Article Text

Abstract
Background Most patients with high-grade serous ovarian cancer (HGSC) lack an effective response to immune checkpoint blockade, highlighting the need for more knowledge about what is required for successful treatment. As follicular cytotoxic CXCR5+CD8+ T cells are maintained by reinvigoration by immune checkpoint blockade in tumors, we attempted to reveal the relationship between CXCR5+CD8+ T cells and the tumor microenvironment to predict immunotherapy responses in HGSC.
Methods 264 patients with HGSC from two cohorts and 340 HGSC cases from The Cancer Genome Atlas cohort were enrolled. Ex vivo and in vivo studies were conducted with human HGSC tumors and murine tumor models. The spatial correlation between CXC-chemokine ligand 13 (CXCL13), CXCR5, CD8, and CD20 was evaluated by immunohistochemistry and immunofluorescence. Survival was compared between different subsets of patients using Kaplan-Meier analysis. The therapeutic effect of CXCL13 and programmed cell death-1 (PD-1) blockade was validated using human HGSC tumors and murine models.
Results High CXCL13 expression was associated with prolonged survival. Tumors with high CXCL13 expression exhibited increased infiltration of activated and CXCR5-expressing CD8+ T cells. Incubation with CXCL13 facilitated expansion and activation of CXCR5+CD8+ T cells ex vivo. CXCR5+CD8+ T cells appeared in closer proximity to CXCL13 in tumors and chemotaxis towards CXCL13 in vitro. The combination of CXCL13, CXCR5, and CD8+ T cells was an independent predictor for survival. In addition, CXCL13 was associated with clusters of CD20+ B cells. CD20+ B cells predicted better patient survival in the presence of CXCL13. Histological evaluation highlighted colocalization of CXCL13 with tertiary lymphoid structures (TLSs). TLSs carried prognostic benefit only in the presence of CXCL13. CXCL13 in combination with anti-PD-1 therapy retarded tumor growth in a CD8+ T-cell-dependent manner, resulting in increased infiltration of cytotoxic CD8+ T cells and CXCR5+CD8+ T cells.
Conclusions These data define a critical role of CXCL13 in shaping antitumor microenvironment by facilitating the maintenance of CXCR5+CD8+ T cells in TLSs and support a clinical investigation for a combination of CXCL13 and PD-1 blockade therapy in HGSC.
- biomarkers
- tumor
- cytokines
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
As the most prevalent histological subtype of epithelial ovarian cancer, high-grade serous ovarian cancer (HGSC) usually presents diffuse carcinogenesis in the advanced stage, and its 5-year survival rate is only 20%–30%.1 Immune checkpoint blockade targeting programmed cell death-1 (PD-1) and its ligand (PD-L1) has revolutionized contemporary treatment in a variety of tumors, but only a subset of patients with HGSC achieved encouraging clinical benefit.2 3 Characterization of tumor immune microenvironment is critical for patient’s stratification to guide optimal immune checkpoint interventions.
CXCR5+CD8+ T cells, also known as follicular cytotoxic T cells (Tfc), are localized similarly to CXCR5+ follicular helper T (Tfh) cells in B-cell follicles.4 In the tumor microenvironment (TME), Tfc cells maintain self-renewal capability during prolonged antigen exposure and might play an important role in sustaining antitumor immunity and responding to PD-1/PD-L1 blockade.5 6 A high frequency of tumor-infiltrating Tfc cells is positively correlated with survival in lung,7 colorectal4 and pancreatic8 cancers. Taken together, these findings suggest that tumor-infiltrating Tfc cells might be used as a potential biomarker to identify patients most likely to benefit from PD-1/PD-L1 blockade and predict the clinical outcomes in patients with HGSC.
B cells and tertiary lymphoid structures (TLSs) could be a potential biomarker and therapeutic target in response to immune checkpoint blockade therapy in patients with melanoma and renal cell carcinoma.9 10 Consistently, the presence of B cells in TLSs was associated with improved survival and a high response rate to PD-1 blockade in patients with soft-tissue sarcoma.11 However, due to the variable functional status of TLSs, main contributors to induce favorable TLSs that augment antitumor efficacy remain unknown.
CXC-chemokine ligand 13 (CXCL13) exclusively binds to the chemokine receptor CXCR5, which is expressed on B cells, Tfh cells, and Tfc cells.5 6 CXCL13 helps to recruit specific B cells and CXCR5+ T cells to the site of chronic inflammation to coordinate both humoral and cell-mediated adaptive immune responses.12 13 CXCL13 plays a critical role in immune cell recruitment, activation, and adaptive immune response regulation, with its helper functions for B cells and cytotoxic T cells.14 CXCL13 also influences innate immunity by shaping the character and magnitude of inflammatory response. Consistently, CXCL13 expression has been confirmed to be strongly correlated with adaptive immune cell infiltration within colorectal cancer (CRC), as well as prolonged disease-free survival in patients with CRC.15 Effects of CXCL13 on CXCR5+CD8+ T-cell maintenance in the TME of HGSC and its prognostic significance remain elusive. Here, our purpose was to elucidate the prognostic significance of CXCL13 and its functional relevance to the maintenance of CXCR5+CD8+ T cells and immune contexture in HGSC.
Methods
Patients and specimens
Tissue specimens from 185 patients with HGSC (training cohort; online supplemental table 1) who had undergone surgery between 2008 and 2015 were obtained from Gynecology and Obstetrics Hospital of Fudan University. All patients were followed up until April 2019. Tissue specimens from 79 patients with HGSC (validation cohort; online supplemental table 1) who had undergone surgery between May 2013 and August 2018 were obtained from Suzhou Municipal Hospital. All patients were followed up until March 2019. None of the patients had autoimmune disorder or a history of prior cancer. None of the patients was treated with chemotherapy, radiation, or any other antitumor medicines before tumor resection. Overall survival (OS) was calculated from the date of surgery to the date of death or last follow-up. Progression-free survival (PFS) was calculated from the date of surgery to the date of any type of progression diagnosis. Fresh tumor tissue samples were obtained from 36 patients with HGSC (online supplemental table 1) during surgery at the Department of Gynecology of our hospital, including 16 HGSC tissues used for ex vivo stimulation and immunofluorescence. For immunohistochemical and immunofluorescence studies, once the surgical specimens were obtained, all specimens were snap-frozen immediately in liquid nitrogen. Peripheral blood samples from 16 healthy donors and 46 patients with HGSC were collected from our hospital. For further research measurements, blood samples were collected to isolate plasma and stored in a freezer at −80℃ until further analysis. Normal ovarian tissues (n=24) were obtained from Bank of Tumor Resources at our hospital. The clinical characteristics of all tissue samples from patients with HGSC are summarized in online supplemental table 1. The number of samples used in each experiment is provided in online supplemental table 2.
Supplemental material
Tissue microarray preparation and immunohistochemistry
Sections of formalin-fixed paraffin-embedded (FFPE) tissues were prepared and stained with H&E. Two representative tumor areas per sample were confirmed and selected to generate tissue microarrays (TMAs). Tissue cores (3 mm in diameter) were collected from the individual FFPE blocks and arranged in recipient paraffin blocks (TMA blocks). All TMA blocks were confirmed to contain suitable tumor lesions representing more than 50% of the core area based on H&E staining.
Primary antibodies against the following antigens were used: CXCL13 (dilution 1:400; R&D Systems), CD4 (dilution 1:200; Sigma-Aldrich), CD8 (dilution 1:200; Cell Signaling Technology), CD20 (dilution 1:800; Invitrogen), and CXCR5 (dilution 1:750; Sigma-Aldrich). Biotin–streptavidinHorseradish Peroxidase (HRP) detection systems (Origene) were applied for the following steps. Sections of lymph nodes that were previously shown to be positive for CXCL13 and CXCR5 were used as positive controls. Isotype antibody instead of primary antibody was used as a negative control (online supplemental figure 1C).
Immunohistochemical evaluation
To avoid bias and subjective interpretation, immunohistochemistry (IHC) was evaluated independently and blindly by two investigators (JL and GZ) who were blinded to the clinical data. The sections with an interobserver variation of >10% were reassessed using a double-headed light microscope to achieve consensus. Immunohistochemical staining of CXCL13 and CXCR5 was scored based on staining intensity on a scale of 0 to 3 (absent=0, weak=1, moderate=2, or strong=3), and a percentage of tumor cells was stained (range, 0%–100%) using an algorithm developed for ImageJ (NIH), as previously described.16 Average CXCL13 or CXCR5 IHC score (range, 0–300)=% positive staining×staining intensity/5 random high-power fields (HPFs). Intratumor staining and stroma CXCL13 staining were evaluated separately in all TMA cores determined by histological verification using H&E (online supplemental figure 1B). The intratumor compartment was defined as the area encompassing all the cancer lesions, whereas the stroma compartment was the field along with the interface between the lesions and its neighborhood. Immunohistochemical staining of CD4+, CD8+, and CD20+ cellular infiltrates was counted based on the number of positively stained tumor infiltrated lymphocytes (TILs) by examining five representative HPFs under 200× magnification, and the cell counts were averaged. Optimal cut-offs for each protein were obtained using medium value—CXCL13: 115 stroma, 75 intratumor; CXCR5: 215; CD8: 6 cells/HPF; CD20: 7 cells/HPF. These cut-offs were used to stratify the tumors into low versus high for each protein.
