Article Text

Abstract
Background Interleukin (IL) 1 released from monocytes/macrophages is one of the critical determinants in mediating the adverse events of chimeric antigen receptor T cell (CAR-T) therapy, including cytokine release syndrome and neurotoxicity. However, the molecular mechanisms of IL-1 production during CAR-T therapy remain unknown.
Methods The roles of AIM2 and α1-adrenergic receptor (α1-AR) in CAR-T treatment-induced IL-1β release were evaluated by gene silencing, agonist or antagonist treatment. The phenotype switch of macrophages in response to CAR-T treatment was analyzed concerning cytotoxicity of CAR-T cells and proliferation of activated T cells.
Results This study provided the experimental evidence that CAR-T treatment-induced activation of AIM2 inflammasome of macrophages resulted in the release of bioactive IL-1β. CAR-T treatment-induced α1-AR-mediated adrenergic signaling augmented the priming of AIM2 inflammasome by enhancing IL-1β mRNA and AIM2 expression. Meanwhile, tumor cell DNA release triggered by CAR-T treatment potentiated the activation of AIM2 inflammasome in macrophages. Interestingly, an apparent phenotypic switch in macrophages occurred after interacting with CAR-T/tumor cells, which greatly inhibited the cytotoxicity of CAR-T cells and proliferation of activated T cells through upregulation of programmed cell death-ligand 1 (PD-L1) and indoleamine 2,3-dioxygenase (IDO) in the macrophages. Blockade of AIM2 inflammasome or α1-AR reversed the upregulation of PD-L1 and IDO and the phenotypic switch of the macrophages.
Conclusion Our study implicates that CAR-T therapy combined with the blockade of AIM2 inflammasome or α1-AR may relieve IL-1β-related toxic side effects of CAR-T therapy and ensure antitumor effects of the treatment.
- cell engineering
- cytokines
- macrophages
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Adoptive immunotherapy with chimeric antigen receptor T (CAR-T) cells has shown promising clinical impact on the survival of patients with cancer, most notably those with hematologic malignancies.1 2 However, CAR-T therapy can also cause dangerous side effects, including cytokine release syndrome (CRS) and neurotoxicity, hindering its therapeutic application. CRS is the most common acute toxicity of CAR-T therapy, characterized by fever, hypotension, and respiratory insufficiency. Patients with neurotoxicity induced by CAR-T therapy exhibit a diverse array of neurologic symptoms, such as tremor, dysgraphia, expressive aphasia, apraxia, and impaired attention. The precise mechanism remains unclear.3–5
Currently, there are many unanswered questions regarding the optimal clinical management of CRS. It has been shown that the administration of monoclonal antibodies against the interleukin (IL) 6 receptor (tocilizumab) is effective in the clinical management of adverse events of CAR-T therapy.6 However, it does not attenuate the neurotoxicity, partially due to the inability of tocilizumab to cross the blood-brain barrier thereby inhibiting the IL-6 signaling in the CNS. The current recommendations prefer the use of corticosteroids for the treatment of patients with high-grade CRS or neurologic adverse effects. Unfortunately, corticosteroids may impair the effectiveness of CAR-T therapy.7–9
Recent studies have demonstrated that IL-1 released from monocytes/macrophages is one of the critical determinants mediating the adverse events of CAR-T therapy.10 11 However, the mechanisms of IL-1 production during CAR-T therapy remain unknown. Understanding these underlying mechanisms may provide an effective strategy for clinical management of the side effects of CAR-T therapy during the early stage of CRS.
Accumulating evidence suggests that macrophages are critical regulators of tumor immunity and immunotherapy.12 Macrophages are able to polarize into different phenotypes in response to cues from the local tissue microenvironment. Within a brief period of time after CAR-T therapy, rapid and massive death of target cells occurs. Whether the abrupt change in the microenvironment causes a shift in macrophage polarization is largely unexplored.
