from these tumors displayed that the knockout of A20 induced significantly increased the number of Ki-67-positive CD8+ T cells and Granzyme B-positive CD8+ T cells (figure 2E,F), indicating that tumorous A20 suppressed the cytotoxicity and infiltration of CD8+ T cells in tumor microenvironment after anti-PD-1 antibody treatment. In order to investigate the effect of tumorous A20 deficiency on the function of infiltrated CD8+ T cells, we cocultured B16F10 melanoma cells with murine Peripheral blood mononuclear cells (PBMCs) extracted from C57BL/6 mice that were burdened with B16F10 cells and received anti-PD-1 antibody treatment (figure 2G). Flow cytometry analysis on PBMCs in the coculture system revealed that the knockout of A20 in melanoma cells increased the percentages of both Ki-67-positive CD8+ T cells and Granzyme B-positive CD8+ T cells (figure 2H,I). Moreover, the deficiency of A20 induced more apoptosis of melanoma cells in the coculture system (figure 2J), while the apoptosis rate in single culture of B16F10 cells was minimally influenced by A20 knockout (online supplemental figure 1C). These results reiterated that tumorous A20 affected the functional status of infiltrated CD8+ T cells and thereby participated in the regulation of anti-PD-1 immunotherapy efficacy. To further demonstrate that CD8+ T indeed mediated the effect, specific antibodies targeting CD8 were then injected intraperitoneally to block CD8+T cells systemically in xenograft mouse model (online supplemental figure 1D). CD8 antibody cotreatment could repotentiate the tumor growth with increased tumor growth and tumor weight when the mice received anti-PD-1 antibody treatment (figure 2K,L). Therefore, CD8+T cells were required for the observed synergistic effect of anti-PD-1 antibody treatment and the knockout of A20 on melanoma regression.
A20 modulates the expression of PD-L1
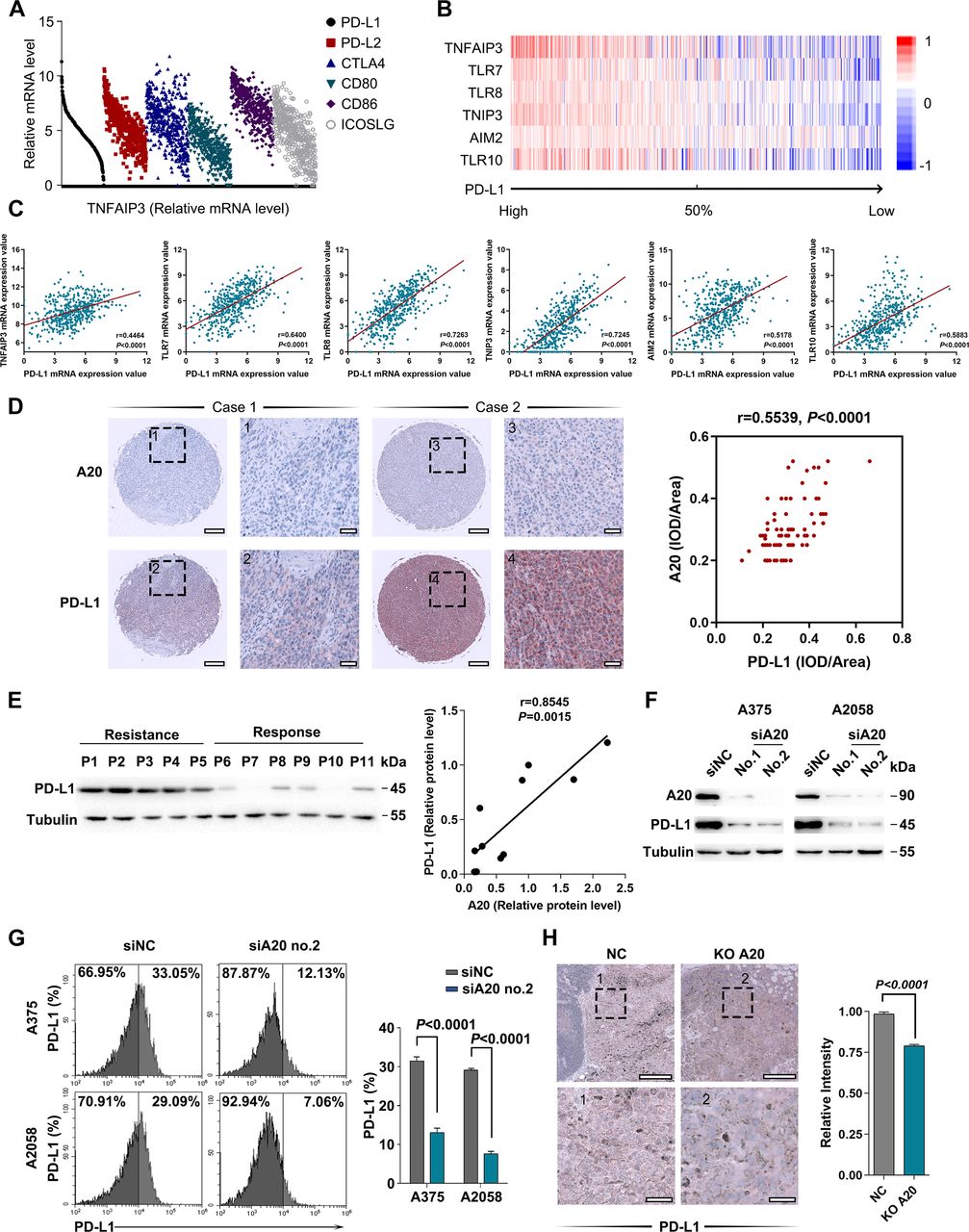
Accumulative evidence has revealed that the dysregulation of immune checkpoints is highly related with tumor immune escape and resistance to immunotherapy.3 31 32 In particular, IFN-γ-induced feedback up-regulation of PD-L1 in tumor microenvironment after anti-PD-1 antibody treatment has been regarded as a crucial mechanism for resistance to immunotherapy.10 Therefore, we proposed that A20 might manipulate the expression of PD-L1 or other immune checkpoints to affect the antitumor capacity of infiltrated lymphocytes, thereby affecting the treatment response to immunotherapy. To clarify this, we analyzed the relationship between TNFAIP3 mRNA level and the expressions of several immune checkpoints in The Cancer Genome Atlas (TCGA) Skin Cutaneous Melanoma (SKCM) database. TNFAIP3 mRNA level was in positive correlation with CD274 (encoding PD-L1) compared with PDCD1LG2 (encoding PD-L2), CTLA-4, CD80, CD86 or ICOSLG (figure 3A; online supplemental figure 1E). In addition, gene set enrichment analysis was performed in TCGA SKCM database to identify distinctively expressed genes and biological pathways in melanoma correlated with PD-L1 mRNA expression (figure 3B; online supplemental table 2). Inflammation-related pathways (including Toll pathway, TNFR1 pathway and TNFR2 pathway) were among the most significant pathways that positively correlated with PD-L1 expression (online supplemental table 2). We analyzed the relationship between TNFAIP3 mRNA level and several inflammation-related molecules, which revealed that PD-L1 mRNA level was in markedly positive correlation with the mRNA expressions of TNFAIP3 and other proinflammatory genes (figure 3B,C). In order to further confirm the relationship between tumorous A20 expression and PD-L1 expression, immunohistochemical staining analysis in tumor tissue microarray (TMA) that consists of 82 melanoma cases was carried out. In line with the result from TCGA SKCM database, A20 expression was proportional to PD-L1 expression in melanoma tissues (figure 3D). Subsequent immunoblotting analysis of tumors from 11 patients reached a common conclusion (figures 3E and 1B).