Preparation of single cells from HGSC tissues
Fresh tissues were washed 3 times with cold phosphate-buffered saline (PBS) containing 1% fetal bovine serum before being minced into small pieces. The specimens were collected in RPMI 1640 medium containing 1 mg/mL collagenase IV and 10 mg/mL deoxyribonuclease I and mechanically dissociated using MACS Dissociator (Miltenyi Biotec). Dissociated cell suspensions were further incubated for 1 hour at 37°C under continuous rotation. The cell suspensions were then filtered through a 100 µm cell strainer (BD), washed once with PBS, and resuspended in cell staining buffer.
Ex vivo tumor stimulation assay
Single-cell suspensions were cultured in 3D assay medium (RPMI 1640 medium prepared with 2% Matrigel (BD) and supplemented with 100 IU/mL rhIL-2, 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% fetal bovine serum). For stimulation assay, the medium was present with or without rhCXCL13 (R&D Systems; 1 µg/mL) for 48 hours at 37℃ and 5% CO2. Cells were collected by digestion with Liberase DH (Sigma-Aldrich) and subjected to flow cytometry analysis.
Immunofluorescence
For immunofluorescence staining, slides were incubated with goat anti-human CXCL13, mouse anti-human CD8, rabbit anti-human CXCR5, or mouse anti-human CD20. Alexa Fluor 488-conjugated, 594-conjugated, or 647-conjugated donkey anti-mouse/rabbit/goat secondary antibodies were used (Jackson ImmunoResearch). Cell nuclei were counterstained with 4',6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) (Sigma-Aldrich). Sections were sealed with Fluoromount Aqueous Mounting Medium (Sigma-Aldrich). Images were taken using confocal microscope (TCS SP8; Leica Microsystems). To measure the spatial relationship between CXCR5+CD8+ or CXCR5−CD8+ T cells and CXCL13, 16 samples from patients with HGSC were used for multiplex immunofluorescence, and at least five fields of view were taken for each sample using a 400-fold field of view. Leica LASX software was used to create a maximum projection image to be applied for subsequent analyses in ImageJ. The XY location of CD8+ objects and CXCL13+ objects, and fluorescence colocalization of CXCR5 staining in CD8+ objects were determined in ImageJ. The distance from CXCR5+CD8+ or CXCR5−CD8+ T cells to the nearest CXCL13+ objects was calculated.
Flow cytometry
Single-cell suspensions were immunostained with a panel of fluorochrome-tagged monoclonal antibodies (online supplemental table 3). Red cells were lysed with an ammonium chloride solution, and samples were incubated with Live/Dead Fixable Dead Cell Staining Kit (ThermoFisher) before staining. Samples stained with isotype-matched antibodies were used as negative controls. For sample acquisition, a Beckman Coulter cytoflex flow cytometer with FACS CytExpert software was used (Beckman Coulter), and FlowJo software (Tree Star) was used for analyses.
ELISA
Serum concentrations of soluble CXCL13 protein were determined using a commercially available Quantikine kit (R&D Systems) according to the manufacturer’s instructions. The concentration of CXCL13 in the sera was interpolated from a standard curve, which was generated using the respective recombinant protein.
TLS quantification
TLSs were quantified using both H&E and CD20 IHC staining.17 Structures were identified as aggregates of lymphocytes having histological features with analogous structures to lymphoid tissue with B cells (CD20) and T cells (CD4, CD8) appearing in the tumor area.
Chemotaxis assay
Single cells from fresh collected HGSC tissues were resuspended in RPIM 1640 and placed at a concentration of 1×107 cells/mL in the upper chamber of Transwell plates (Corning). For CXCR5 neutralizing group, cell suspension was incubated with 5 µg/mL anti-human CXCR5 (R&D Systems) at 37°C for 30 min. The lower chamber of Transwell plates contained RPIM 1640 with 1 µg/mL CXCL13 (R&D Systems) to serve as a chemoattractant. Cells were allowed to migrate for 2 hours at 37°C. Cells in the top and bottom of the transwell were harvested and analyzed using flow cytometry. Chemotaxis percentage was calculated as the migration percentage of the cell numbers of lower chamber to that of the total cell numbers and normalized to control migration.
Animal experiments
Female B6C3F1 mice were obtained from Charles River Laboratories (4–6 weeks old). All experiments were carried out in accordance with the guidelines for experimental animals at the Fudan University of China. OV2944-HM1 (HM-1) cells (1×106) were subcutaneously injected into the right flank using a 27 g needle. Tumor volume (mm3) was measured every second day and calculated as (length×width2)/2. rmCXCL13 (5 µg per mouse) was injected intraperitoneally on the day of tumor implantation and every other day thereafter until the end of the experiment. Mice were injected intraperitoneally with Invivo plus rat IgG2a isotype control (100 µg, 2A3; BioXCell) or Invivo plus anti-mouse PD-1 (200 µg, J43; BioXCell) or anti-mouse CD8α (200 µg, BE0061; BioXCell) every 3 days. For the combination therapy, mice were treated with anti-murine PD-1 with or without CXCL13 from 4 days after tumor implantation and readministered every 3 days for six maximum treatments. On the indicated days, tumors were harvested and either digested into single-cell suspension for flow cytometry analysis or formaldehyde-fixed for IHC of mouse CD8 (1:2000; Abcam), CD20(1:800; Invitrogen), CD4 (1:2000; Abcam), and cleaved-caspase 3 (1:1500; CST).
Statistical analyses
Significant differences between two groups were determined by either Mann-Whitney U test or unpaired t-test or paired t-test. Significant differences between three or more groups were determined by one-way analysis of varaince (ANOVA) or two-way ANOVA followed by multiple comparisons with the Bonferroni post hoc test. Simple correlations were summarized using the Pearson’s correlation coefficient. Kaplan-Meier analysis and log-rank test were used to analyze the OS and PFS of patients with HGSC . Univaraite and multivariate analyses were performed by Cox regression survival analyses (Enter for univariate and Backward and Forward methods for multivariate analyses). All statistical analyses were performed using GraphPad Prism V.8.0.0 (GraphPad Software) and MedCalc V.19.0.5 (MedCalc Software). Asterisks indicate the significance level of p value: *p<0.05, **p<0.01, and ***p<0.001.
Results
Prognostic impact of CXCL13 in HGSC
CXCL13 staining was significantly high in HGSC tissues and mainly located in stroma in specimens from patients with advanced-stage HGSC (figure 1A; online supplemental figure 1A–D). Compared with healthy donors (n=16), CXCL13 concentrations in serum were significantly higher in patients with HGSC (n=46, (online supplemental table 1; figure 1B). After an average follow-up of 46.04 months, follow-up information was available on 26 patients, and higher CXCL13 concentration was indicative of prolonged OS (p=0.0456; figure 1C). CD45+ lymphocytes were the main source of CXCL13 in HGSC (figure 1D). Furthermore, CD20+ B cells and CD68+ macrophages represented the predominant source of CXCL13 (figure 1E). Patients with high levels of stroma CXCL13 staining exhibited significantly longer PFS and OS compared with their low CXCL13 counterparts (median PFS: >120 months vs 79 months; p=0.0012; median OS: >120 months vs 59 months; p=0.0009) (figure 1F). This phenomenon was more obvious in patients with advanced tumors (online supplemental figure 2A). We also identified a trend toward improved PFS (but not OS) for patients with HGSC with high levels of intratumoral CXCL13 (figure 1G). Univariate Cox analysis confirmed the prognostic impact for PFS of CXCL13 in both stroma (p=0.0018) and intratumor (p=0.0223). However, CXCL13 as a continuous variable failed to reach a statistical significance in PFS and OS (online supplemental tables 4 and 5). To validate these findings, we evaluated the prognostic role of CXCL13 in an independent validation cohort. Stroma CXCL13 failed to predict survival in the validation cohort (online supplemental figure 1E). Also, patients in The Cancer Genome Atlas (TCGA) cohort were stratified by the median value of CXCL13 messenger RNA (mRNA) expression and predicted better survival especially at advanced stage (online supplemental figure 2B,C).
