Article Text

Abstract
Background Hematopoietic progenitor kinase 1 (HPK1 or MAP4K1) has been demonstrated as a negative intracellular immune checkpoint in mediating antitumor immunity in studies with HPK1 knockout and kinase dead mice. Pharmacological inhibition of HPK1 is desirable to investigate the role of HPK1 in human immune cells with therapeutic implications. However, a significant challenge remains to identify a small molecule inhibitor of HPK1 with sufficient potency, selectivity, and other drug-like properties suitable for proof-of-concept studies. In this report, we identified a novel, potent, and selective HPK1 small molecule kinase inhibitor, compound K (CompK). A series of studies were conducted to investigate the mechanism of action of CompK, aiming to understand its potential application in cancer immunotherapy.
Methods Human primary T cells and dendritic cells (DCs) were investigated with CompK treatment under conditions relevant to tumor microenvironment (TME). Syngeneic tumor models were used to assess the in vivo pharmacology of CompK followed by human tumor interrogation ex vivo.
Results CompK treatment demonstrated markedly enhanced human T-cell immune responses under immunosuppressive conditions relevant to the TME and an increased avidity of the T-cell receptor (TCR) to recognize viral and tumor-associated antigens (TAAs) in significant synergy with anti-PD1. Animal model studies, including 1956 sarcoma and MC38 syngeneic models, revealed improved immune responses and superb antitumor efficacy in combination of CompK with anti-PD-1. An elevated immune response induced by CompK was observed with fresh tumor samples from multiple patients with colorectal carcinoma, suggesting a mechanistic translation from mouse model to human disease.
Conclusion CompK treatment significantly improved human T-cell functions, with enhanced TCR avidity to recognize TAAs and tumor cytolytic activity by CD8+ T cells. Additional benefits include DC maturation and priming facilitation in tumor draining lymph node. CompK represents a novel pharmacological agent to address cancer treatment resistance.
- adaptive Immunity
- CD8-positive T-Lymphocytes
- dendritic cells
- lymphocytes
- tumor-Infiltrating
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- adaptive Immunity
- CD8-positive T-Lymphocytes
- dendritic cells
- lymphocytes
- tumor-Infiltrating
- tumor microenvironment
Introduction
Cancer immunotherapy has become one of the major pillars of cancer care, complementing surgery, chemotherapy, targeted and radiation therapies. Immune checkpoint inhibitors (CPIs) that target the PD-1 and CTLA-4 pathways have transformed therapeutic outcomes across various tumor types via the revitalization of exhausted cytotoxic T cells (CTLs). Immunotherapy promises to be the most impactful form of treatment for patients whose tumors have already metastasized.1 Despite the encouraging success of CPI, roughly 60%–70% of tumors are unresponsive to single-agent CPI therapy,2 whereas those that do respond can acquire resistance over time. Significant challenges remain to identify effective ways to unleash the immune system to fight cancer and to overcome the diverse array of immune-evasion mechanisms. Multiple inhibitory feedback mechanisms have a role in suppressing T cells in the tumor microenvironment (TME), which diminish the activities of CTLs against tumor cells through the recruitment of immunoregulatory cells and induction of inhibitory signals to hamper T-cell infiltration, function, expansion, and survival. The presence of suppressive immune cell populations and associated immunosuppressive factors in the TME, including PGE2, adenosine, and transforming growth factor beta (TGF-β), represents a major source of treatment resistance whereby normal immunoregulatory mechanisms are hijacked by tumor cells.3 Hence, there is an urgent need to develop novel therapeutic agents that act in synergy with existing CPI. Hematopoietic progenitor kinase 1 (HPK1) is of particular interest as it has been implicated in several important steps that are thought to limit T-cell responsiveness, particularly in cancer. HPK1 is predominantly expressed in hematopoietic cell linages with high expression observed in T cells, B cells, and dendritic cells (DCs), and low expression in monocytes/macrophages (human protein atlas). HPK1 was shown to serve as a negative regulator in T cells, B cells and DCs.4–9 The signal transduction pathway of HPK1 was mostly studied and best understood in T cells. HPK1 is essential in negatively regulating T-cell activation with involvement of the linker of activated T cells (LAT) and associated downstream signaling molecules, including adaptor protein Src homology 2 domain containing leukocyte protein of 76 kDa (SLP76), phospholipase Cγ1 and extracellular signal-regulated kinase signaling pathway.4 Previous work has shown that knockout (KO) of germline HPK1 decreased the threshold for T-cell receptor (TCR) signaling and rendered T cells resistant to the suppressive effects of PGE2.4 5 Kinase activity is critical in mediating the negative regulatory function of HPK1 as revealed by studies from genetically engineered mice containing catalytically inactive HPK1 (kinase dead (KD)).6 7 HPK1 KD mice exhibited normal bone marrow development and immune cell homeostasis.7 No overt autoimmunity was associated with HPK1 KO or KD, in contrast to the lethal inflammation associated with genetic deletions of other negative regulators such as CTLA410 11 or Cbl-b.12–15 Intriguingly, despite the lack of autoimmunity findings, the HPK1 KO or KD animals showed improved immune response as demonstrated by significant beneficial effects in combination with blockade of PD1 pathway in both antiviral and antitumor immunity,6 7 suggesting that HPK1 inhibition with a pharmacological agent may offer a superior balance of efficacy and safety at tolerated doses. However, it remains elusive if observations from the engineered mouse models were the result of modification of immune system during the developmental stage, and the role of HPK1 in human immune cells is still to be investigated. To understand the potential HPK1 as a valid target for therapeutic intervention, it is critical to assess HPK1 biology in matured immune systems and the translatability of antitumor immunity from mice to humans. The development of a small molecule HPK1 inhibitor is the optimal solution to elucidate the role of HPK1 in human disease with potential clinical application. Efforts to identify a potent and selective HPK1 inhibitor suitable for proof-of-concept studies have been made more difficult owing to the diversity of function and high sequence homology among MAP4K family members. Herein, we report the identification and characterization of a novel, potent and selective HPK1 small molecule inhibitor with drug-like pharmaceutical properties, compound K (CompK). Human primary T cells were exploited to investigate the effect of CompK on T-cell functions under a variety of settings. We observed improved functions of T cells in the context of immunosuppressive factors, increased TCR sensitivity against viral and tumor antigens, and enhanced antigen-specific T-cell response in synergy with anti-PD1, with additional benefit on DC maturation. Substantial antitumor efficacy with CompK, in combination with anti-PD1, was demonstrated in a mouse syngeneic tumor model, which for the first time shows that enhanced immune response by HPK1 ablation is not a result of developmental alteration and can be recapitulated by pharmacological inhibition of HPK1 in the context of a fully matured immune system. In addition, augmented immune responses by CompK were observed in patient tumor-explant studies, providing mechanistic translation which supports our findings in the animal model.
Materials and methods
Reagents
Prostaglandin E2 (Santa Cruz, Dallas, Texas, USA), 5′-(N-ethylcarboxamido) adenosine (NECA) (Sigma, St. Louis, Missouri, USA), TGF-β (Peprotech, Rocky Hill, New Jersey, USA), AlphaLISA kits (PerkinElmer, Waltham, Massachusetts, USA), isolation kit for CD3+ T cells (Stemcell Biotechnologies, Cambridge, Massachusetts, USA), X-linker/goat anti-mouse IgG (Pierce, Appleton, Wisconsin, USA), dimethyl sulfoxide (DMSO) (Sigma), anti- human CD8 FITC (eBiosciences, San Diego, California, USA), anti-human CD4-PE (BD Biosciences, Franklin Lakes, New Jersey, USA), pSLP76 Alexa 647 (Cell Signaling Technology, Danvers, Massachusetts, USA), 5X Lyse/Fix buffer (BD Biosciences), Perm III Buffer (BD Biosciences), messenger RNA (mRNA) catcher purification kit (Thermo Fisher, Waltham, Massachusetts, USA), SuperScript IV VILO master mix (Invitrogen, Carlsbad, California, USA), POWER SYBR Green PCR Master Mix (Invitrogen), interferon gamma (IFN-γ) and interleukin (IL)-2 KiCq Start SYBR Green primers (Sigma), T2 cells (ATCC, Manassas, Virginia, USA), anti-human papillomavirus (HPV) T cells (Astarte Biologics, Bothell, Washington, USA), HPV E7 (11–20) peptide (Astarte Biologics), CMV pp65 peptide, A2 restricted (Astarte Biologics), recombinant soluble dimeric human HLA-A2:Ig fusion protein (BD Biosciences), IL-7 and IL-15 (Peprotech), One-Go EX reagent (Promega, Fitchburg, Wisconsin, USA), tumor necrosis factor alpha (TNF-α) (BUV395) and CD11c (PE-DZ) (BioLegend, San Diego, California, USA), and PepMix human (HLA class I Ig-like C1 domain and PepMix CEFT Pool (JPT Peptide Technologies, Berlin, Germany).