Supplemental material

A20 regulates the expression of PD-L1. (A) Correlation analysis between TNFAIP3 mRNA level and several immune checkpoints in TCGA SKCM database. (B, C) GSEA analysis of PD-L1-associated molecules in TCGA SKCM database, and the correlation analysis between PD-L1 and several inflammation-related molecules. (D) Representative immunohistochemical (IHC) staining images of A20 and PD-L1 expressions in TMA. The association between A20 and PD-L1 expression in melanoma tissues was analyzed. (E) Immunoblotting analysis of PD-L1 expression in melanomas derived from 11 patients related to figure 1A. The scatter diagram shows the linear correlation between A20 and PD-L1 protein expression. (F) Immunoblotting analysis of the expression of PD-L1 in melanoma cell lines after the intervention of A20. (G) Flow cytometry analysis of membrane PD-L1 expression in melanoma cells after the intervention of A20. (H) Immunohistochemical staining analysis of PD-L1 in xenograft tumors with the intervention of A20. Scale bars=100 µm. Data represent the mean±SEM of at least triplicates. P value was calculated by two-tailed Student’s t-test. GSEA, gene set enrichment analysis; PD-L1, programmed death ligand 1; SEM, SE of the mean; SKCM, Skin Cutaneous Melanoma.
We went on to observe whether A20 could regulate PD-L1 expression in melanoma. The immunoblotting analysis of whole cell lysis in two human melanoma cell lines showed that the knockdown of A20 significantly suppressed PD-L1 expression (figure 3F), so was the transcriptional level of PD-L1 (online supplemental figure 1F). In addition, flow cytometry analysis revealed diminished membrane PD-L1 expression after the knockdown of A20 (figure 3G). Moreover, PD-L1 staining intensity in previous B16F10 xenograft tumors was also decreased with A20 deficiency (figure 3H), which proved that A20 could promote PD-L1 expression in melanoma.
Tumorous A20 induces therapeutic resistance to anti-PD-1 immunotherapy via PD-L1
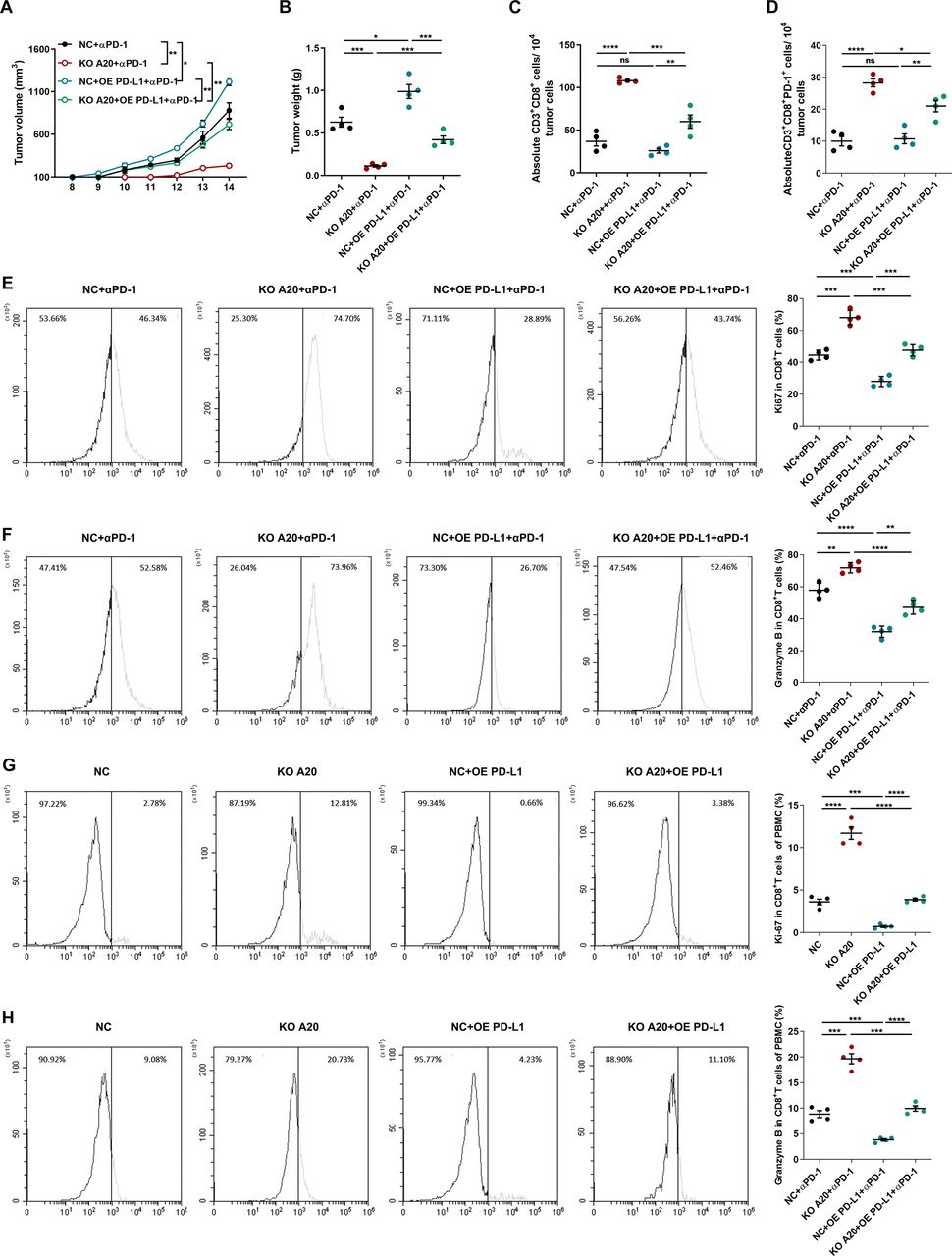
We continued to explore whether the increase of PD-L1 expression was responsible for A20-induced resistance to anti-PD-1 antibody treatment. To this end, C57BL/6 mice were subcutaneously injected with B16F10 melanoma cells with the interventions of both A20 and PD-L1, followed with anti-PD-1 antibody treatment. In response to anti-PD-1 antibody treatment, although the lack of A20 significantly inhibited melanoma progression, concurrent overexpression of PD-L1 reversed the inhibitory effect of A20 knockout (figure 4A,B). Through flow cytometry analysis of the cell suspension of implanted tumors, we discovered that the knockout of tumorous A20 could increase the number of CD8+ T cells in tumor microenvironment after anti-PD-1 antibody treatment, whereas the concurrent over-expression of PD-L1 reversed the infiltration of CD8+ T cells (figure 4C). Since that PD-L1 on tumor cells mainly affects the function of immune cells expressing PD-1,33 we examined the number of infiltrated CD8+PD-1+T cells in tumor. PD-L1 overexpression inhibited the increase of CD8+PD-1+ T cells caused by A20 deficiency as well (figure 4D). Moreover, the knockout of A20 in melanoma elevated the expressions of Ki-67 and Granzyme B in CD8+ T cells in tumor microenvironment, which were resuppressed after PD-L1 overexpression (figure 4E–F), indicating that the role of tumorous A20 in regulating the response to anti-PD-1 immunotherapy was dependent on PD-L1. For further verification, we cocultured B16F10 melanoma cells with murine PBMCs that were extracted from C57BL/6 mice burdened with implanted B16F10 cells and received anti-PD-1 antibody treatment (figure 2G). The knockout of A20 in B16F10 cells led to more apoptosis in the co-culture system, whereas cocurrent overexpression of PD-L1 could repress cell death caused by cocultured PBMCs (online supplemental figure 2A). Subsequent flow cytometry analysis revealed that the deficiency of A20 in B16F10 cells increased the expressions of Ki-67 and Granzyme B in CD8+ T cells of co-culture system, while the co-current overexpression of PD-L1 eliminated the alteration (figure 4G,H), which was consistent with the results obtained in implanted tumors. Collectively, tumoral A20 could contribute to the resistance to anti-PD-1 antibody treatment via the regulation of PD-L1 expression.