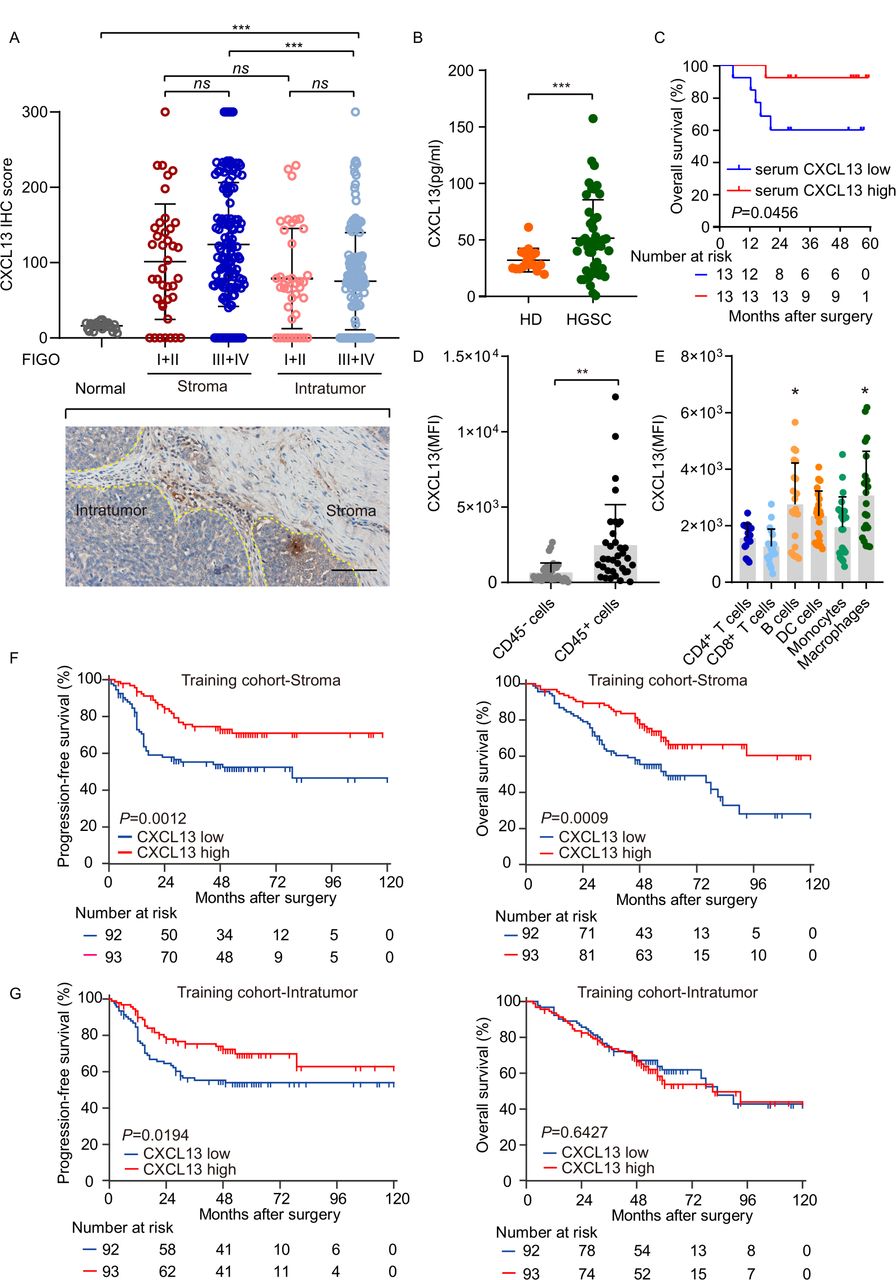
CXCL13 is associated with improved survival in patients with HGSC. (A) H score of CXCL13 expression in normal ovarian tissue and stroma, and intratumor of HGSC tissues (n=185). Bar=100 µm. Results are expressed as mean±SD. (B) Concentration of CXCL13 of peripheral blood was compared between healthy donors (n=16) and patients with HGSC (n=46; Mann-Whitney U test). (C) Kaplan-Meier survival curves for OS of patients with HGSC according to serum CXCL13 concentration (n=26; Gehan-Breslow-Wilcoxon test and p values are shown). (D) CXCL13 expression of CD45− or CD45+ cells was analyzed in HGSC tissues (n=36; Mann-Whitney U test). (E) The mean fluorescence intensity of CXCL13 on different subgroups of lymphocytes (n=20; one-way ANOVA with Bonferroni post hoc test; B cells: CD20+, DC: CD11b+CD11c+, monocytes: CD11b+C11c−, and macrophages: CD68+). (F, G) Kaplan-Meier survival curves for PFS and OS of patients with HGSC from the training cohort according to stroma or intratumor CXCL13 expression (n=185; log-rank test and p values are shown). *p<0.05; **p<0.01; ***p<0.001; ns, no significant difference. ANOVA, analysis of variance; CXCL13, CXC-chemokine ligand 13; HD, healthy donors; DC, dendritic cells; HGSC, high-grade serous ovarian cancer; IHC, immunohistochemistry; OS, overall survival; PFS, progression-free survival.
CXCL13 associates with antitumor immune microenvironment
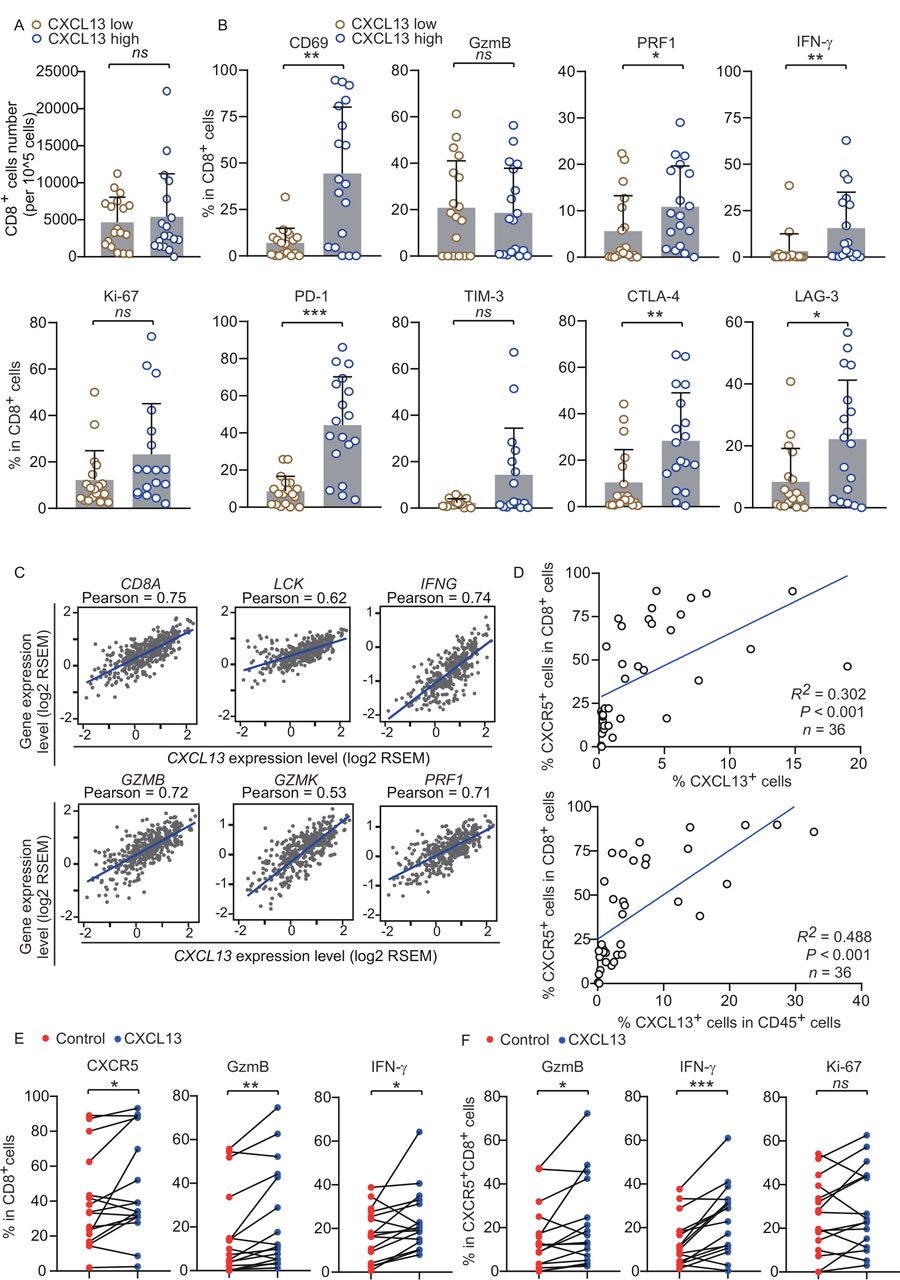
CD8+ cytotoxic T cells are a key component of effective antitumor immunity, which can yield favorable prognosis in HGSC.18 Thus, we explored the functional status of CD8+ T cells in 36 HGSC tumors, which was stratified by the median value of mean fluorescence intensity (MFI) of CXCL13 (online supplemental figure 4A). The counts of tumor-infiltrating CD8+ T cells showed no difference between the two groups (figure 2A; online supplemental figure 3A), whereas the elevation of CD69, interferon-γ (IFN-γ), and perforin-1 (PRF1) expression in CD8+ T cells in the CXCL13 high group was confirmed (figure 2B; online supplemental figure 3A). The expression of immune checkpoint molecules in CD8+ T cells, including PD-1, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and lymphocyte activation gene-3 (LAG-3), was also increased in the CXCL13 high group (figure 2B; online supplemental figure 3A). We found no obvious difference in the number of CD4+ T cells as well as effector molecule (CXCR5, GzmB, IFN-γ, and tumor necrosis factor-α (TNF-α)) expression in CD4+ T cells between the two groups (online supplemental figure 4D,E). Further analyses revealed a positive correlation between CXCL13 mRNA and individual gene transcripts related to T-cell infiltration and antitumor immunity, including CD8A, LCK, IFNG, GZMB, GZMK, and PRF1 from TCGA (figure 2C). These data suggested that CXCL13 is associated with enhanced cytotoxic activity of CD8+ T cells. Given that CXCL13 is an exclusive ligand for CXCR5, we hypothesized that CXCR5+CD8+ T cells may participate in CXCL13-mediated responses. To address this, we analyzed the correlation between these two factors. The percentage of CXCL13+ cells was positively associated with CXCR5+CD8+ T cells in HGSC (figure 2D), and no correlation between CXCL13 and CXCR5−CD8+ T cells was found (online supplemental figure 4B), while the abundance of CXCR5+CD8+ T cells and CXCR5−CD8+ T cells exhibited similar trend (online supplemental figure 4C). To verify the effects of CXCL13 on CXCR5+CD8+ T cells, we stimulated HGSC tissues using CXCL13 ex vivo. A significant increase in the percentage of CXCR5+CD8+ T cells was observed after stimulation with CXCL13. CXCL13 also promoted the expression of GzmB and IFN-γ in CD8+ T cells (figure 2E; online supplemental figure 2B). In addition, ex vivo CXCL13 stimulation significantly upregulated GzmB and IFN-γ expression in CXCR5+CD8+ T cells (figure 2F; online supplemental figure 2B), whereas no effect was observed on the proliferation or effector molecule (GzmB and IFN-γ) expression in CXCR5−CD8+ T cells (online supplemental figure 4F). Effector molecule (CXCR5, IFN-γ, and TNF-α) expression in CD4+ T cells was unaffected by CXCL13 stimulation (online supplemental figure 4G). These data were consistent with the hypothesis that CXCR5+CD8+ T cells contribute to CXCL13-mediated antitumor activity. Furthermore, TIM-3 expression was lower on CXCR5+CD8+ T cells compared with CXCR5−CD8+ T cells (online supplemental figure 4H), but TIM-3 and PD-1 expression in CXCR5+CD4+ T cells showed a conversely high level (online supplemental figure 4I). Taken together, these observations suggest that CXCL13 may influence antitumor immune responses by acting on CXCR5+CD8+ T cells.