Animals
C57BL/6 (Charles River, Wilmington, Massachusetts, USA) and Balb/c mice (Envigo, Indianapolis, Indiana, USA) were maintained at 70–74°F and 40%–60% relative humidity, with a 12:12-hour light:dark cycle. Animals were fed Teklad Global Diets 2918 (Envigo, Madison, Wisconsin, USA) ad libitum and were housed on ALPHA-dri (Shepard Specialty Papers, Milford, New Jersey) bedding in ventilated caging. All animal procedures were approved by the Bristol Myers Squibb (BMS) Institutional Animal Care and Use Committee. The animal care and use program at BMS is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Biochemical assay to evaluate HPK1 inhibitors
The kinase assay carried out in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 10 mM MgCl2, 0.015% Brij-15 and 2 mM Dithiothreitol (DTT) containing 0.625 nM recombinant HPK1 (SingalChem M23-11G), 3 µM fluorescently labeled peptide substrate (NH2-fluorescein-RFARKGSLRQKNV-COOH), 22 µM ATP, and various concentrations of HPK1 inhibitor. The reactions were incubated for 3 hours and quenched with 1 mM EDTA solution, followed by capillary electrophoresis on a Caliper LabChip EZ Reader (PerkinElmer) to determine peptide substrate conversion and IC50 values.
Human T-cell assays under immunosuppressive conditions
Human CD3 +T cells were treated for 24 hours with 2 nM PGE2 and/or 2 µM NECA in the presence of anti-CD3 and anti-CD28. CompK was then added followed by incubation for additional 48 hours. IL-2 and IFN-γ secretion was analyzed by AlphaLISA. For proliferation studies, Human CD3 +T cells were treated with 3 nM TGF-β in the presence/absence of CompK under stimulation with anti-CD3 and anti-CD28 for 72 hours. 3H Thymidine was added 6 hour prior to harvest, followed by the measurement of incorporation of 3H Thymidine.
Antigen-specific T-cell studies
Peripheral blood mononuclear cells (PBMCs) from healthy donors were stimulated in vitro with peptide pools encoding for cytomegalovirus, Epstein-Barr virus, influenza and tetanus toxin (CFET) or the negative control peptide at 0.1 µg each peptide/mL, with fresh media containing IL-7 and IL-15 at final 5 ng/mL added on day 4 followed by an additional 3D culture. Antigen-presenting cells (APCs) from the same donor were pulsed with control or CEFT peptides at final 0.1 µg each peptide/mL followed by coculturing with the primed PBMC at 1:1 ratio in the presence of peptides, CompK, and/or anti-PD1 for 24 hours. Supernatants were collected to measure IFN-γ using AlphaLISA.
For HPV-E7-specific CD8+ T-cell studies, different concentrations of E7 or control peptide were added to T2 cells, followed by addition of HPV specific CD8+ T cells, along with 0.5 µM CompK. The coculture was incubated for 72 hours. The supernatants were assessed for IFN-γ by AlphaLISA.
NY-ESO tumor associated antigen T cell receptor (NY-ESO-TCR) or green fluorescent protein (GFP) overexpressing primary T cells were generated by transducing the activated cells with lentiviral vectors, encoding either the 1G4 TCR-recognizing NY-ESO-1-derived Major histocompatibility complex I (MHC-I)-specific antigenic peptide SLLMWITQC16 17 or GFP. The transduced cells were expanded and expression of the recombinant TCR or GFP were quantified by flow cytometry. The engineered T cells were then cocultured with the SAOS2 sarcoma cells, engineered to constitutively express a luciferase reporter gene and with endogenous expression of NY-ESO-1 protein and HLA-A2 surface molecules, for 72 hours in the presence or absence of CompK. Surviving tumor cells were quantified through their luminescence signal using the One-Go EX reagent and reading samples in an Envision multimode reader (PerkinElmer). The percentage of lysed tumor cells was calculated through the following formula, in which the low control corresponds to wells containing tumor cells only (no T cell-mediated killing), and the high control equates to wells containing only T cells (no luminescence signal):  .
.
Measurement of pSLP76 and IFN-γ in human whole blood
Heparinized blood was pretreated with either DMSO or CompK for 60 min. Ten microgram/millilitre10 µg/mL of anti-CD3/anti-CD28 plus X-linker was used to stimulate the samples for 15 min, followed by treatment with BD lysis/fix buffer. The cells were washed and stained for surface markers. All samples were then stained with pSLP76 antibody after being washed and permeabilized with BD Perm Buffer III, followed by data acquisition with fluorescence activated cell sorting (FACS) Canto I with DIVA V.6.1.1 software. For IFN-γ measurement, heparinized human whole blood was pretreated with various concentrations of CompK and then stimulated with 0.5 µg/mL of anti-CD3/anti-CD28 plus X-linker for 3 hours. At the end of the treatment, mRNA was extracted and complementary DNA was prepared. Quantitative PCR (qPCR) was performed with SYBR standard protocol on ABI 7900 sequence detector followed by calculation for ∆∆CT.
Evaluation of CompK dose required to elicit in vivo response
C57BL/6 mice bearing 1956 sarcoma at ~100 mm3 were used in this study. CompK, formulated as a solution in 80% polyethylene glycol 400, 10% tocopheryl polyethylene glycol succinate, and 10% ethanol, was administered at 30 mg/kg body weight (mpk) and 100 mpk two times per day for 5 days and 10 µL dried blood spots were obtained from the tail vein at 1, 3, and 7 hours postdose on days 1 and 4, and prior to the morning dose on days 2 and 5. Drug exposure was quantified by liquid chromatography with tandem mass spectrometry. The blood levels of CompK were determined by non-compartmental analysis of plasma concentration versus time data (Phoenix WinNonlin software V.6.4; Certara, Princeton, New Jersey). The peak concentration, Cmax, and the time for Cmax, Tmax, were recorded directly from experimental observations. At the end of the study, blood pSLP76 was measured using flow cytometry. IFN-γ, CD69 and CD25 were evaluated using qPCR.
Evaluation of CompK in MC38 syngeneic tumor model
Mice were given a subcutaneous injection of MC38 cells at 1×107/mL into the right flank on day 0. CompK was dosed at 100 mpk two times per day right after implanting tumor cells. Anti-PD1 was dosed when tumor volume reached 100 mm3 (days 7, 10, and 13 post-tumor cell implantation). Tumor volumes were measured using Vernier scale calipers and were calculated using the formula 1/2(length×width2). Groups were weighed and measured two times per week until individual tumors or groups reached 1000 mm3.
Human tumors
Fresh human colorectal carcinoma (CRC) tissues were obtained from the Cooperative Human Tissue Network. Tumor specimens were collected with patient consent and shipped immediately after surgery at 4°C for next day delivery in the tubes containing fresh media (RPMI-1640, 10% fetal bovine serum (FBS), 100 µg/mL penicillin and 100 µg/mL streptomycin).