Supplemental material

tumorous A20 contributes to therapeutic resistance to anti-PD-1 antibody treatment via PD-L1. (A, B) Tumor growth (A) and tumor weight (B) of implanted B16F10 melanomas with indicated interventions of A20 and PD-L1 expression followed by anti-PD-1 antibody treatment (n=4 per group). (C, D) Absolute number of CD3+CD8+T cells and CD3+CD8+PD-1+T cells in implanted melanomas related to (A, B) (n=4 per group). (E, F) Flow cytometry analysis of the percentage of Ki-67 (E) and Granzyme B (F) in infiltrated CD8+T cells from the indicated melanomas related to (A, B) (n=4 per group). (G, H) Flow cytometry analysis of the percentage of Ki-67 and Granzyme B in PBMC in melanoma cell-PBMC coculture system (n=4 per group). P value was calculated by two-tailed Student’s t-test. Data represent the mean±SEM. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001. ns, not significant; PD-1, programmed cell death; PD-L1, programmed death ligand 1; SEM, SE of the mean.
A20 promotes PD-L1 expression through STAT3 signal
We then investigated the potential mechanism underlying the regulation of A20 on PD-L1 expression. Bioinformatics analysis on TCGA SKCM database suggested that the genes positively correlated with TNFAIP3 mRNA level were enriched in multiple signaling pathways (figure 5A,B; online supplemental table S3), among which we focused on JAK-STAT signaling pathway due to its canonical regulatory effect on PD-L1 expression.34 Although the analysis of TCGA SKCM database revealed that JAK2 mRNA expression was associated with TNFAIP3 mRNA expression, the alteration of neither JAK2 protein expression nor its downstream STAT3 protein expression was observed in melanoma with A20 knockdown (figure 5C; online supplemental figure 2B). Nonetheless, the phosphorylation of STAT3 at Y705 was significantly weakened in case of A20 deficiency and increased after A20 overexpression (figure 5C). The immunofluorescence staining analysis revealed that A20 knockdown induced the decrease of STAT3 phosphorylation as well (figure 5D). Consistently, the mRNA levels of the transcriptional targets of STAT3 including c-myc and Mcl-1 were remarkably reduced (figure 5E).35 36 Therefore, the knockdown of A20 suppressed the transcriptional function of STAT3.
Supplemental material

A20 promotes PD-L1 expression through STAT3 signal. (A, B) Representative pathways and corresponding genes identified to be associated with A20 expression in TCGA SKCM database. (C) Immunoblotting analysis of the expression of total STAT3 and phosphor-STAT3 (Y705) in melanoma cells after the intervention of A20. (D) Immunofluorescence staining analysis of phosphorylated STAT3 after the intervention of A20. Scale bar=20 µm. (E) Immunoblotting analysis of Mcl-1 and c-myc in melanoma cells after the intervention of A20. (F) Immunoblotting analysis of PD-L1 expressions in melanoma cells after the overexpression of A20, followed by the treatment of STAT3 inhibitor WP1066. (G) Flow cytometry analysis of PD-L1 membrane expression in melanoma cells after the overexpression of A20 followed by the treatment of STAT3 inhibitor WP1066 (n=3 per group). (H) Chromatin immunoprecipitation and qRT-PCR analysis displayed the mRNA level of PD-L1 promoter region that interacted with anti-STAT3 antibody in melanoma cells after the intervention of A20 (n=3 per group). Data represent the mean±SEM of triplicates. P value was calculated by two-tailed Student’s t-test. ES, enrichment score; PD-L1, programmed death ligand 1.