CXCL13 associates with antitumor immune microenvironment. (A) Difference in CD8+ T-cell numbers per 105 cells in HGSC tissues between CXCL13 low group and CXCL13 high group (n=36). (B) Difference in effector cytokines (CD69, IFN-γ, GzmB, and PRF1), proliferation (Ki-67), and immune checkpoint molecule (PD-1, Tim-3, CTLA-4, LAG-3) expression on CD8+ T cells in HGSC tissues between CXCL13 low group and CXCL13 high group (n=36). (C) Pearson’s correlation of messenger RNA expression for CXCL13 with CD8A, LCK, IFNG, GZMB, GZMK, and PRF1 from TCGA. (D) Correlation analysis of CXCR5+CD8+ T-cell percentage with CXCL13+ cell percentage (upper) and CXCL13+CD45+ cell percentage (lower) in HGSC tissues (n=36). (E) Effects of rhCXCL13 on CD8+ T cells from HGSC tissues ex vivo (n=16). (F) Effects of rhCXCL13 on CXCR5+CD8+ T cells from HGSC tissues ex vivo (n=16) (Mann-Whitney U test in (A, B); Pearson’s correlations in (C); linear regression in (D); paired t-test in (E, F)). Bar plots show mean±SD; *p<0.05; **p<0.01; ***p<0.001; ns, no significant difference. CXCL13, CXC-chemokine ligand 13; HGSC, high-grade serous ovarian cancer; IFN-γ, interferon-γ; PD-1, programmed cell death-1; PRF-1, perforin-1; RSEM, accurate quantification of gene and isoform expression from RNA-Seq data; TCGA, The Cancer Genome Atlas.
CXCL13-expressing and CXCR5-expressing CD8+ T cells colocalize and are associated with prolonged survival in HGSC
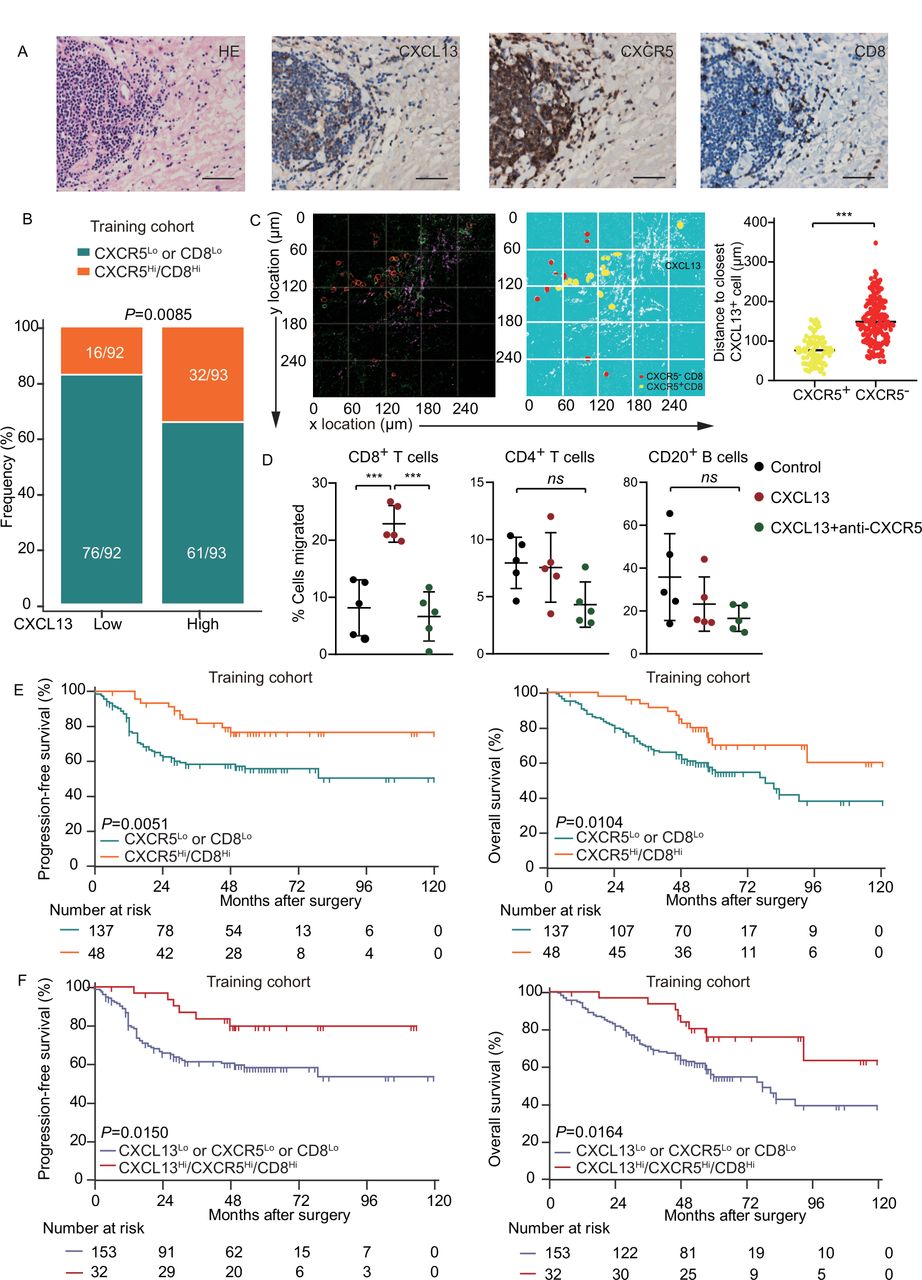
As CXCL13 contributes to activities of CXCR5+CD8+ T cells in the TME, they would be expected to be located in close proximity. IHC in consecutive tissue sections showed that the distribution of CXCL13 was close to CD8+ cells, together with CXCR5 expression (figure 3A; online supplemental figure 5A). There was a strong correlation between CXCL13 and CXCR5Hi/CD8Hi expression in both training and validation cohorts (figure 3B; online supplemental figure 5B). To further confirm this spatial correlation, we carried out three-color immunofluorescence with antibodies to CXCL13, CXCR5, and CD8. The distance between CXCR5+CD8+ T cells and CXCL13 was significantly closer than CXCR5−CD8+ T cells (figure 3C). Consistently, CXCL13 induced chemotaxis for CD8+ T cells, while its effect had been blocked by anti-CXCR5 neutralizing antibody (figure 3D). CXCR5 alone was not sufficient to distinguish survival of patients (online supplemental figure 5C,D). Confirming prior observations,18 CD8+ T cells had a positive impact on PFS of patients (p=0.018), but not on OS in the training or validation cohort (online supplemental figure 5C,D). Importantly, CXCR5Hi/CD8Hi was associated with prolonged survival (median PFS and OS for CXCR5Hi/CD8Hi were >120 months and >120 months) in the training cohort (figure 3E), which was confirmed in the validation cohort (online supplemental figure 5E). These data indicate that CXCR5 might potentially facilitate immune response of CD8+ T cells in HGSC. Next, we investigated the prognostic value of combined CXCL13, CXCR5, and CD8 and revealed that CXCL13Hi/CXCR5Hi/CD8Hi predicted superior survival compared with their counterpart in both training and validation cohorts (figure 3F; online supplemental figure 5F). Multivariate analysis demonstrated that CXCL13Hi/CXCR5Hi/CD8Hi represented an independent prognostic predictor in the training cohort (table 1). All the above findings indicate that the concomitant assessment of CXCL13, CXCR5, and CD8+ T cells in the tumor of patients with HGSC conveys robust prognostic information.