In this study, we provide the first experimental evidence that CAR-T treatment-induced activation of the AIM2 inflammasome results in the release of bioactive IL-1β. α1-adrenergic receptor (α1-AR)-mediated adrenergic signaling further promotes AIM2 inflammasome activation and IL-1β production. The cooperation of the AIM2 inflammasome pathway and adrenergic signaling induces macrophage polarization towards an immunosuppressive phenotype by upregulating the expression of programmed cell death-ligand 1 (PD-L1) and indoleamine 2,3-dioxygenase (IDO). Blockade of the AIM2 inflammasome or α1-AR reduces the production of bioactive IL-1β and reverses macrophage phenotype switch triggered by CAR-T therapy.
Materials and methods
Cell culture, flow cytometry, quantitative real-time PCR, ELISA, western blot, co-immunoprecipitation, AIM2 shRNA (short hairpin RNA)-mediated silencing, immunofluorescence and cytotoxicity of tumor-specific CAR-T cells are described in detail in the online supplemental materials and methods.
Supplemental material
Viral vector construction
The construction of the anti-CD19, anti-BCMA or Her2 chimeric antigen receptor (CAR) has been reported by us and other groups.13–16 The lentiviral transfer plasmid contains an anti-CD19, anti-BCMA or anti-Her2 single chain variable fragment, human CD8α hinge and transmembrane region and human 4-1BB and human CD3ζ signaling moieties. The lentivirus was manufactured by Genechem Shanghai, China).
CAR-T cell production
Primary peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood of the healthy donor and the patients with diffuse large B cell lymphoma or B cell acute lymphoblastic leukemia (B-ALL). T lymphocytes were isolated using the EasySep human T Cell Isolation Kit (STEMCELL) according to the manufacture’s instruction. The procedure for the production of CAR-T cells has been described in our previous studies.13–15
Primary macrophage production and M2 macrophage polarization
To obtain the primary human macrophages, PBMCs were treated with 100 ng/mL macrophage colony-stimulating factor (PeproTech) in RPMI (Roswell Park Memorial Institute) 1640 medium supplemented with 20% fetal bovine serum for 7 days. To generate M2 macrophages, the cells were treated with 20 ng/mL IL-4 (PeproTech) and 20 ng/mL IL-13 (PeproTech) for an additional 72 hours.
The polarization of M2 macrophages derived from THP-1 cells is described in our previous study.17 Briefly, THP-1 cells were treated with 320 nM phorbol-12-myristate-13-acetate (PMA, Sigma) for 6 hours and then with PMA plus 20 ng/mL IL-4 (PeproTech) and 20 ng/mL IL-13 (PeproTech) for 18 hours to obtain the M2 phenotype.
Generation of the coculture system
The 5×105 THP-1 cells/well or 1×106 PBMCs were seeded into the six-well plates and polarized into M2 macrophages. After thoroughly washing to remove all PMA and cytokines, the macrophages were cocultured with CAR-T cells and Raji cells. The ratio of CAR-T cells, tumor cells, and macrophages was 1:3:1. In some experiments, the cells were treated with thalidomide (1 µM), VX-765 (20 µM), Ac-YVAD-CMK (10 µM), prazosin (10 µM), propranolol (10 µM), or epinephrine (1 µM) (Selleck Chemicals).
Tumor cell DNA labeling
Raji cells were incubated in complete medium supplemented with 0.5 µg/mL 5-ethynyl-2’-deoxyuridine (EdU) for 3 days to label tumor cell DNA. Afterwards, the cells were rigorously washed by PBS and subjected to the coculture with CAR-T and macrophages.