Mouse and human tumor explant studies
Fresh tumor was sectioned into 0.5–1.0 mm3 pieces followed by randomization of explants via mixing. The explants were laid to the plate and incubated in a tissue culture incubator for 3 hours to allow the firm attachment of the explants to the tissue culture plate, using the extracellular matrix (ECM) derived from the tumor. Growth media (100 µL) containing Roswell Park Memorial Institute (RPMI) with glutamax, 10% FBS, 1× penicillin and streptomycin, 25 mM HEPES, 50 µg/mL gentamycin, and 55 µM 2-mercaptoethanol were then added to each well of the 96-well plate, and the explants were cultured in the incubator for 72 hours under 37°C and 5% CO2.
Quantitative PCR
The studies were conducted with Thermo Fisher ViiA 7 Real-Time PCR System with 384-Well Block. The primers (vendor preferred choice) were used in these studies (Sigma).
Luminex analysis
The supernatants from the studies were collected after treatment and analyzed using protocol described by the manufacturer (Luminex Corporation, Austin, Texas, USA).
Statistical analysis
Data were plotted as mean±SEM. Student t-test was carried out using GraphPad Prism (GraphPad Software, La Jolla, California, USA) and statistical significance was considered meaningful at p<0.05.
Results
Discovery of HPK1 small molecule inhibitor, CompK
To identify small molecule HPK1 kinase inhibitors, we conducted a high-throughput screen with a biochemical assay to determine compound inhibition of HPK1. Extensive lead optimization efforts led to the identification of CompK (figure 1A). Selectivity was assessed in a panel of over 300 kinase assays and a panel of diverse non-kinase targets. CompK was found to be highly selective for HPK1, demonstrating over 50-fold selectivity against other members of the MAP4K family (online supplemental table S2). The general kinase selectivity of CompK was confirmed in the kinome analysis (online supplemental figure S1 and table S1). The selectivity was further evaluated in Jurkat cells with HPK1 KO using CRISPR (clustered regularly interspaced short palindromic repeats) (online supplemental figure S2A). Significantly higher IL-2 production was observed in HPK1 KO compared with wild-type (WT) Jurkat cells (online supplemental figure S2B). Compound B, a less optimized HPK1 inhibitor with related structure and selectivity profile to CompK, was evaluated in HPK1 WT and KO Jurkat cells. Compound B treatment only increased IL-2 production in WT Jurkat cells without effect on HPK1 KO Jurkat cell (online supplemental figure S2C), indicating that compound-induced IL-2 release was due to specific HPK1 inhibition rather than off-target activity. To investigate the effect of HPK1 inhibition on the function of human primary T cells, CompK was evaluated for IFN-γ secretion with human CD8+ T cells. The compound enhanced IFN-γ secretion in a concentration-dependent manner (figure 1B). To determine the compound activities in a more physiologically relevant setting, human whole blood was exploited to evaluate the influence of CompK on IFN-γ production and phosphorylation of serine 376 of SLP76, a proximal downstream target of HPK1. In a concentration-dependent fashion, CompK increased IFN-γ production and reduced the phosphorylation of SLP76 (pSLP76) as revealed by qPCR and flow cytometry analysis, respectively (figure 1C,D). The pSLP76 IC50 was calculated around 5 µM in human whole blood, consistent with the EC50 generated for IFN-γ production under the whole blood setting. Enhanced IL-2 and IFN-γ induced by treatment with CompK was also observed in whole blood from patients with pancreatic cancer, similar to that seen in healthy human subjects (online supplemental figure S3).
Supplemental material
Supplemental material
Supplemental material
Supplemental material
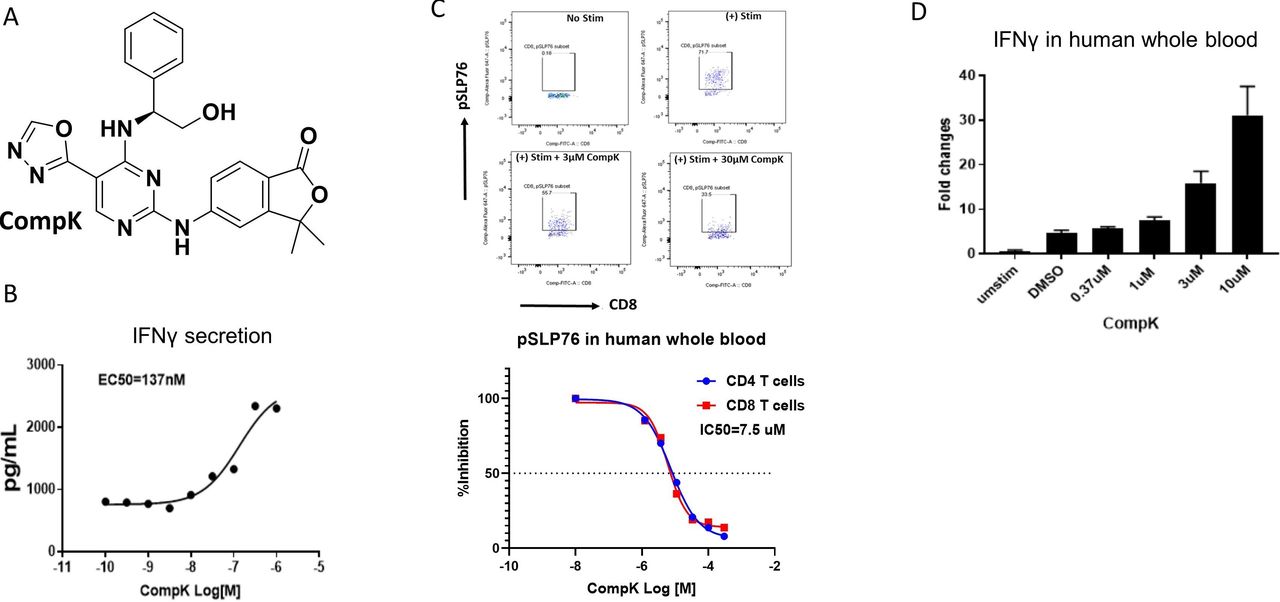
Identification and characterization of selective HPK1 inhibitor, CompK. (A) Chemical structure of CompK. (B) Enhanced IFN-γ production in human CD8+ T cells by CompK. Cells were stimulated by anti-CD3 and anti-CD28 for 48 hours in the presence of different concentrations of CompK followed by measurement of IFN-γ release. (C) Reduction of pSLP76 in CD4+ and CD8+ T cells by CompK in human whole blood. Heparinized fresh human whole blood was treated with CompK for 60 min followed by stimulation with anti-CD3 and anti-CD28 for 15 min. Cells were stained and pSLP76 was measured by flow cytometry. (D) Increased production of IFN-γ by CompK treatment using human whole blood. Human whole blood was stimulated with anti-CD3 and anti-CD28 in the presence of different concentrations of CompK for 3 hours. Messenger RNA and complementary DNA were generated followed by quantitative PCR analysis. Studies were conducted with multiple donors; representative graphs are shown here. CompK, compound K; DMSO, dimethyl sulfoxide; IFN-γ, interferon gamma.
CompK reversed the inhibitory activities by immunosuppressive factors
Immunosuppressive factors, such as PGE2, adenosine and TGF-β, exhibit potent suppressive activities on T cells in the TME. To interrogate the potential benefit of a HPK1 inhibitor in attenuating the influence of these suppressive factors, secretion of IL-2 and IFN-γ was evaluated in human CD3+ T cells in the presence of PGE2 and a stable adenosine analog (NECA) on treatment with CompK. Reduced production of IL-2 and IFN-γ was observed in the presence of PGE2 and NECA. Strikingly, CompK treatment fully reversed the suppressive activities of the combined factors (figure 2A,B), similar results were also demonstrated with single-agent treatment of PGE2 or NECA (online supplemental figure S4A–D). Moreover, TGF-β showed attenuated T-cell proliferation, whereas CompK treatment led to diminished TGF-β-induced inhibitory effect (figure 2C). These findings support the potential benefit of CompK in restoring T-cell polyfunctionality under immunosuppressive conditions relevant to the TME.