Multivariable analyses of PFS and OS of patients with HGSC in the training cohort
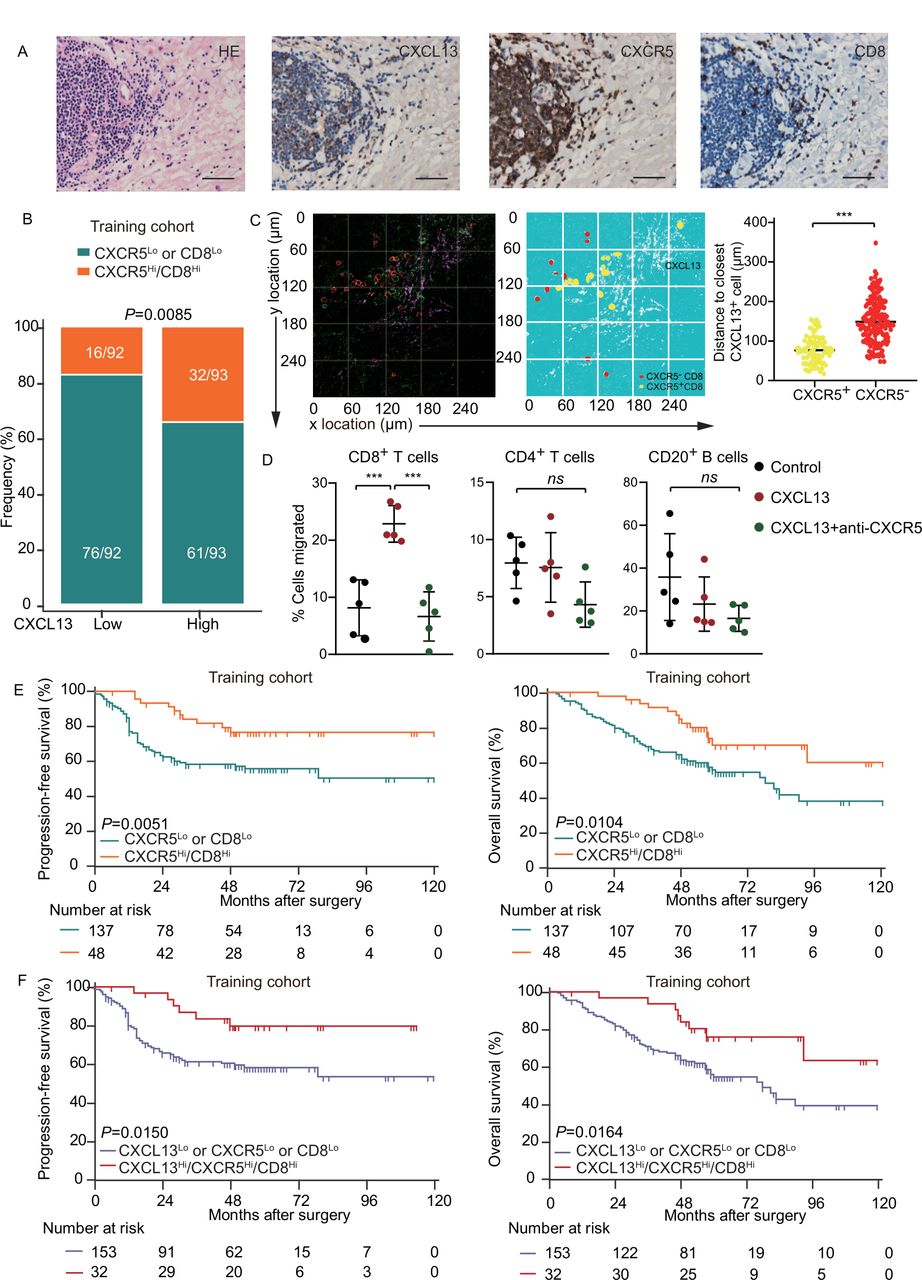
CXCL13-expressing and CXCR5-expressing CD8+ T cells colocalize and are associated with prolonged survival in HGSC. (A) Representative images of CXCR5, CD8, CXCL13 staining and H&E staining in HGSC tissue. Bar=50 µM, H&E stain. (B) Frequency of CXCR5Lo or CD8Lo and CXCR5Hi/CD8Hi patients from the training cohort was compared according to CXCL13 expression (n=185; χ2 test and p value is shown). (C) Spatial relationship map and distance from CXCR5− and CXCR5+ CD8+ T cells to the closest CXCL13+ cell (unpaired t-test). (D) Migration of CD8+ T cells, CD4+ T cells, and CD20+ B cells from single-cell suspension from HGSC towards CXCL13 with or without CXCR5 neutralizing antibody (n=5, one-way ANOVA with Bonferroni post hoc test). (E) Kaplan-Meier survival curves for PFS (left) and OS (right) of patients with HGSC from the training cohort according to CXCR5-CD8 stratification (n=185; log-rank test and p values are shown). (F) Kaplan-Meier survival curves for PFS (left) and OS (right) of patients with HGSC from the training cohort according to CXCL13-CXCR5-CD8 stratification (n=185; log-rank test and p values are shown). Bar plots show mean±SD; *p<0.05; **p<0.01; ***p<0.001; ns, no significant difference. ANOVA, analysis of variance; CXCL13, CXC-chemokine ligand 13; HGSC, high-grade serous ovarian cancer; OS, overall survival; PFS, progression-free survival.
CXCL13 correlates with conglomerated CD20+ B cells and prolongs survival
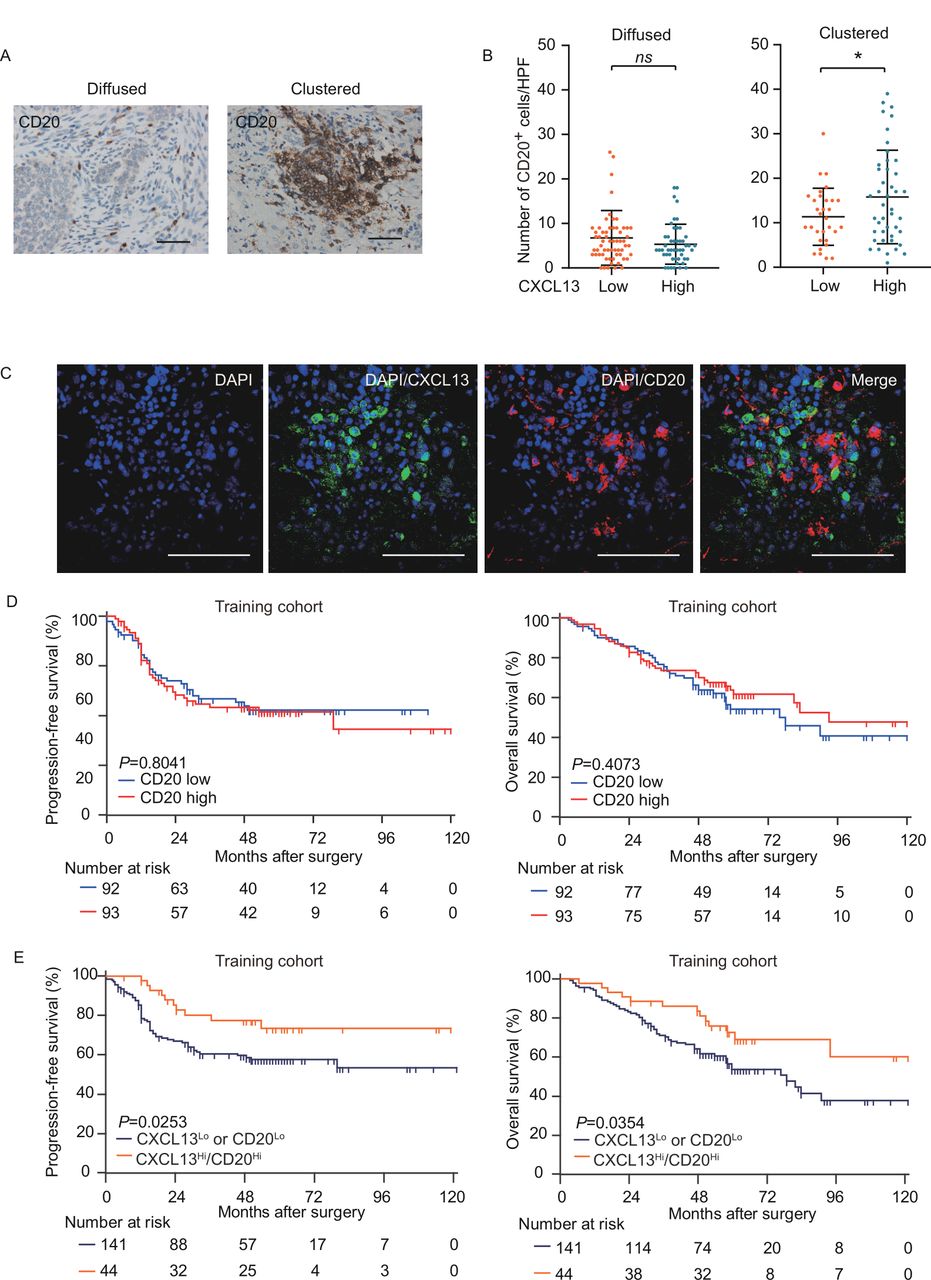
CXCL13 can recruit not only CXCR5+CD8+ T cells but also CD20+ B cells and has an auxiliary effect on its functions.14 Thus, we assumed that CXCL13 may correlate with B cells in the tumor of HGSC. We observed CD20+ B cells diffused or clustered in the stroma (figure 4A). Contrary to expectation, there was no correlation between CXCL13 and CD20+ B cells. Nevertheless, the conglomerate of CD20+ B-cell density increased with high CXCL13 level (figure 4B), indicating that CXCL13 may contribute to CD20+ B-cell agglomeration in TME of HGSC. The colocalization between CXCL13 and conglomerated CD20+ B cells was confirmed by dual-color immunofluorescence (figure 4C). The prognostic significance of CD20+ B cells remains inconsistent,19 20 and no prognostic significance of CD20+B cells was observed in the training cohort (figure 4D). As the presence of both CD20+ and CD8+ TILs correlated with improved survival in patients with HGSC,21 we assessed PFS and OS on stratifying patients into two subsets (CXCL13Hi/CD20Hi, CXCL13Lo or CD20Lo). CXCL13Hi/CD20Hi exhibited better OS (median >120 months) than counterparts of CXCL13Lo or CD20Lo (median=78 months, p=0.0354). Meanwhile, CXCL13Hi/CD20Hi patients exhibited better PFS (p=0.0253) (figure 4E). Collectively, these results suggest that CXCL13 may contribute to the survival benefit of CD20+ B cells by promoting its agglomeration in the TME.