T cell proliferation assay
Autologous T cells were labeled with 5 µM carboxyfluorescein succinimidyl ester (CFSE, Selleck Chemicals) for 25 min at 4°C and preactivated with plate-bound 2 µg/mL anti-CD3 (Thermo Fisher) at 37°C, 5% CO2 for 16–18 hours. Three days after activation, the T cells were incubated with the macrophages for 2 days at a T cell:macrophage ratio of 5:1. The proliferation of T cells was measured by CFSE staining and flow cytometry. In some experiments, 5 mg/mL antihuman PD-L1 neutralizing antibody (Biolegend), 5 mg/mL isotype control antibody (Biolegend) or 4 mg/mL 1-MT (Selleck Chemicals) was added in the coculture.
Clinical samples
All clinical tissue samples were obtained from the Affiliated Hospital of Xuzhou Medical University with the informed consent of patients and with approval for experiments from the hospital.
Statistical analysis
All of the data are presented as mean±SEM. The data were analyzed by comparing the means using one-way analysis of variance (ANOVA) followed by Dunnett’s test or two-way ANOVA followed by Bonferroni’s post hoc test or t-test. A value of p<0.05 was considered statistically significant.
Results
CAR-T treatment induces IL-1β release by macrophages through activating the AIM2 inflammasome pathway
IL-1 is a putative mediator of inflammation released by activated monocytes or macrophages. Previous studies demonstrated that CAR-T cell-induced CRS, which usually occurs within days of T cell infusion, is mediated by macrophages and abated by IL-1 blockade.10 11 To explore the mechanisms of IL-1 release in response to CAR-T treatment, we used the in vitro cocultue system, which was modified according to a previous study.18 We established the model by coculturing CD19 CAR-T cells, Raji cells, which are human lymphoblast-like cells expressing high levels of CD19, and M2 macrophages. Since the proportion of M2-like macrophages is significantly increased in patients with leukemia and lymphoma,19–22 we used M2 macrophages induced from the PBMCs derived from the same healthy donor of CAR-T cells. IL-1β in the supernatants of the coculture system was evaluated by ELISA assay. As shown in figure 1A, the level of IL-1β was