We further examined whether STAT3 acted as an intermediate signal in the expression of PD-L1 regulated by A20. WP1066 that is a selective STAT3 inhibitor was used to inhibit STAT3 activity. Immunoblotting analysis showed that WP1066 treatment reversed the upregulation of PD-L1 caused by A20 overexpression (figure 5F). Flow cytometry analysis also displayed that WP1066 impeded membrane PD-L1 expression in melanoma cells with the overexpression of A20 (figure 5G). Chromatin immunoprecipitation assay showed impaired combination of STAT3 protein to PD-L1 promoter region after the knockdown of A20 (figure 5H). Together, A20 promoted PD-L1 transcription and expression in a STAT3-dependent way.
Prohibitin mediates the regulatory role of A20 in STAT3 phosphorylation
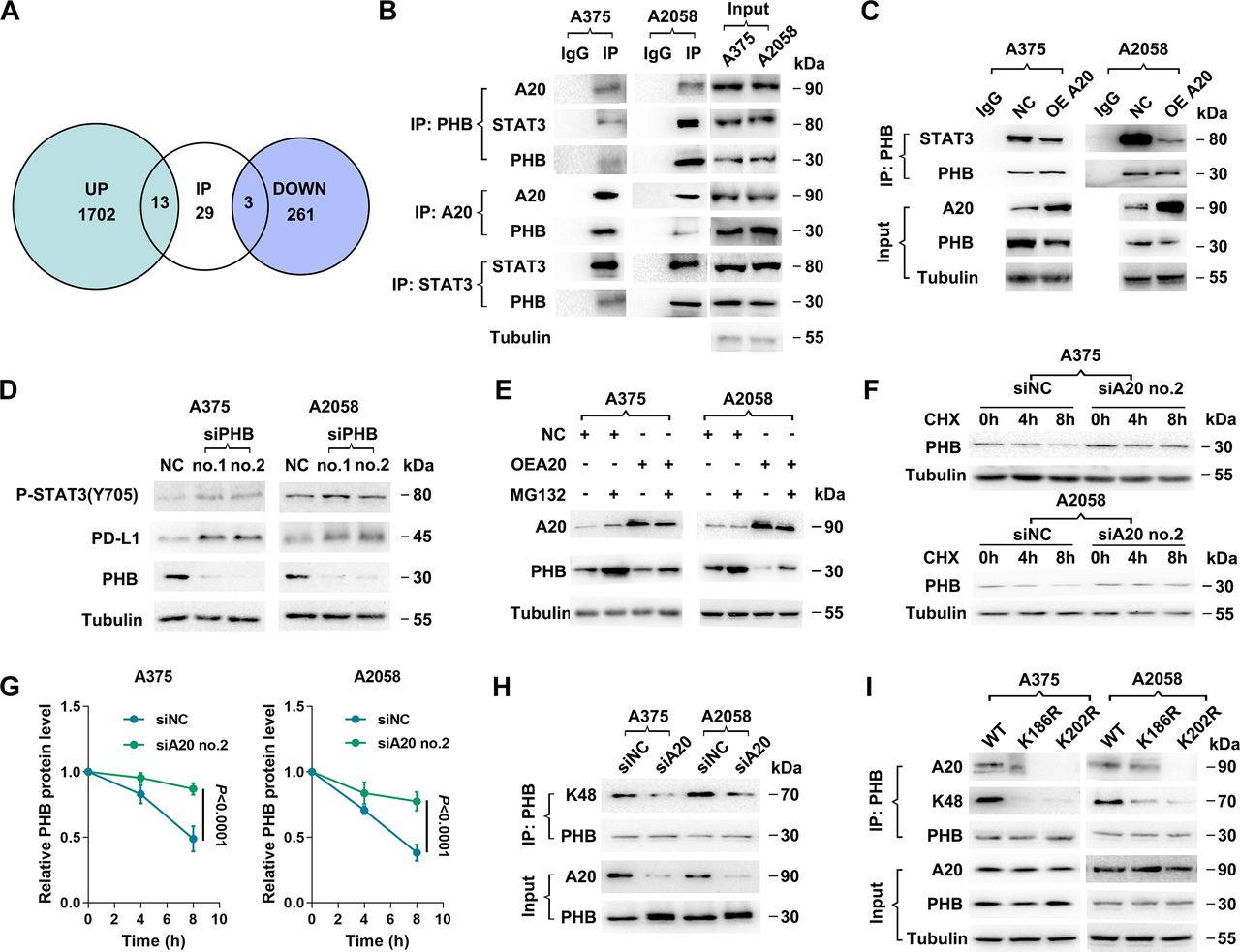
Thereafter, the potential mechanism by which A20 regulated STAT3 phosphorylation was investigated. We first supposed that IL-6, a classic upstream cytokine of STAT3, may be regulated by A20 in melanoma cells. However, we failed to observe significant alteration of IL-6 secretion after the knockdown of A20 (online supplemental figure 2C), indicating that A20 was not capable of activating STAT3 by promoting autocrine IL-6 signal. Meanwhile, SOCS3, the crucial negative regulator of STAT3, was not significantly altered after A20 knockdown (online supplemental figure 2D), excluding the responsibility of SOCS3 in the phosphorylation of STAT3 induced by A20. Afterwards, mass spectrum analysis was used to identify differentially-expressed proteins after the knockdown of A20 in two melanoma cell lines A2058 and A375, respectively (figure 6A). As a result, the expressions of 1715 proteins were upregulated in both A375 and A2058 cells, while the expressions of 264 proteins were downregulated (figure 6A). Since that A20 could regulate protein expressions with its ubiquitin-editing enzymatic activity via direct interaction with its substrates,19 we performed coimmunoprecipitation (co-IP) assay, followed by mass spectrum analysis to uncover the proteins that directly interacted with A20, and examined whether the interacted proteins were enlisted in upregulated or downregulated proteins after the intervention of A20. Conspicuously, 13 of the 1715 upregulated proteins and 3 of the 264 downregulated proteins were enlisted in A20-interacted proteins (figure 6A; online supplemental table 4), among which prohibitin (PHB) had been previously reported as a regulator of STAT3 phosphorylation.37 Afterwards, we performed co-IP assay by precipitating STAT3, A20 and PHB in cell lysate respectively, and confirmed that PHB directly interacted with A20 and STAT3 (figure 6B). Moreover, the knockdown of A20 led to upregulation of PHB expression, while the overexpression of A20 suppressed the expression of PHB (online supplemental figure 2E), which was consistent with the result of previous mass spectrum analysis. Subsequent immunoprecipitation assay denoted that elevated A20 expression inhibited the interaction between PHB and STAT3, along with the decline of PHB expression (figure 6C). Furthermore, the knockdown of PHB in A2058 and A375 cell lines was established, which brought about prominent increase of STAT3 phosphorylation at Y705. Meanwhile, PD-L1 expression was upregulated (figure 6D), implicating the negative regulatory role of PHB in STAT3 activation and PD-L1 expression. In summary, A20 was capable of alleviating the inhibitory effect of PHB on STAT3 phosphorylation and activation, thus activating PD-L1 transcription and expression.