CXCL13 correlates with conglomerated CD20+ B cells and prolongs survival. (A) Representative images of CD20+ B cells in HGSC. Diffused (left) and clustered (right). Bar=50 µM. (B) Numbers of CD20+ B cells were compared between patients with low and high CXCL13 expression. Diffused (n=107, left) and clustered (n=78, right) (Mann-Whitney U test). (C) Representative immunofluorescence staining of HGSC. The sample was stained for CXCL13 (magenta), CXCR5 (red), CD8 (green), and DAPI (blue). Bar=50 µM. (D) Kaplan-Meier survival curves for PFS (left) and OS (right) of patients with HGSC from the training cohort according to CD20+ B-cell density (n=185; log-rank test and p values are shown). (E) Kaplan-Meier survival curves for PFS (left) and OS (right) of patients with HGSC from the training cohort according to CXCL13-CD20 stratification. (n=185; log-rank test and p values are shown). CXCL13, CXC-chemokine ligand 13; DAPI, 4',6-Diamidino-2-Phenylindole, Dihydrochloride; HGSC, high-grade serous ovarian cancer; OS, overall survival; PFS, progression-free survival.
CXCL13 localizes in the context of TLSs and prolongs survival
In our previous results, we observed colocalization between CXCL13 and CXCR5+CD8+ T cells, as well as CD20+ B-cell clusters in the HGSC TME, which have been regarded as markers of TLS formation.10 We evaluated the presence of TLSs in the HGSC and found 50 of 185 specimens contained TLSs (27%) (figure 5A; online supplemental figure 6A), consistent with the previous study.22 To our surprise, there was no correlation between CXCL13 and TLSs (figure 5B), and TLSs showed no significant correlation with clinical outcomes in patients with HGSC (online supplemental figure 6B). Intriguingly, we observed a correlation between CXCL13 and prolonged OS exclusively in patients with HGSC who presented with TLSs, but not counterparts without TLSs (figure 5C). These data imply that CXCL13 may rely on the TLSs to exert antitumor function in HGSC. Moreover, immunofluorescence staining of two known TLS markers (CXCR5 and CXCL13), in combination with CD8 and CD20, supported the notion that CXCL13 promoted CXCR5+CD8+ T-cell recruitment to TLSs (figure 5D). Statistics confirmed the correlation between CXCL13 and CXCR5Hi/CD8Hi in patients with HGSC with TLSs (figure 5E). These data suggest that the combination of CXCL13 and TLSs probably represents an immune activation status associated with CXCR5+CD8+ T cells for patients with HGSC .
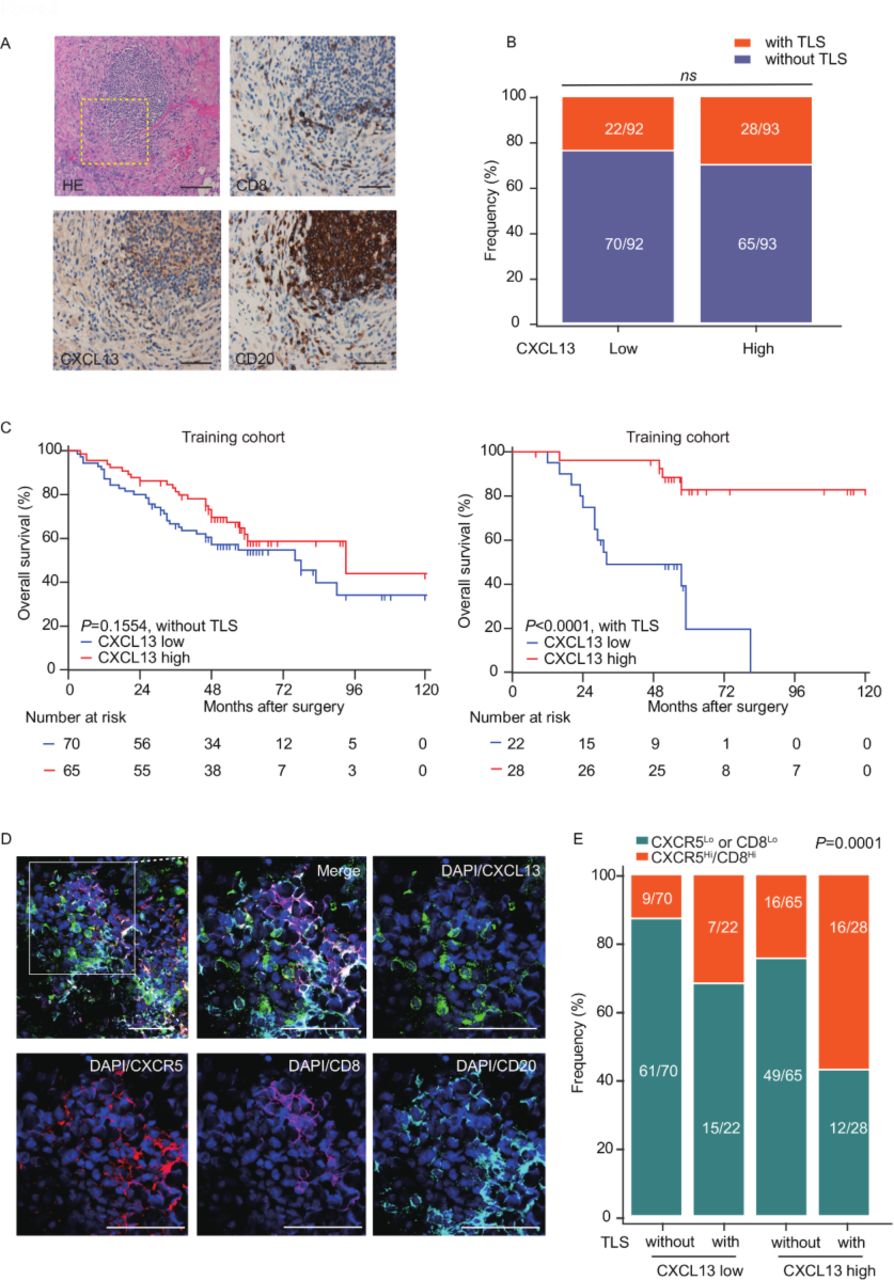
CXCL13 localizes in the context of TLSs and prolongs survival. (A) Representative images of immunostaining with H&E, CD20, CD8, and CXCL13 in HGSC. Bar=50 µM. (B) The frequency of patients with or without TLSs was compared according to CXCL13 expression (n=185, χ2 test). (C) Kaplan-Meier survival curves for OS of patients with HGSC from the training cohort without (n=135, left) or with TLSs (n=50, right) according to CXCL13 expression (log-rank test and p values are shown). (D) Representative images of immunofluorescence staining of HGSC. The sample was stained for CXCL13 (green), CXCR5 (red), CD8 (magenta), CD20 (cyan-blue) and DAPI (blue). Bar=50 µM. (E) The frequency of CXCR5Lo or CD8Lo and CXCR5Hi/CD8Hi patients from the training cohort was compared according to the TLS-CXCL13 group. (n=185; χ2 test and p value is shown). CXCL13, CXC-chemokine ligand 13; HGSC, high-grade serous ovarian cancer; OS, overall survival; DAPI, 4',6-Diamidino-2-Phenylindole, Dihydrochloride; TLSs, tertiary lymphatic structures.