very low in the cultural supernatants of tumor cells, CAR-T, or macrophages alone. The production of IL-1β was significantly increased when macrophages were cocultured with CAR-T cells, while the level of IL-1β is the highest in the supernatants of CAR-T cell/tumor cell/macrophage coculture. Similar results were observed in coculturing CAR-T cells, tumor cells, and M2 macrophages derived from the human monocyte‐like cell line THP‐1 (figure 1B). These data are fully consistent with the observations in the previous studies, demonstrating that IL-1β is released predominantly by the macrophages.10 11 Clearly, the macrophages were activated after coculturing with CAR-T and Raji cells. Our data also indicate that the in vitro model is reasonable and feasible.
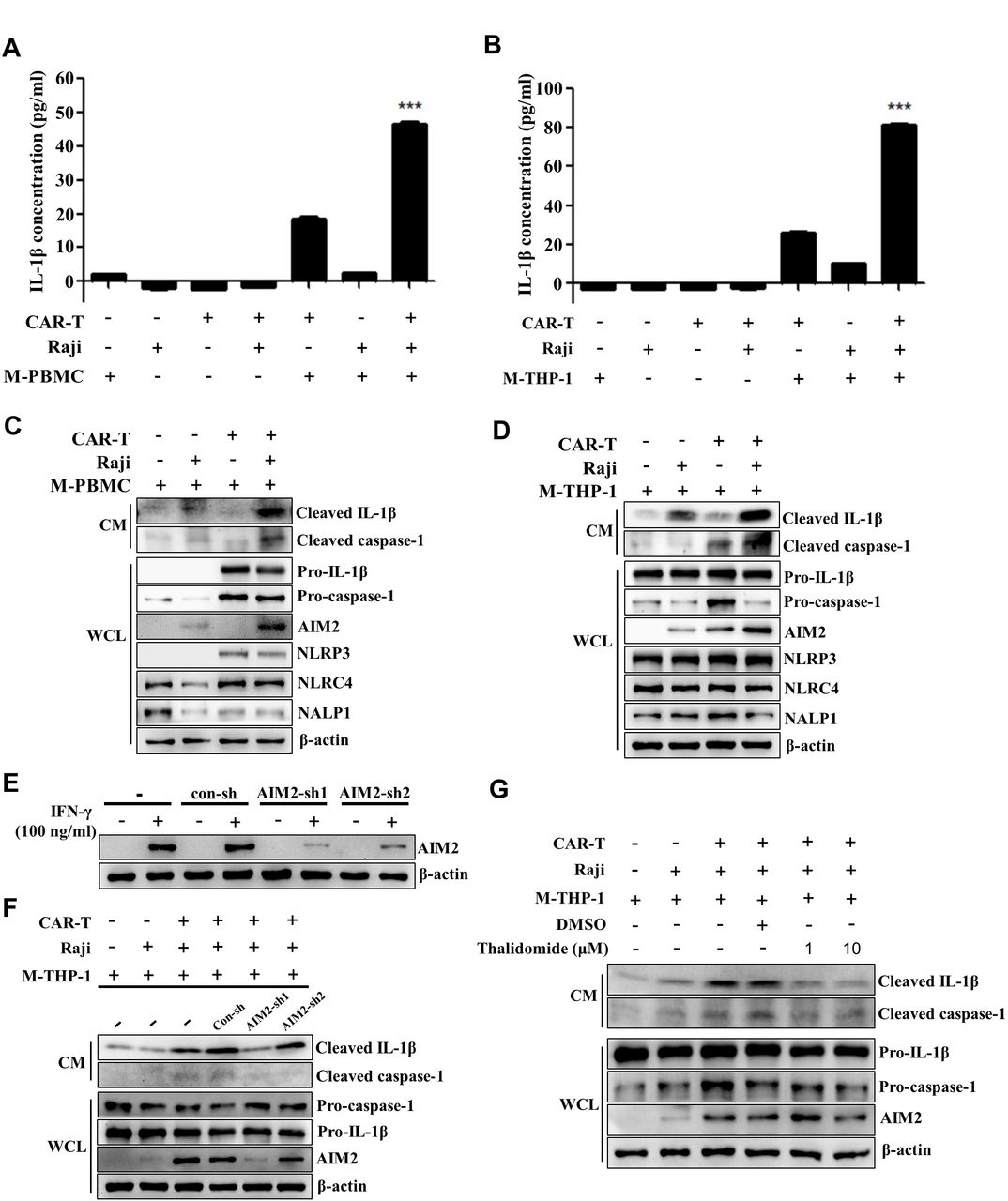
CAR-T treatment induces IL-1β release by macrophages through activating AIM2 inflammasome pathway. (A,B) The CD19 CAR-T cells were cocultured with Raji cells and M2 macrophages derived from PBMCs (M-PBMCs) (A, ***p<0.001) or macrophages derived from THP-1 cells (M-THP-1) (B, ***p<0.001). The concentration of IL-1β in the supernatants of the coculture system was evaluated by ELISA assay. (C,D) The expression of inflammasomes (NLRP1, NLRP3, NLRC4, AIM2), pro-caspase-1, and pro-IL-1β in the whole cell lysate (WCL) of adherent M-PBMC (C) or M-THP-1 (D) in response to the coculture was examined by western blot. Meanwhile mature IL-1β and cleaved caspase-1 in the culture medium (CM) were determined by western blot. (E) The expression of AIM2 in THP-1 cells was knocked down by the specific shRNAs. The transfected THP-1 cells were then induced with IL-4/IL-13 for M2 polarization. The expression of AIM2 in the presence of IFN-γ (100 ng/mL) was evaluated by western blot. (F,G) The expression of mature IL-1β and cleaved caspase-1 in the coculture supernatants and pro-caspase-1, pro-IL-1β, and AIM2 in the M-THP-1 in response to AIM2 knocking down or AIM2 inhibitor (thalidomide) was examined by western blot. Data are presented as mean±SEM and analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s test. CAR-T, chimericantigen receptor T; PBMCs, peripheral blood mononuclear cells.