Supplemental material

Prohibitin mediates the regulatory role of A20 on STAT3 phosphorylation. (A) Venn diagram displayed the overlap between A20-interated proteins and differentially-expressed proteins after the intervention of A20. (B) Reciprocal coimmunoprecipitation among PHB, A20 and STAT3, followed by immunoblotting analysis in A375 and A2058 melanoma cell lines. (C) Coimmunoprecipitation assay followed by immunoblotting analysis shows the changes of the interaction between STAT3 and PHB in melanoma cells after the overexpression of A20. (D) Immunoblotting analysis of phosphor-STAT3 (Y705), PD-L1 and PHB after the knockdown of PHB. (E) Immunoblotting analysis of PHB expression in melanoma cells with or without A20 overexpression followed by the treatment with MG132. (F, G) Immunoblotting analysis of PHB expression in melanoma cells with or without the knockdown of A20 followed by the treatment with CHX (n=3 per group). (H) Coimmunoprecipitation analysis of the interaction between K48-linked ubiquitin chain and PHB after the knockdown of PHB. (I) Co-immunoprecipitation analysis of interaction between PHB and K48-linked ubiquitin chain or A20 in melanoma cells with the knockdown of endogenous PHB followed by the overexpression of WT PHB, PHB K186R and PHB K202R. Data represent the mean±SEM of triplicates. P value was calculated by two-tailed Student’s t-test. CHX, cycloheximide; PD-L1, programmed death ligand 1; PHB, prohibitin; SEM, SE of the mean.
We further explored how A20 regulated PHB expression. The intervention of A20 did not change the mRNA level of PHB (online supplemental figure 2F), suggesting that A20 regulated PHB at the post-translational level. A20 has been generally recognized as an ubiquitin-editing enzyme with integrated function of deubiquitinating and ubiquitinating its substrates. For this assessment, we then investigated whether A20 acted as a deubiquitinase or ubiquitin ligase of PHB. After the treatment with proteasome inhibitor MG132, A20-induced reduction of PHB expression was reversed in melanoma cell lines (figure 6E), indicating that proteasomal pathway was involved in the degradation of PHB mediated by A20. Furthermore, cycloheximide pulse-chase analysis showed that the turnover of intracellular PHB was significantly delayed after the knockdown of A20 (figure 6F,G). Moreover, immunoprecipitation assay stated that K48-linked ubiquitin chain bonded to PHB, which was markedly attenuated after the knockdown of A20 (figure 6H), implying that A20 may act as an E3 ubiquitin ligase of PHB and regulate its ubiquitination and degradation.
We continued to identify the ubiquitination site of PHB. Through the analysis in two websites that predict ubiquitination site, CKSAAP Ubsite and Ubpred (online supplemental table 5), 2 lysine residues (K186 and K202) with relative higher ubiquitination potential were suggested. We constructed two PHB mutants in which the K186 and K202 residues were replaced by arginine, respectively (named K186R and K202R), and then transfected vectors harboring WT PHB or the two PHB mutants into melanoma cells with the knockdown of endogenous PHB expression. Consequently, either K186R or K202R could give rise to significant reduction of K48-linkage ubiquitination of PHB (figure 6I). Moreover, the interaction between A20 and PHB was dampened after the transfection of either K186R or K202R mutant similarly (especially K202R) (figure 6I), indicating that K202 and K186 were both required for the ubiquitination of PHB by A20. Together, PHB was a direct ubiquitination substrate of A20, which at least partially mediated the proteasomal degradation of PHB. Additionally, A20 overexpression could promote PHB ubiquitination and proteasomal degradation, thereby restoring STAT3 activity to promote PD-L1 transcription.