Supplemental material

Improved function of T cells by CompK under immunosuppressive conditions. (A) Recovery of IL-2 release from human primary T cells by CompK in the presence of PGE2 and NECA. Purified human T cells were prereated with PGE2 and NECA for 24 hours in the presence of anti-CD3 and anti-CD28. CompK was then added to the cell culture for an additional 48 hours followed by measurement of IL-2 production. (B) Reinvigoration of IFN-γ production by CompK under suppressive conditions created by PGE2 and NECA. The treatment conditions were the same as IL-2 evaluation. (C) Attenuation of TGF-β inhibition on T-cell proliferation by CompK. Purified human T cells were treated with 3 ng/mL TGF-β for 72 hours in the presence of 0.5 µM CompK. The cells were pulsed with 3H-labeled thymidine 6 hours prior to harvest. All studies were repeated with T cells from multiple donors. The representative graphs are shown here. Student t-test was used for statistical analysis. *P≤0.05, **P<0.01, ***P<0.001, ****P<0.0001. CompK, compound K; DMSO, dimethyl sulfoxide; IFN-γ, interferon gamma; IL, interleukin; NECA, 5′-(N-ethylcarboxamido) adenosine; TGF-β, transforming growth factor beta.
CompK improved the functions of antigen-specific human T cells
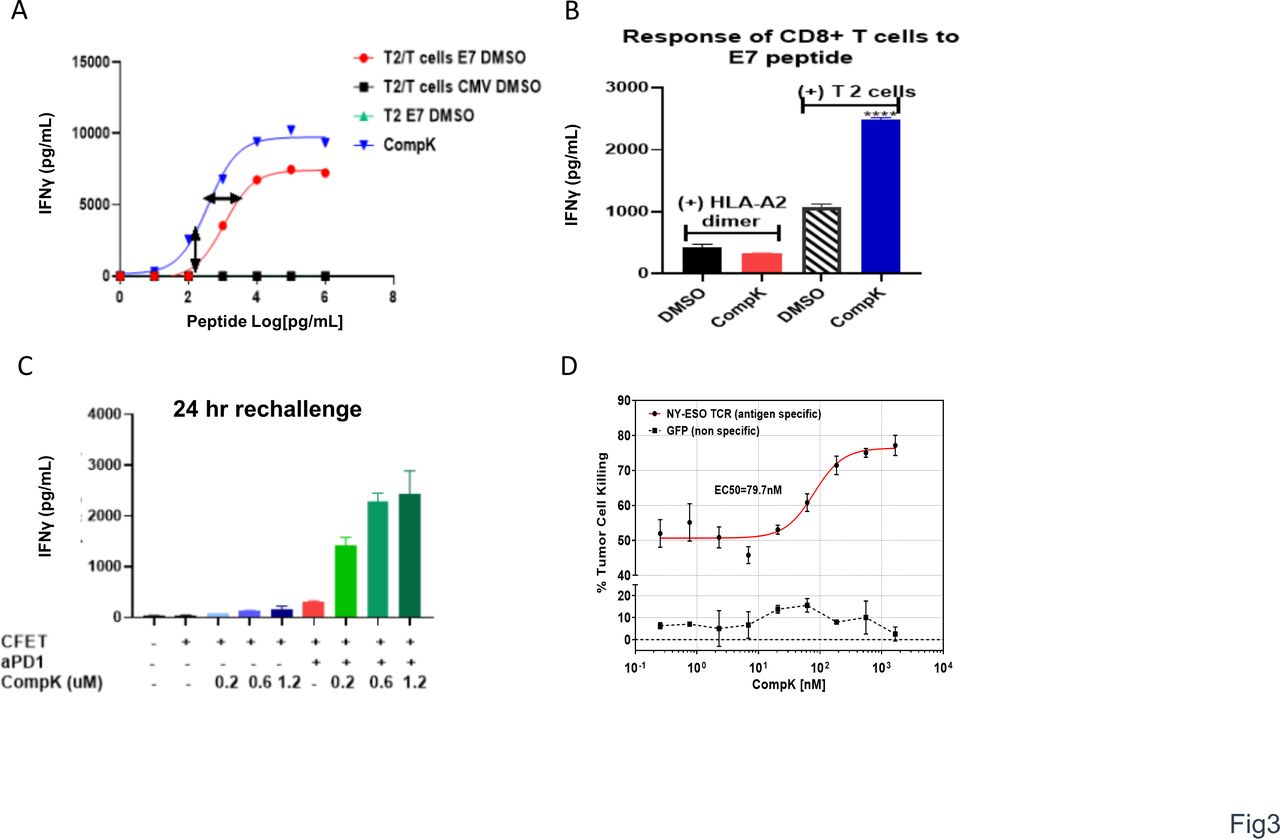
With the enhancement of T-cell polyfunctionality observed on treatment with CompK via engagement of TCR with anti-CD3 and anti-CD28, we next decided to investigate the impact of CompK on T-cell response to a specific antigen at various doses aiming to understand whether HPK1 inhibition leads to augmented functional avidity of T cells.18 We hypothesized that inhibition of HPK1 kinase activity could reduce the TCR activation threshold and increase antigen sensitivity of TCR to recognize lower concentrations of antigens. By using HLA-A2-restricted T2 cells as the antigen-presenting cells with no antigen processing capability, MHC-I is stabilized on T2 cell surface by the presented cognate peptide in a concentration-dependent manner. The strength of TCR-pMHC interaction can then be regulated by the concentration of the cognate peptide. HPV E7 epitope, a tumor-associated antigen (TAA) that is present in a significant proportion of head and neck as well as cervical cancers,19 was used in this study. E7-specific CD8+ T cells were cocultured with T2 cells at varying concentrations of E7 peptide. The secretion of IFN-γ by CD8+ T cells was enhanced in an E7 dose-dependent manner. Treatment with 0.5 µM CompK led to a threefold leftward shift of the E7 EC50 response curve (figure 3A), suggesting that CompK reduced the threshold required to trigger TCR responses of antigen-specific CD8+ T cells, and increased their ability to recognize weak viral/tumor associated antigens. To investigate the necessity of interaction between T cells and APCs, T cells were stimulated with HLA-A2 dimer or T2 cells in the presence of E7 peptide. Enhanced IFN-γ production by CompK was only observed in the presence of T2 cells (figure 3B), suggesting that cell–cell interplay is required. The expression of negative regulators, such as PD-1, increases on TCR triggering.20 We therefore reasoned that HPK1 expression is likely upregulated on T-cell activation. To confirm this, we stimulated human T cells with various concentrations of anti-CD3 and anti-CD28. HPK1 expression was enhanced on stimulation in effector and memory CD8+ T cells (online supplemental figure S5), implying that HPK1 may play a more active role with its increased expression levels in the context of inflammatory environments such as tumor-draining lymph node (TDLN) and TME. To reduce the negative regulatory function of HPK1 and PD1 in activated CD8+ T cells, the treatment effect may be magnified by combining a HPK1 inhibitor and anti-PD1. We therefore investigated the combined effect of CompK and anti-PD1. The study was conducted to assess the impact of treatment on T-cell responses specific to CFET antigens. T cells were cocultured with antigen-pulsed, irradiated APCs, along with CompK, anti-PD1 or combination. Treatment with CompK or anti-PD1 as a single agent increased IFN-γ release, whereas the combination of CompK and anti-PD1 yielded remarkable synergy with robust IFN-γ secretion (figure 3C), suggesting the combined utility of a HPK1 inhibitor and anti-PD1 in the clinic. To further demonstrate improved function of CD8+ T cells in the context of tumor antigen on HPK1 inhibition, we conducted a study with T cells transduced with TCR specific to NY-ESO-1, a cancer testis antigen. SAOS2 sarcoma cells were used as the target cells with endogenous expression of HLA-A2 restricted NY-ESO-1. CompK, in a concentration-dependent manner, markedly enhanced NY-ESO-1-specific tumor lytic activity of the engineered T cells (figure 3D). Collectively, our results revealed that HPK1 inhibition by CompK led to improved function of CD8+ T cells against specific viral and tumor antigens, reduced TCR activation threshold with increased sensitivity to antigens, as well as combination benefit with anti-PD1.