CXCL13 enhances the response of anti-PD-1 therapy in subcutaneous ovarian cancer mouse model
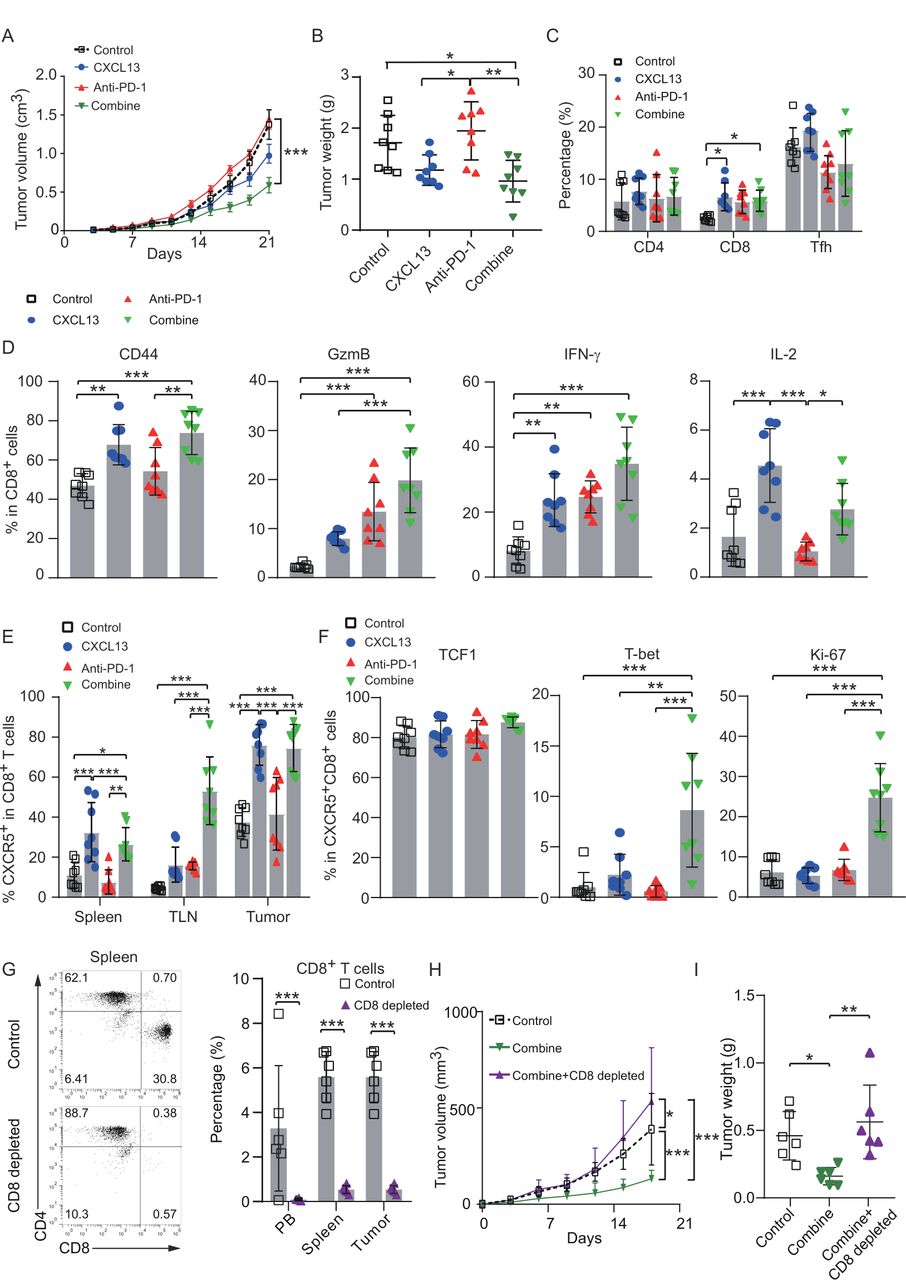
As CXCR5+CD8+ T cell was reported to be the main responding factor during immune checkpoint blockade,6 we explored the effect of CXCL13 on the response of checkpoint blockade therapy in vivo. Treatment of subcutaneous ovarian cancer mice with the combination of CXCL13 and anti-PD-1 significantly retarded tumor growth (figure 6A,B), which could probably derive from the increased CD8+ T-cell infiltration in mouse tumors (figure 6C; online supplemental figure 7A). The percentages of CD44, IFN-γ, and interleukin 2 (IL-2) in CD8+ T cells were significantly higher in the CXCL13 treatment group (figure 6D; online supplemental figure 7B). In the meanwhile, the combination treatment promoted the expression of CD44 and IL-2 compared with anti-PD-1 treatment alone (figure 6D; online supplemental figure 7B). Consistent with the results of in vitro experiments, CXCL13 alone or combined with anti-PD-1 increased the proportion of CXCR5+CD8+ T cells in the spleen, tumor-associated lymph nodes, and tumors (figure 6E; online supplemental figure 7C). The Ki-67 and T-bet expression in CXCR5+CD8+ T cells was significantly enhanced by combination treatment (figure 6F, online supplemental figure 7D). On combination treatment, the increased counts of CD8+ TILs and the expression of cleaved-caspase 3 were confirmed by IHC (online supplemental figure 7E). To verify the critical role of CD8+ T cells in the antitumor effects during the combination therapy, CD8+ T cells were eliminated by neutralizing antibodies (figure 6G). The antitumor effect of CXCL13 plus anti-PD1 therapy had been reversed by CD8+ T-cell depletion (figure 6H and I). These data suggest that CXCL13 may potentiate response of anti-PD-1 therapy through expansion and activation of CXCR5+CD8+ T cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CXCL13 enhances response of anti-PD-1 therapy in subcutaneous ovarian cancer mouse model. (A) Tumor growth curves of subcutaneous tumor model treated with CXCL13 or anti-PD-1 or combination of CXCL13 and anti-PD-1. Dotted lines indicate the time point at which tumor sizes were compared between control and treatment groups (n=8 per group). (B) Tumor weights of subcutaneous tumor among the indicated four groups (n=8 per group). (C) Percentage of CD8+ T cells, CD4+ T cells and Tfh in TILs from subcutaneous tumor among the indicated four groups (n=8 per group). (D) Percentage of effector cytokines (CD44, GzmB, IFN-γ, and IL-2) in CD8+ T cells from subcutaneous tumor among the indicated four groups (n=8 per group). (E) Percentage of CXCR5+CD8+ T cells from spleen, TLN, and subcutaneous tumor among the indicated four groups (n=8 per group). (F) Percentage of transcription factors (TCF1, T-bet) and Ki-67 in CXCR5+CD8+ T cells from subcutaneous tumor among the indicated four groups (n=8 per group). (G) Percentage of CD8+ T-cell percentage in mouse spleens from control and CD8+ T-cell-depleted groups (n=6 per group). (H) Tumor growth curves of subcutaneous tumor model treated by combination therapy with or without CD8+ T-cell depletion. Dotted lines indicate the time point at which tumor sizes were compared between control and treatment groups (n=6 per group). (I) Tumor weights of subcutaneous tumor model treated by combination therapy with or without CD8+ T-cell depletion (two-way ANOVA with Bonferroni post hoc test in (A, H); one-way ANOVA with Bonferroni post hoc test in (B–F) and (I); paired t-test in (G)). Bar plots show mean±SD; *p<0.05; **p<0.01; ***p<0.001; ns, no significant difference. ANOVA, analysis of variance; CXCL13, CXC-chemokine ligand 13; HGSC, high-grade serous ovarian cancer; IFN-γ, interferon-γ; IL-2, interleukin 2; OS, overall survival; PD-1, programmed cell death-1; Tfh, follicular helper T cells; TILs, tumor infiltrated lymphocytes; TLN, tumor-associated lymph nodes.
Discussion
This work provides new insights into the potential role of CXCL13 in the maintenance of CXCR5+CD8+ T cells that contribute to immune checkpoint blockade therapy in HGSC. Elevated CXCL13 expression in tumors suggested a more immunoactive microenvironment and tended to prolong patient’s survival. CXCL13 was located adjacent to CXCR5+CD8+ T cells in vivo and promoted recruitment and activation of CXCR5+CD8+ T cells in vitro. Combination of CXCL13, CD8, and CXCR5 was confirmed as an independent prognostic factor for OS and PFS in patients with HGSC. Combining CXCL13 with PD-1 blockade therapy enhanced antitumor response and retarded tumor growth, which was abolished on depletion of CD8+ T cells (online supplemental figure 8).