Supplemental material
Tumorous A20 expression is associated with the invigoration of circulating exhausted-phenotype CD8+ T cells in response to anti-PD-1 antibody treatment
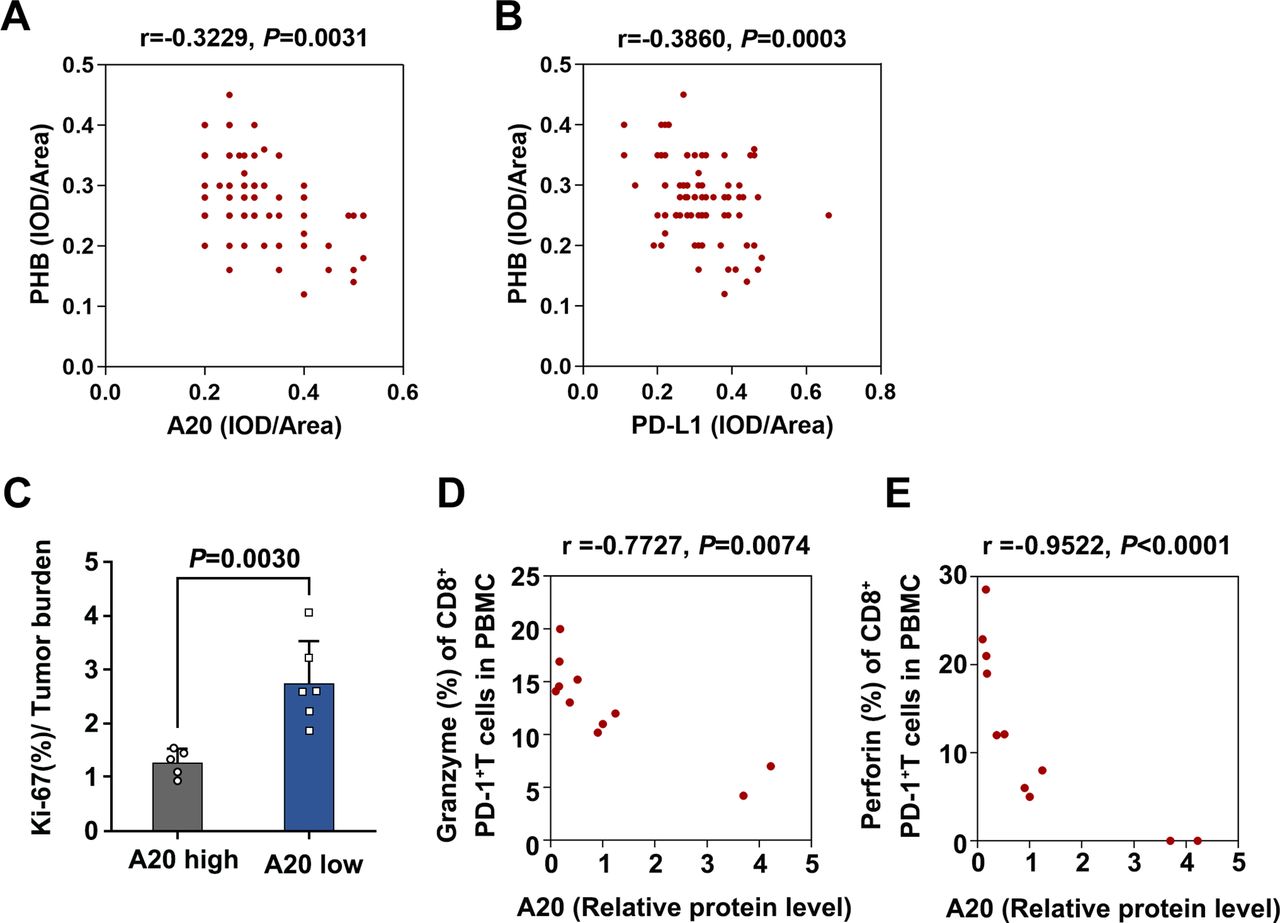
After confirming the involvement of A20-PHB-STAT3-PD-L1 axis in the regulation of immunotherapy resistance, we performed immunohistochemical analysis in TMA to examine the association between A20 and PHB expression and the relationship between PHB and PD-L1 expression, so as to verify this axis further. PHB expression was significantly downregulated in melanoma tissues compared with nevus tissues (online supplemental figure 2G). Moreover, PHB expression was in negative correlation with PD-L1 expression, and the expression of A20 was also negatively correlated with PHB expression in melanoma in TMA (figure 7A,B). Our analysis on TCGA SKCM database showed that the level of PHB was negatively correlated with TNFAIP3 mRNA expression and CD274 mRNA level respectively (online supplemental figure 2H,I). These results provided in vivo evidence for the regulatory effect of A20 on PHB and PD-L1 expressions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
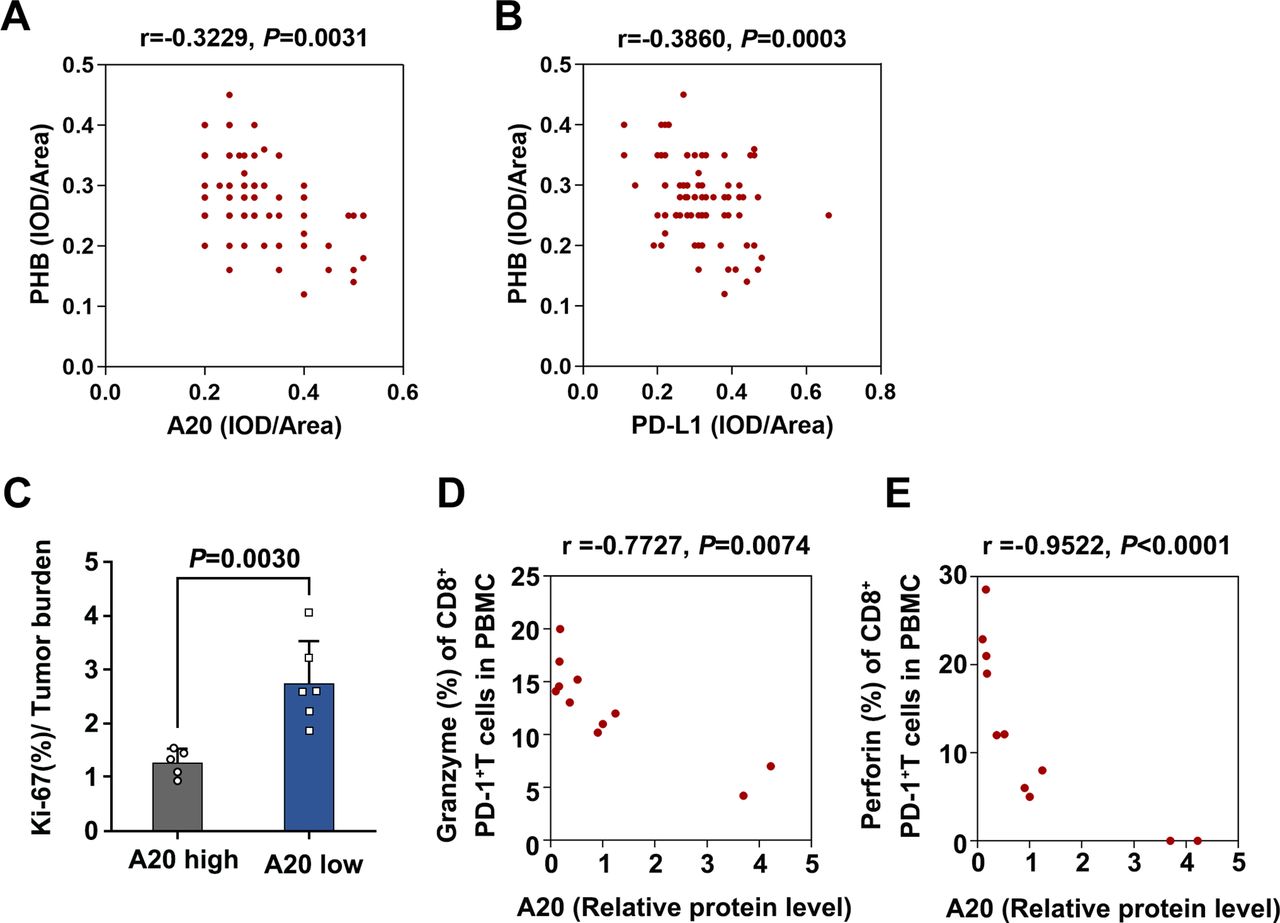
tumorous A20 expression is associated with the invigoration of exhausted-phenotype CD8+T in response to anti-PD-1 antibody treatment. (A) The correlation between A20 and PHB expression in melanoma tissues in TMA, r value was calculated by Spearman correlation, p value was calculated by two tailed Student’s t-test. (B) The correlation between PD-L1 expression and PHB expression in melanoma tissues in TMA. r value was calculated by Spearman correlation, p value was calculated by two tailed Student’s t-test. (C) The ratio of the Ki-67 percentage in CD8+PD-1+T cells to tumor burden in melanomas with high expression of A20 (n=5, Resistance group) and low expression of A20 (n=6, Response group). (D, E) The correlation between relative A20 protein expression and Ki-67 percentage or Granzyme B percentage of CD8+PD-1+T cells in melanomas. r value was calculated by Spearman correlation, p value was calculated by two-tailed Student’s t-test. PD-L1, programmed death ligand 1; PHB, prohibitin.