Supplemental material

Reduced TCR activation threshold and enhanced antigen-specific T-cell response by CompK. (A) Decreased threshold of TCR activation by viral antigen via CompK treatment. CD8+ T cells specific to HPV E7 antigen were cocultured with E7 or control peptide pulsed T2 cells for 18 hours in the presence of different concentrations of E7 peptides and 0.5 µM CompK; IFN-γ release was measured at the end of the study. (B) Required presence of APCs (T2 cells) in increased IFN-γ production by CompK. HPV E7 peptide was presented with either HLA-A2 dimer or T2 cells to stimulate E7-specific CD8+ T cells for 72 hours followed by measurement of IFN-γ release. (C) Significant synergy of IFN-γ production by CFET-specific T cells by combination of CompK and anti-PD1. CFET-specific T cells were cocultured with CFET pulsed APCs in the presence of different concentrations of CompK, along with 5 µg/mL anti-PD1 for 24 hours, followed by measurement of IFN-γ secretion. (D) Augmented tumor lysis by tumor antigen-specific T cells. Study was conducted by coculturing T cells with engineered TCR specific to NY-ESO-1 and sarcoma cells with endogenous expression of HLA-A2 restricted NY-ESO-1. IFN-γ secretion was analyzed at the end of the study. All studies were repeated three times and representative graphs are shown here. APCs, antigen-presenting cell; CFET, cytomegalovirus, Epstein-Barr virus, influenza and tetanus toxin; CompK, compound K; DMSO, dimethyl sulfoxide; HPV, human papillomavirus; IFN-γ, interferon gamma; TCR, T-cell receptor; aPD1, anti PD1.
CompK treatment promoted maturation of human DCs
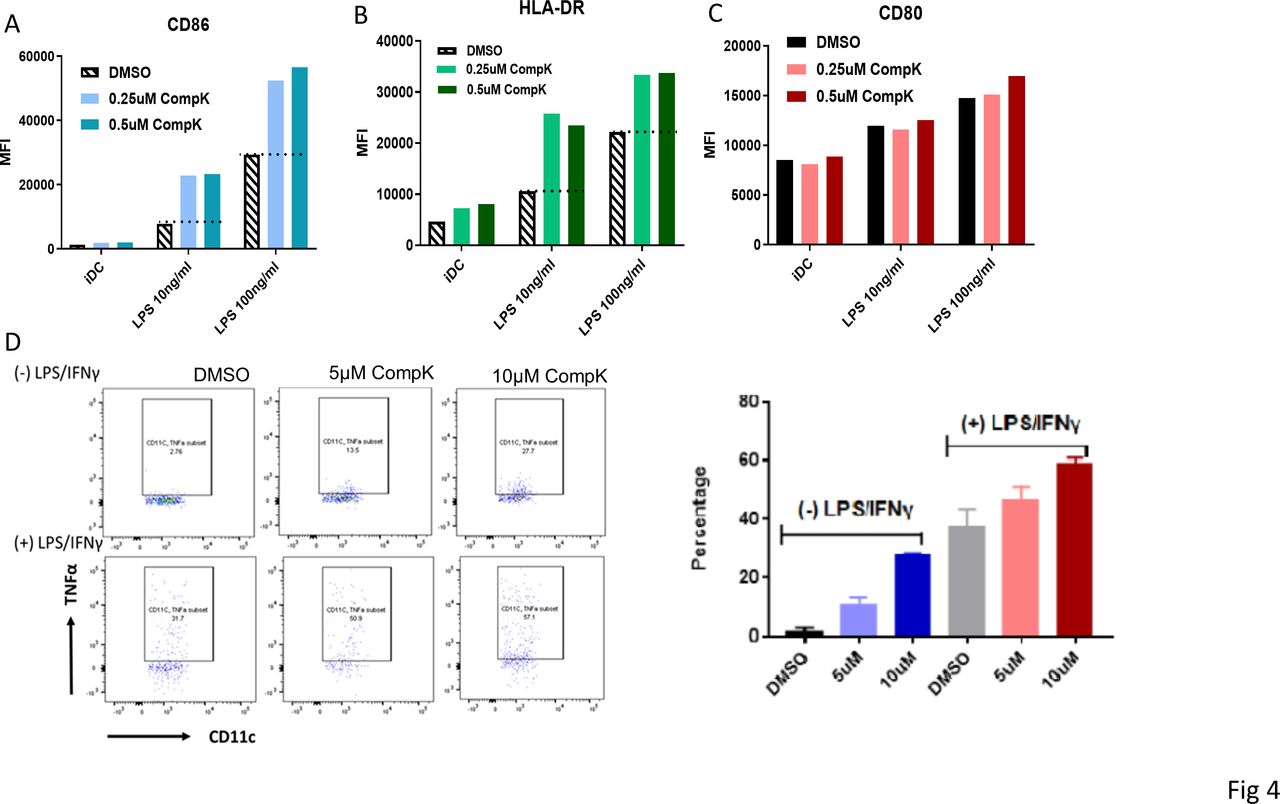
Improved maturation was observed with bone marrow-derived DCs from HPK1 KO mice on stimulation, and these DCs were associated with enhanced antigen-presenting ability to elicit increased antitumor immunity.8 To investigate if HPK1 kinase inhibition can result in similar benefit to promote human DC maturation, we elected to investigate the treatment effect of CompK by using human monocyte-derived DCs and DCs from peripheral blood. The expression levels of CD86 and Human Leukocyte Antigen- DR isotype (HLA-DR), the costimulator and antigen-presenting complex, were increased on maturation by lipopolysaccharides (LPS). CompK treatment led to a marked enhancement of both CD86 and HLA-DR compared with LPS alone, with minimal effect on CD80 (figure 4A–C). Further studies were conducted to assess the effect of CompK on endogenous DC from peripheral blood. Human whole blood was stimulated with LPS and IFN-γ in the presence and absence of CompK for 18 hours. CompK treatment resulted in augmented TNF-α+DC population in blood (figure 4D). Higher concentrations of CompK were exploited in the whole blood studies to compensate for the protein-binding properties of the compound in this setting. Our results indicate that HPK1 kinase inhibition is beneficial to promote cancer immunity via modulation of human DCs and associated network.

Enhanced maturation of human DC by CompK. (A–C) Improved maturation of monocyte-derived DC by CompK. iDCs were generated from purified human monocytes in the presence of GM-CSF and IL-4. IDCs were then matured by adding LPS in the presence of CompK. the cell surface markers were analyzed by FACS after maturation. D. increased TNF-α+DC population after stimulation on treatment with CompK. human whole blood was stimulated with 10 ng/mL LPS plus 100 ng/mL IFN-γ in the presence or absence of CompK for 18 hours. TNF-α+DC population was analyzed by FACS. CompK, compound K; DC, dendritic cell; DMSO, dimethyl sulfoxide; GM-CSF, granulocyte-macrophage colony-stimulating factor; iDC, immature dendritic cell; IFN-γ, interferon gamma; TNF-α, tumor necrosis factor alpha.
Investigation of CompK dose required in vivo response using sarcoma-bearing mice
To interrogate the CompK dose required to elicit significant in vivo responses, a study was conducted with mice bearing 1956 sarcoma. Vehicle, 30 or 100 mpk (two times per day) doses of CompK were administered to the mice for five consecutive days. At the end of treatment, blood samples were collected to measure pSLP76, IFN-γ, CD25 and CD69 after stimulation with anti-CD3 and anti-CD28 ex vivo. CompK treatment reduced pSLP76 levels and increased the expression of IFN-γ, CD25 and CD69 (figure 5A–D). The IC50 of CompK to inhibit pSLP76 production in mouse whole blood is ~6 µM (figure 5E). Based on the compound levels measured over time, CompK covered the whole blood IC50 for approximately 6 hours at the 100 mpk dose with two times per day administration (figure 5E,F), which was associated with increased in vivo pharmacological response as revealed by the measurement of pSLP76, IFN-γ, CD25 and CD69 relative to the 30 mpk dose. Our data support that CompK treatment can enhance immune response in vivo under tumor-bearing conditions.