Recent studies reported that follicular dendritic cells and Tfh cells were major sources of CXCL13.13 23 In addition, CD8+ TIL was a potent producer of CXCL13 in human melanoma, non-small cell lung cancer, breast cancer, and hepatocellular carcinoma.24–27 In rheumatoid arthritis and ulcerative colitis, most cells that express CXCL13 were of the macrophage lineage,28 whereas in chronic obstructive pulmonary disease, they were B cells.29 Consistently, our results demonstrated that B cells and macrophages are the main source of CXCL13 in HGSC.
CXCL13 acts through its cognate receptor, CXCR5, to recruit lymphocytes to B-cell zones of secondary lymphoid tissues. Normally, mature B cells and a subset of memory T cells expressed CXCR5 and migrated in response to CXCL13.30 Our results revealed that CXCR5 in CD8+ T cells is strongly associated with prolonged survival. As CXCR5-expressing B cells were not associated with outcomes in the HGSC, this may interfere with the prognostic value of CXCR5 alone.
CXCL13 induced chemotaxis of B cells and upregulation of B-cell lymphotoxin-α1β2 to drive a positive feedback loop that is important in germinal center (GC) development and homeostasis.31 In addition, CXCL13 orchestrated B-cell clustering, lymphoid aggregation, and antibody production in TLSs.32 CXCL13-producing CXCR5+ Tfh cells were primarily located in TLSs and distinguished extensive immune infiltrates in breast cancer.13 CXCL13-producing CXCR5−CD4+ Tfh cells (TFHX13) potentially triggered TLS formation and generated GC B-cell responses at the breast cancer site.27
Three subsets of CXCR5+CD8+ T cells have been defined by immune context as progenitors of exhausted CD8+ T cells,5 regulatory CD8+ T cells,33 and follicular helper CD8+ T cells.34 In hepatitis B virus-associated hepatocellular carcinoma, CXCR5+CD8+ T cells could induce tumor cell death more effectively than CXCR5− counterparts.35 In pancreatic cancer, CXCR5+CD8+ T cells represented a promising subset of CD8+ T cells and could respond to anti-PD1/anti-TIM3 blockade.8 In CRC, CXCR5+CD8+ T cells potentially contributed to antitumor immunity and were highly activated in tumor-draining lymph nodes to achieve prognostic benefit.4 36 In our results, CXCR5+CD8+ T cells demonstrated higher IFN-γ and granzyme B expression and a more potent proliferation capability following CXCL13 exposure ex vivo. CXCL13 plus anti-PD1 blockade similarly promoted the proliferation of CXCR5+CD8+ T cells in tumor-bearing mice.
The differentiation of CXCR5+CD8+ T cells required the transcription factors TCF1, Bcl6, and E2A but was inhibited by the transcription regulators Blimp1, Id2, Id3, Irf4, Runx3, and STAT4.34 37–40 However, less is known about the specific cytokines that participate in the maintenance of these cells. Interleukin 12 (IL-12) downregulated the expression of TCF1 in CD8+ T cells via STAT4 signaling, suggesting IL-12 may suppress CXCR5+CD8+ T-cell differentiation.40 Cell-intrinsic type I interferon (IFN-I) signaling suppressed TCF1high progenitor-like CD8+ T-cell differentiation, but blockade of IFN-I receptor led to CXCR5+CD8+ T-cell expansion in an interleukin 27-dependent and STAT1-dependent manner.41 42 Our data confirmed the positive correlation between CXCL13 and CXCR5+CD8+ T cells. CXCL13 stimulated activation and expansion of CXCR5+CD8+ T cells in HGSC tumors ex vivo and promoted recruitment of CXCR5+CD8+ T cells in vitro. Still the detailed molecular mechanism underlying CXCL13-driven expansion of CXCR5+CD8+ T cells needs further investigation.
Compared with the circulating population, tumor-infiltrating CXCR5+CD8+ T cells expressed higher levels of PD-1 and TIM-3. On PD-1 and TIM-3 blockade, the function of CXCR5+CD8+ T cells was further improved.8 In muscle-invasive bladder cancer, CXCR5+CD8+ T cells expressed elevated PD-1, LAG-3, and CTLA-4 and reduced TIM-3 compared with CXCR5−CD8+ T-cell counterparts.43 TCF1+CD8+ T cells with PD-1 and LAG-3 expression were crucial for the T-cell expansion that occurred in response to inhibitory receptor blockade during chronic infection.37 Our results indicated that CXCR5+CD8+ T cells expressed lower TIM-3 expression compared with CXCR5−CD8+ T-cell counterparts. CXCR5+CD8+ T cells produced more effector molecules (such as GzmB and IFN-γ) on CXCL13 incubation in HGSC tissues, but CXCR5−CD8+ T cells remained unresponsive.
During HIV and Epstein-Barr virus infection, CXCR5+CD8+ T cells selectively entered B-cell follicles and eradicated the infected CD4+ Tfh cells and B cells.34 44 In rheumatoid synovitis, perifollicular CD8+ T cells facilitated the structural function of GC in ectopic lymphoid follicles.45 The association of B-cell-rich TLSs and survival and anti-PD1 immunotherapy response in soft-tissue sarcoma and melanoma has been recently established.9 11 In addition, co-occurrence of tumor-associated CD8+ T cells and CD20+ B cells is associated with improved survival in melanomas. Furthermore, B-cell-rich tumors were accompanied by increased numbers of TCF7+-naïve and/or memory T cells.10 Similarly, immunofluorescence staining of CXCL13, CXCR5, and CD8 in combination with CD20 displayed the formation of TLSs in HGSC. CXCR5+CD8+ T cells were adjacent to CXCL13 and B-cell follicles, suggesting a clue that CXCR5+CD8+ T cells may take part in the formation of lymphoid follicles. Our results indicate that the combination of CXCL13 and TLSs is associated with prolonged OS. CXCL13 potentiated PD-1 blockade therapy against tumors, implying its potential utility as a biomarker for stratification and response to immune checkpoint blockade therapy.
Our study has several limitations. First, the number of patients in the validation cohort was relatively small. Second, this study included patients with HGSC of all stages from three cohorts (training, validation, and TCGA), which resulted in a large but clinically heterogeneous retrospective study cohort. Third, CXCL13 staining is heterogeneous in some TMA cores, and the predominant pattern was taken into account for scoring. Still, the TMA method could provide meaningful information when used in a large cohort. Fourth, the molecular mechanisms by which CXCL13 induced maintenance of CXCR5+CD8+ T cells in HGSC need to be further explored. Considering the above-mentioned limitations, a multicenter prospective cohort study should be taken.
Conclusion
Our results demonstrate that CXCL13 confers antitumor effects via recruitment, activation, and expansion of CXCR5+CD8+ T cells, and its effect could be dependent on immune contexture shaping including TLSs. Combined CXCR5-CD8-CXCL13 IHC score represents a potential biomarker for stratification of clinical outcomes in patients with HGSC. Our findings also provide a perspective on CXCL13 promoting the response of HGSC to PD-1 blockade. Accordingly, CXCL13 and CXCR5+CD8+ T cells should be considered as a potential prognosticator or response biomarker for immune checkpoint blockade therapy.
Acknowledgments
We thank Dr Jing Zhao (Department of pathology, Obstetrics and Gynecology Hospital of Fudan University, Shanghai, China) for her excellent pathological technology help and Dr Hongbo Zhao (Bank of Tumor Resources, Obstetrics and Gynecology Hospital of Fudan University, Shanghai, China) for her help.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MY, JL and GZ contributed equally.
Contributors HL performed study concept and design; acquisition, analysis, and interpretation of data; and study supervision. CX obtained funding and study supervision. MY, JL, and GZ performed acquisition, analysis, and interpretation of data and statistical analysis. YW, MH, and QX provided technical and material support. All authors read and approved the final manuscript.
Funding This study was funded by grants from National Key Research and Development Program of China (2016YFC1303100), National Natural Science Foundation of China (82072881, 31570803 81773090), Natural Science Foundation of Shanghai (20ZR1409000), and Natural Science Foundation of Xinjiang Uygur Autonomous Region (2019D01C088).
Disclaimer All these study sponsors have no roles in the study design and in the collection, analysis, and interpretation of data.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All samples used in this study were anonymously coded in accordance with Helsinki Declaration. Written informed consent was obtained from each patient included in this study, and the protocol of all study cohorts was approved by the Ethical Board of the Institutional Review Board of the Gynecology and Obstetrics Hospital of Fudan University (Kyy2016-49, Kyy2017-27).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. TCGA-OV ovarian cancer mRNA expression data from 340 cases with clinical data were downloaded from GDC legacy archive (https://protal.gdc.cancer.gov/) in November 2017. All data generated that are relevant to the results presented in this article are included in this article.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.