Of note, a previous study has proved that exhausted-phenotype CD8+ T cells (Tex), a subset of CD8+ T cells with restrained function because of inhibitory receptors like PD-1, is the major target of anti-PD-1 antibody treatment.2 The invigoration of circulating Tex characterized by increased ratio of Ki-67 percentage to tumor burden in particular the ratio of Ki-67 in circulating PD-1+CD8+ T cells to tumor burden, is highly associated with the clinical response to anti-PD-1 antibody immunotherapy.5 By using flow cytometry analysis of PBMCs from the previously enrolled 11 melanoma patients receiving anti-PD-1 antibody treatment, we observed that the ratio of Ki-67 percentage in circulating PD-1+CD8+ T cells to tumor burden was increased more prominently in patients with low tumorous A20 expression than those with high tumorous A20 expression (figure 7C). Evidently, the expression of tumorous A20 was in negative correlation with both Granzyme B and Perforin in circulating PD-1+CD8+ T cells of 11 melanoma patients after anti-PD-1 antibody treatment (figure 7D,E). These results convey that tumorous A20 was significantly associated with the reactivation of circulating exhausted-phenotype CD8+ T in response to anti-PD-1 antibody treatment, which is consistent with the biological effect of A20 on the therapeutic efficacy of anti-PD-1 immunotherapy.
Discussion
So far, approved anti-PD-1 antibodies are widely applied in clinical practice, which significantly prolongs the survival of patients with advanced melanoma. However, low response rate and the establishment of drug resistance significantly restrain the durable effect of anti-PD-1 antibodies.2 6 7 Some studies have signified that tumorous PD-L1 expression is closely associated with clinical response and therapeutic efficacy of immune checkpoint blockade in melanoma.38 39 Nevertheless, the role of tumorous PD-L1 in the efficacy of immunotherapy remains controversial. For one thing, high expression of PD-L1 in tumor cells before treatment is regarded as a biomarker of better response.40 For the other, in response to anti-PD-1 antibody treatment, the re-activation of CD8+ T cell is in position to promote the secretion of IFN-γ, which induces feedback upregulation of PD-L1 that limits the effect of PD-1/PD-L1 blockade.10 12 Hence, the role of tumorous PD-L1 in the regulation of therapeutic outcome of immunotherapy is equipped with a bearing on the time phase of treatment (before or during the treatment), and suppressing the sustained PD-L1 upregulation in tumor during treatment could be helpful for obtaining durable therapeutic effect and elevating the therapeutic efficacy. In the present study, the increased A20 expression contributed to the upregulation of PD-L1 expression in melanoma after anti-PD-1 antibody treatment, indicating that A20 is required for sustained PD-L1 expression that causes treatment resistance. Therefore, controlling A20 expression in tumor during anti-PD-1 antibody treatment is a valuable strategy to increase therapeutic efficacy and provide clinical benefits for patients with melanoma and other cancer types.
Inflammation-related signals can trigger the eradication of cancer immune escape and influence the outcome of immunotherapy,13 14 16but the underlying mechanism needs to be further elucidated. A20 has always been recognized as an important modulator of inflammation and immune response.19 Through the ubiquitination of RIPK1 and TRAF6, A20 is capable of inhibiting proinflammatory NF-κB pathway and the expressions of downstream cytokines.41 Moreover, the loss of function of A20 could cause inflammatory response and even lead to the onset and development of many autoimmune diseases.19 21 41 Recently, the regulatory role of A20 in immune cells has been expanded to be associated with antitumor immunity. Precisely, the overexpression of A20 in CD8+ T cells can dampen the nuclear accumulation of NF-κB components and impose a brake on the antitumor activity of CD8+ T cells.23 In addition, in vivo silence of A20 expression in DCs could potentiate the antitumor immune response and delay tumor progression.22 It is necessary to note that A20 is also abundantly expressed in tumor cells that are dominant in the tumor microenvironment. Still, previous studies mainly paid attention to the role of tumorous A20 in the regulation of tumor cell behavior instead of surrounding immune cells. Extending to this, our present study demonstrated that A20 was proved as a novel upstream regulator of PD-L1 and was responsible for attenuated activity of infiltrated CD8+ T cell and thereby tumor progression and immunotherapy resistance. Therefore, A20 is capable of exerting its immunosuppressive effect against tumor immunity in various ways, not only by regulating the function of immune cells directly, but also by activating suppressive signals in immune cells indirectly through tumorous immune checkpoints like PD-L1.