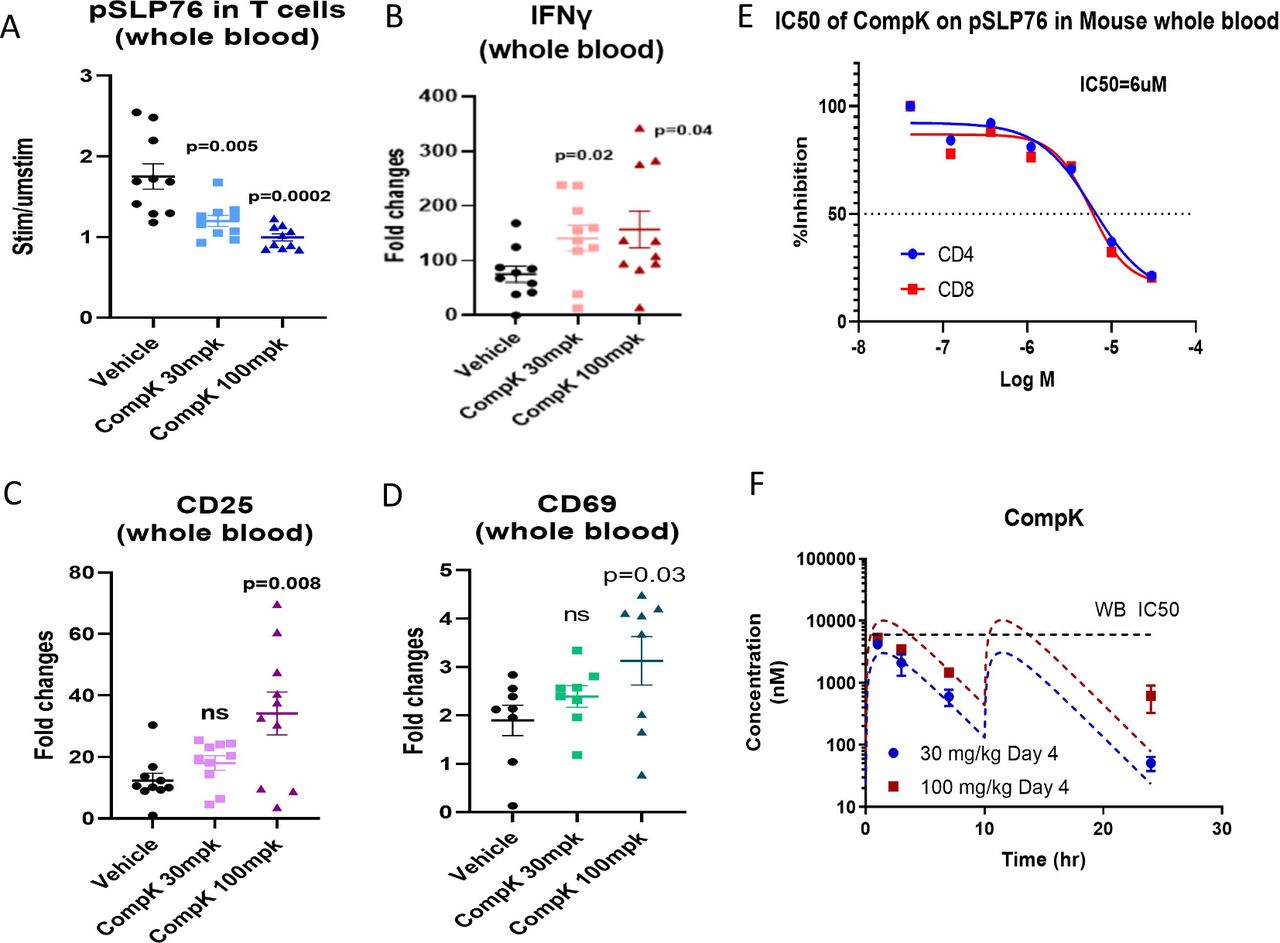
Improved immune response by CompK in dose finding study with tumor-bearing mice. (A) Inhibition of SLP76 phosphorylation by CompK. Compound was administrated to mice bearing 1956 sarcoma at 30 mpk and 100 mpk doses, two times per day for 5 days. Blood was harvested at the end of the study followed by analysis of pSLP76 in T cells by flow cytometry. (B–D) Enhanced levels of IFN-γ and T-cell activation markers by CompK treatment. Blood samples were used to prepare messenger RNA and complementary DNA. Quantitative PCR analysis was conducted to evaluate IFN-γ, CD25, and CD69 expression after treatment with vehicle or CompK. (E) IC50 of CompK on SLP76 phosphorylation in mouse whole blood. (F) Analysis of blood levels of CompK over time. Blood samples were collected at 1, 3, 7 and 24 hours post compound dosing. CompK levels were measured by liquid chromatography with tandem mass spectrometry. CompK, compound K; IFN-γ, interferon gamma; ns, not significant; WB, whole blood.
Antitumor immunity with combined treatment of CompK and anti-PD1
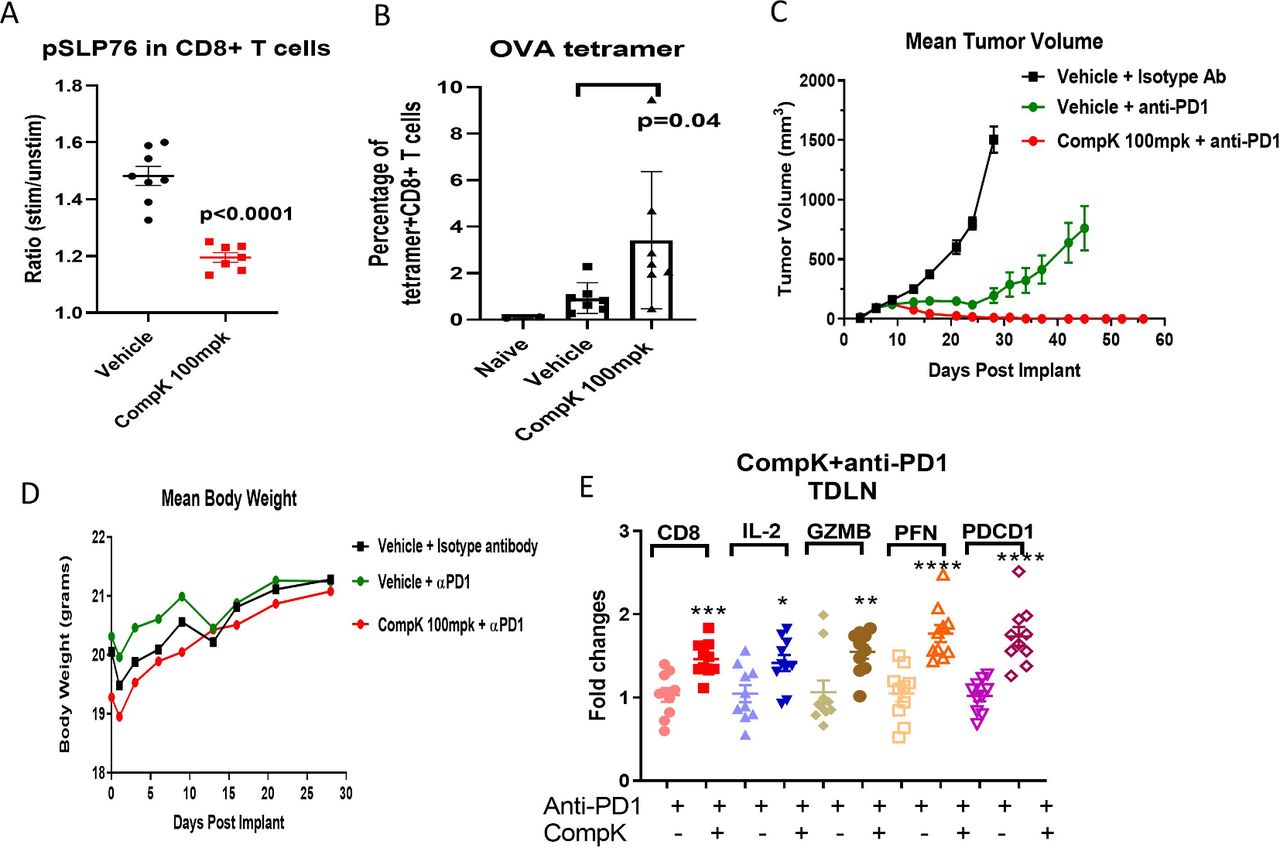
Having identified the effective CompK dose required to improve immune response in vivo, we then assessed if the same compound dose led to elevation of tumor antigen-specific CD8+ T-cell populations in the blood. The study was conducted in mice bearing ovalbumin (OVA) expressing MC38 tumors. After 5 days of compound treatment, blood samples were collected to investigate pSLP76 and OVA +CD8+ T cells by flow cytometry. CompK reduced pSLP76 levels and increased the number of OVA+CD8+ T cells (figure 6A,B), suggesting that CompK treatment promoted the expansion of the tumor antigen-specific CD8+ T-cell population. Based on the strength of these results, CompK was advanced into a MC38 syngeneic tumor efficacy study with primary focus on tumor free response. Previous studies in the MC38 model using HPK1 KD mice (wherein HPK1 kinase activity was maximally inhibited) showed no complete tumor-free responses in the absence of anti-PD1 (online supplemental figure S6B), whereas synergistic efficacy was demonstrated in combination of anti-PD1 treatment with HPK1 KD (online supplemental figure S6D), suggesting that an HPK1 inhibitor alone is not sufficient to generate any complete responses in the MC38 model. For this reason, we focused on investigating the synergy of CompK with anti-PD, and the CompK-alone group was not included in this study. Remarkably, a 100% complete tumor free response was observed with the combination treatment of CompK and anti-PD1, whereas only 20% complete response was achieved with anti-PD1 single therapy (figure 6C), suggesting a synergistic benefit of combining CompK and anti-PD1. The body weights of the mice were similar between vehicle and CompK-treated groups, and no overt treatment toxicity was observed (figure 6D). To investigate if CompK treatment influences the priming of T-cell response, we assessed TDLNs with qPCR to compare anti-PD1 alone versus anti-PD1 in combination with CompK using IFN-γ signature genes. Significant enhancement of these genes was observed in the group that received the combined treatment compared with the group treated with anti-PD1 alone, consistent with the improvement of tumor regression (figure 6E). Collectively, our results revealed critical roles of HPK1 and PD1 in mediating tumor immunity with synergistic benefit achieved via simultaneous blockade of HPK1 and PD1 pathways.
Supplemental material

Enhanced antitumor immunity by CompK treatment in a mouse syngeneic tumor model. (A) Inhibition of pSLP76 by CompK in MC38 OVA tumor-bearing mouse model. CompK was administrated to mice bearing MC38 OVA tumor at 100 mpk, two times per day for five consecutive days. Blood samples were collected to analyze pSLP76 in T cells by FACS. (B) Increased tumor antigen-specific CD8+ T cells by CompK. OVA+CD8+ T cells in the blood were measured by OVA tetramer staining followed by FACS analysis. (C) Promoted tumor regression by treatment with combination of CompK and anti-PD1. C57BL/6 mice were implanted with MC38 tumor cells followed by treatment with isotype control antibody, anti-PD1, and combination of CompK plus anti-PD1. All groups contain the same amount of vehicle used to formulate CompK. Tumor volumes were monitored up to 58 days. Compound dosing was discontinued on day 28. (D) No significant difference of body weights among study groups. Body weights were monitored during the time period of compound administration. (E) Improved priming of immune cells by CompK plus anti-PD1. Draining lymph nodes were collected at the end of study. Quantitative PCR analysis was conducted to investigate proinflammatory markers. Comparison was made between groups of anti-PD1 alone and anti-PD1 plus CompK. Statistical analysis was conducted using Student t-test. *P≤0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are presented as mean±SEM. CompK, compound K; GZMB, Granzyme B; IL, interleukin; OVA, ovalbumin; PFN, perforin; TDLN, tumor-draining lymph node.
Mechanistic translation from mouse to human tumors
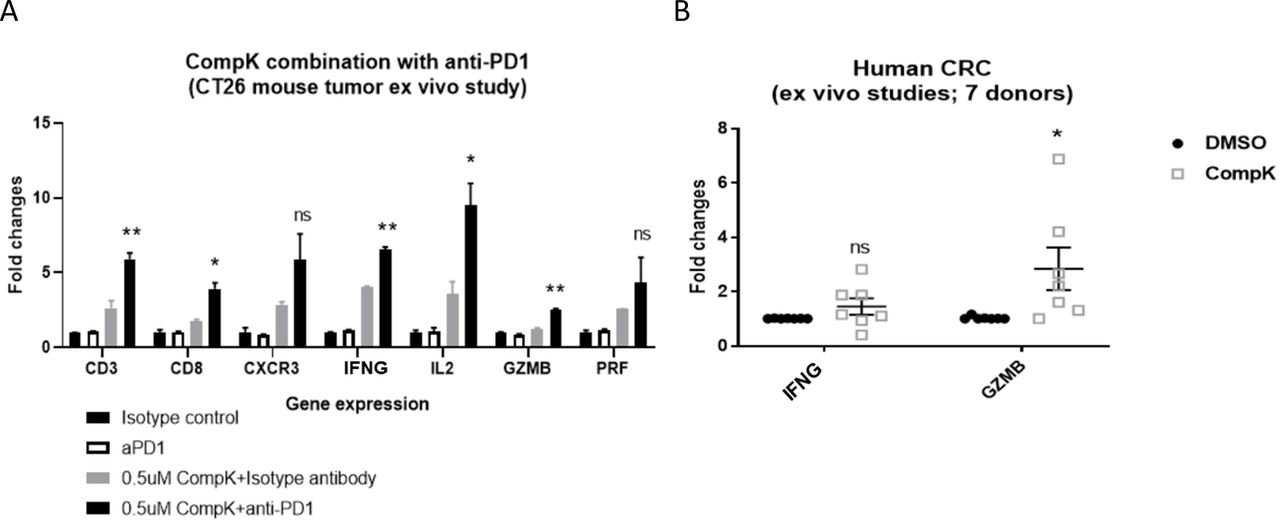
Given the improved antitumor immune response in the mouse syngeneic tumor model, we elected to interrogate whether this observation could be translated from mouse to human tumors. To better mimic and capture the TME-associated cross-talk of various cell types in the relevant spatial context, we developed a platform to study tissue explants from mouse or human tumors using self-derived ECM from the corresponding individual tumor, which provides increased physiological relevance. In a mouse CT26 tumor explant study, we treated the explants with CompK, anti-PD1, and combination of the two agents. Minimal treatment effects were observed with anti-PD1 single agent, consistent with findings from the in vivo study in the same model.21 CompK treatment yielded an improved immune signature, whereas combined treatment revealed synergy with augmented immune response (figure 7A). Further investigation was conducted with CRC explants from seven patients, CompK treatment increased IFN-γ and granzyme B as demonstrated by qPCR analysis (figure 7B), supporting that the improved immune response by CompK can be translated, at least mechanistically, from mouse to human tumors and that HPK1 is a relevant target in mediating tumor immunity in patients.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mechanistic translation from mouse to human tumors. (A) Improved proinflammatory gene signatures by CompK and anti-PD1 in CT26 tumor explant study. Randomized CT26 tumor explants were treated by 0.5 µM CompK, 1 µg/mL anti-PD1 or a combination of CompK plus anti-PD1 for 72 hours. Explants were harvested followed by qPCR analysis. Each data point was generated from eight explants. Statistical significance was generated by comparing groups of anti-PD1 alone and anti-PD1 plus CompK. (B) Enhanced levels of IFN-γ and GZMB by CompK in human CRC explant study. Tumor explants were prepared with fresh human CRC tumors. The randomized tumor explants were treated by 0.5 µM CompK for 72 hours followed by qPCR analysis. Data were generated with tumor samples from seven patients. Student t-test was used for statistical analysis. *P≤0.05, **P<0.01. Data are presented as mean±SEM. CompK, compound K; CRC, colorectal carcinoma; DMSO, dimethyl sulfoxide; IFN-γ, interferon gamma; IL, interleukin; ns, not significant; qPCR, quantitative PCR.