Cumulating evidence has revealed the regulatory mechanisms of PD-L1 expression from the perspective of transcriptional, translational and post-translational modification, respectively, which can be connected to multiple hallmark characteristics of cancer, including oncogene, cell-cycle progression and lipid metabolism.42–44 Inflammation is also crucial for cancer pathogenesis owing to its contribution to tumor cell survival, angiogenesis, cancer metastasis and the eradication of antitumor immunity.45 A recent study demonstrated the proinflammatory cytokine TNF-α as a major factor triggering cancer immunosuppression against T cell surveillance via the stabilization of PD-L1.16 Our data forwardly emphasized the connection between inflammation and antitumor immunity by demonstrating that A20 promoted the expression of PD-L1 to impair the antitumor activity of CD8+ T cells. Mining the publicly available transcriptomic data in TCGA SKCM database also provided us with the notion that inflammation pathway was highly correlated with PD-L1 expression in melanoma. Therefore, targeting A20 or alternative inflammation-related signals could be adopted in a broad spectrum of therapeutic backgrounds in view of the interplay among inflammation, cancer pathogenesis and tumor immune response.
The pathogenic role of A20 has been verified in different kinds of cancers, which is associated with distinct ubiquitin-editing functions. In breast cancer, A20 facilitates TGF-β1-induced tumor metastasis through multi-monoubiquitylation of Snail1,24 whereas in gastric cancer, A20 mediates poly-ubiquitination of RIP1 and inhibits TRAIL-induced cell apoptosis.25 Supplementary to these, our study recognized PHB as a novel substrate of A20. A20 acted as a ubiquitin ligase of PHB by binding K48-linked ubiquitin chain and accelerating its proteasomal degradation, which extended the substrate network of A20 and the regulatory mechanism of PHB. In addition, PHB directly interacted with STAT3 and suppressed its phosphorylation and activation, which was attenuated after A20 overexpression. Our results indicated an A20-PHB-STAT3-PD-L1 regulatory axis in melanoma, which was supported by further bioinformatics analysis on TCGA SKCM database. It ought to be noted that previous studies have demonstrated that PHB mediated the resistance to chemotherapy and BRAF-targeted therapy in tumor.46 47 More than this, our study implicated that PHB also mediated the resistance to immunotherapy by transducing the signal from A20 to STAT3, suggesting PHB as a versatile regulator and a promising therapeutic target for the determination of the treatment outcomes of diverse therapeutic approaches in cancer.
The invigoration of infiltrated cytotoxic CD8+ T cells in tumor microenvironment, especially the exhausted-phenotype CD8+ T cells (Tex) which are restrained by inhibitory receptors like PD-1, is largely responsible for the effect of anti-PD-1 antibody.5 In consistent with this notion, the suppression of tumorous A20 could revive the antitumor capacity of infiltrated CD8+ T cells, synergizing with the effect of anti-PD-1 antibody treatment. In fact, several previous studies have demonstrated the crucial role of tumor intrinsic molecules in the regulation of surrounding immune cells and their antitumor immunity.48 Hence, to obtain the invigoration of dysfunctional immune cells by targeting tumor cells is promising for elevating the efficacy of immunotherapy. What’s more, T cell invigoration to tumor burden ratio is highly associated with the clinical response of anti-PD-1 antibody treatment.5 Since that the expression of tumorous A20 was in negative correlation with the T cell invigoration to tumor burden ratio, the application of tumorous A20 expression status to guiding appropriate use of anti-PD-1 antibody in treating advanced melanomas can be taken into consideration in the future.
Previous studies have expounded the excellent translational potential of targeting A20 in terms of treating various diseases. Precisely, A20 in DCs is able to suppress NF-κB signaling and constrain DCs-mediated T-cell stimulation, mitigating T-cell-dependent blood pressure elevation.49 In addition, the downregulation of A20 contributes to the development of childhood asthma, and acts as a possible biomarker for predicting asthma.50 Consequently, targeting A20 has the translational potential for the prevention and treatment of hypertension and asthma, and it is further expanded for cancer treatment with particular focus on PD-L1-dependent antitumor immunity according to our present investigation, providing a novel method for immunity-based cancer therapy for the crucial regulatory role of A20 in PD-L1 expression and inflammatory response as well. Conspicuously, some clinical trials have enrolled the patients for the test of combined therapies of anti-PD-L1/PD-1 antibodies and the inhibitors of the upstream regulatory molecules of PD-L1 like Akt and STAT3 (NCT03421353, NCT03772561) .34 48 Moreover, several studies demonstrated that the concurrent suppression of the upstream regulator of PD-L1 expression could increase the efficacy of immunotherapy in pre-clinical mouse model.43 51 Given the findings of the present study, it is necessary to testify the translational potential of targeting A20 in cancer immunotherapy in prospective trials.
Conclusions
In summary, our findings delineate the interplay between inflammation-related signal and antitumor immunity by expounding that A20 controls PD-L1 expression via PHB-STAT3 axis so as to confer therapeutic resistance to anti-PD-1 immunotherapy. Targeting tumorous A20 is able to potentiate the antitumor capacity of cytotoxic CD8+ T cells and promote the therapeutic response to PD-1/PD-L1 blockade in melanoma. Given the fact that dysregulated A20 contributes to the pathogenesis of various cancers and A20 is a crucial regulator of inflammation and immunity, our study provides the proof of a principle for the development of immunotherapeutic strategy in cancers that are associated with inflammation.
Acknowledgments
The authors gratefully thank Professor Rui Zhang for the helpful suggestions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
WG, JM, SG, HW and SW contributed equally.
Contributors CL and WG conceived of the study and carried out its design. WG, JM, SG, HW, SW, XY, QN, FY, JM and YY performed the experiments. WG, JM, SG, SW, QS, LL, TZ, JZ, JC and GZ conducted the statistical analyses. CY applied the ethical documents. TG provided crucial suggestions for experimental design. WG and JM wrote the paper. CL and WG revised the paper. SC proofread the paper. All authors read and approved the final manuscript. Moreover, the authors declare no competing interests.
Funding The research leading to these results has received funding from National Natural Science Foundation of China (No. 81625020, No. 81902791, No. 81803112, No. 81803141) and Key Research and Development Program of Shaanxi Province (2019SF-079).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval All animal experiments were approved by the Animal Experiment Administration Committee of Fourth Military Medical University. The use of human samples was approved by the Ethics Committee of Xijing Hospital and conformed to Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as online supplemental information. Data are available in a public, open access repository. All data generated or analyzed during this study are included in this manuscript.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.