Discussion
As an intracellular checkpoint molecule, HPK1 is a key negative regulator in antitumor immunity,9 and the kinase activity is critical in mediating its regulatory functions.6 7 A HPK1 inhibitor is expected to enhance activation of T cells and APC function of DCs to recognize low immunogenic tumor antigens, with potential benefit to treat multiple types of tumors and promote the performance of cell therapy.
MAP4K family kinases exhibit a high degree of similarity in protein structure.22 Diverse roles have been revealed for MAP4K family members in immune cell signaling and inflammatory responses. MAP4K1/HPK1 and MAP4K4/HGK are negative regulators in T-cell activation and inflammatory responses.4 23 In contrast, MAP4K3/GLK plays a positive role in T-cell activation and autoimmune responses.24 25 Given the varied impact of MAP4K family members on immunity, family selectivity is crucial in order to establish the clinical potential of HPK1 inhibitors. We identified CompK with over 50-fold to 400-fold selectivity against the other family members. This level of family selectivity for CompK, along with other drug-like features of the compound, facilitated the investigation of HPK1 pharmacology under a therapeutically relevant setting. We first interrogated the dose and CompK levels over time required to initiate antitumor immune responses in sarcoma-bearing mice; and observed significant elevation of inflammatory markers after 5 days of dosing with target coverage above calculated blood IC50 for approximately 6 hours per 24-hour interval. With the same dosing regimen, we discovered increased tumor antigen-specific CD8+ T-cell population in blood in the MC38 syngeneic tumor model. Remarkable antitumor efficacy was further demonstrated in the same model with the combination therapy of CompK and anti-PD1, consistent with that observed in HPK1 KD mice.6 Importantly, the mechanistic translation was demonstrated in tumor explant study with fresh CRC tumors from multiple patients.
The TDLN is essential for initiating an effective antitumor T-cell immune response. However, the cancer-derived immunosuppressive environment renders the TDLN immune compromised.26 27 Given the strong bias in favor of tolerance in TDLN from patients with cancer, pharmacological agents, which can reshape the TDLN towards more proinflammatory anatomical context, are expected to be beneficial to enhance antitumor immunity. Moreover, TDLNs were reported to function as sites of T-cell invigoration required for anti-PD1/anti-PDL-1 blockade therapy.25 In our MC38 tumor model study, we observed marked improvement of IFN-γ signatures in TDLN with combination treatment of CompK and anti-PD1 compared with anti-PD1 single therapy, suggesting that the combination therapy can create a more optimal TDLN to enhance tumor immune response. With the concept of dominant tolerance in TDLN when tumor-derived antigens are presented by both tolerizing and immunizing routes,26 our findings suggest that CompK treatment could modify the prevailing tolerance for optimal priming of TDLN to tackle the impairment caused by tumors.
Immune responses are compromised by tumor-derived suppressive factors, such as TGF-β, PGE2 and adenosine.28–30 To understand the mechanism of action of CompK to reinvigorate immune response in the context of the suppressive environment, the influence of CompK treatment was studied using human primary T cells under immunosuppressive conditions containing PGE2, NECA (adenosine analog) and TGF-β. We observed significant attenuation of the activities exerted by these suppressive factors on CompK treatment. Our findings suggest potential utility of HPK1 inhibitors to combat the tolerance prone environment inside the tumor, with the enriched levels of PGE2, adenosine and TGF-β observed in multiple tumor types.7 31 Our DC studies demonstrated that HPK1 inhibition by CompK promoted the maturation of monocyte-derived DCs and enhanced the TNF-α+DC population on stimulation in human whole blood, offering a potential underlying mechanism of CompK treatment in creating more optimal microenvironment to elicit an immune response to control tumor progression. A previous study indicated an autocrine effect of TNF-α to sustain the survival of DCs on stimulation via regulating BAK, BCL-2, and FLIPL.32 Reduced LPS-induced apoptosis of DCs was demonstrated in HPK1 KO mouse DCs.8 With the elevated TNF-α+DC population observed on CompK treatment, it is conceivable that CompK could prolong human DC survival in the context of inflammation. Further studies are needed to interrogate this potential benefit and associated mechanism, which may have important clinical implication for HPK1 inhibitors in facilitating the sustainability of optimal tumor antigen presentation.
A significant percentage of tumor antigens are derivatives of normal or altered cellular genes, which typically are not sufficient to mount an effective immune response. The low immunogenicity of many tumor cells can also be attributed to low antigen expression, as well as absence of inflammatory and costimulatory signals. Tumor-reactive T cells generally have a lower functional avidity than what can be observed during acute infection. Hence, a central problem in cancer immunotherapy is how to efficiently prime CTLs against poorly immunogenic tumor antigens.18 33 Induction of higher-avidity CD8 T-cell responses promoted more effective tumor rejection.34 35 Pharmacological agents capable of boosting higher-avidity CD8 T cells may be particularly relevant to overcome the tolerance to tumor antigens. By conducting studies with viral antigen-specific CD8+ T cells and APCs under various antigen (E7) doses, we demonstrated that CompK treatment significantly left-shifted the dose–response curve for IFN-γ production. Over 2000 pg/mL IFN-γ was produced at 100 pg/mL dose of E7 peptides on CompK treatment, whereas vehicle control group showed no detectable level of IFN-γ under identical conditions, suggesting that CompK is involved in sensitizing TCR to recognize antigens at the levels as low as TAAs on malignant cells.36 Although the exact mechanism remains to be elucidated regarding how HPK1 mediates this process, it is plausible that CompK may facilitate the stability of the LAT complex on TCR engagement to the cognate antigen. Interestingly, the enhancement of IFN-γ production by CompK required the presence of APCs, as no difference observed when E7 was presented to CD8+ T cells by HLA-A2 dimer, implying that HPK1 is potentially involved in mediating optimal synapse formation for CD8+ T cells/APCs and functions as a gauge to fine tune TCR signaling strength. Moreover, we demonstrated a strong synergy by combination of CompK and anti-PD1 in elevating CFET-specific T-cell response. Notably, the synergistic effect was observed at the optimal dose of anti-PD1, supporting that HPK1 inhibition likely exerts its benefit independent of PD-1 pathway, and the combination leads to potential cross-talk between the corresponding signaling nodes to generate robust synergistic effects. These data are consistent with our observation in MC38 model study with the combination of CompK and anti-PD1. Low immunogenicity of tumors represents a significant barrier for cancer immunotherapy. We propose that HPK1 inhibition could promote robust T-cell response to TAAs and increase polyclonality/functionality of CTLs to attack tumors, where most viable antigens are expressed at lower levels and more heterogeneously. Further studies are needed to interrogate this hypothesis and the potential of HPK1 inhibition to facilitate epitope spreading, which is considered necessary to stimulate self-sustaining and long-lasting antitumor immune responses. Collectively, CompK produces benefits to multiple nodes critical to the cancer-immunity cycle37 and represents a promising agent to combat treatment resistance to existing therapies.
Acknowledgments
The authors thank Jessica Wong for analyzing immune cell infiltration of tumor tissues; Julie Carman and Michael Ciancanelli for their efforts on translational research related to HPK1; and Alla Pritsker and Mark Fereshteh for investigating the pharmacology of HPK1 inhibitors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DY, SH, GL, CS, YF, YC, SB, JG-B, DZ and JM: data collection and analysis; CC, RB, BMJ, KEP and GG: data collection and analysis, scientific guidance and data analysis; AM, ED, JK, JC and JF: scientific guidance and data analysis; CG: scientific guidance; APD; scientific guidance and data analysis, manuscript editing; GK: compound synthesis and data collection; MW, MQ and LS-C: scientific guidance and resources; JTH and MAS: scientific guidance and resources, manuscript editing; EL: supervision and resources; JL: scientific guidance and data analysis, manuscript writing and editing.
Funding All authors were the employees of Bristol Myers Squibb when the reported work was conducted.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All human tumor tissues were collected during the surgery with the consent of patients.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.