The activation of caspase-1, which is triggered by inflammasomes, is required for proteolytic cleavage of the IL-1β precursor pro-IL-1β. Assembly of the inflammasome complex is initiated by the nucleotide binding domain and leucine-rich repeat-containing (NLR) proteins or absent in melanoma 2 (AIM2)-like receptors, including NLRP1, NLRP3, NLRC4, and AIM2.23 24 To explore whether CAR-T treatment triggers the activation of inflammasomes, the expression of inflammasomes (NLRP1, NLRP3, NLRC4, AIM2), caspase-1, and pro-IL-1β was examined in the adherent macrophages by western blot. Meanwhile mature IL-1β and cleaved caspase-1 in the coculture supernatants were determined. Figure 1C showed that the expression of NLRP1 and NLRC4 was present in the macrophages derived from human PBMCs of all the groups. However, NLRP3 was observed only in the macrophages cocultured with CAR-T cells or CAR-T/tumor cells. Coincidentally, the pro-caspase-1 and pro-IL-1β were also remarkably increased in these two groups. Noticeably, only in the coculture system of macrophage/CAR-T/tumor cells, the expression of AIM2 was remarkably upregulated. Moreover, the cleaved caspase-1 and mature IL-1β released into the supernatant were detected in macrophage/CAR-T/tumor cell coculture. To further verify the finding, the macrophages and CD19 CAR-T cells derived from the PBMCs of seven patients with diffuse large B cell lymphoma or B-ALL were employed. As shown in online supplemental figure 1, in most of the donors examined, the upregulation of AIM2 in the macrophages and mature IL-1β in the supernatant were evidently detected in the coculture of macrophage/CAR-T/tumor cells. The most obvious findings in the macrophages derived from THP-1 cells cocultured with the CAR-T/tumor cells were remarkable upregulation of AIM-2, which is consistent with the above results, and increased release of cleaved IL-1β in the supernatant (figure 1D). The levels of cleaved IL-1β in supernatants and AIM2 in the CAR-T cells and Raji cells cultured alone are shown in online supplemental figure 2. To explore whether the mature IL-1β released from macrophages depends on the specific interaction between CAR-T cells and cancer cells, the BCMA (B-cell maturation antigen) CAR-T and Her2 CAR-T cells were used in the coculture model. Online supplemental figure 3 shows that Raji cells were negative for Her2 or BCMA expression and that Her2 CAR-T or BCMA CAR-T did not upregulate the level of cleaved IL-1β in the cultural supernatants compared with CD19 CAR-T treatment. These data imply that the specific interaction between CAR-T cells and cancer cells triggers AIM2 inflammasome activation and production of bioactive IL-1β from macrophages.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
To verify the role of AIM2 inflammasome in the production of bioactive IL-1β induced by CAR-T/tumor cells, the expression of AIM2 in THP-1 cells was knocked down by the specific shRNAs. The transfected THP-1 cells were then induced with IL−4/IL-13 for M2 polarization. Figure 1E demonstrates that IFN-γ-induced AIM2 expression was effectively inhibited by the shRNA AIM2-sh1. Knockdown of AIM2 greatly reduced the level of cleaved IL-1β in the coculture supernatant of CAR-T cells/tumor cells/macrophages. The production of IL-1β was not significantly affected by AIM2-sh2, which did not silence the expression of AIM2 (figure 1F). The role of the AIM2 inflammasome in inducing IL-1β release from the macrophages triggered by CAR-T/tumor cells was further confirmed by a pharmacological inhibitor of AIM2 inflammasome, thalidomide, as thalidomide did not exert evident effects on the apoptosis and proliferation of CAR-T cells, Raji cells or M-THP-1 cells (online supplemental figure 4), but evidently repressed the release of bioactive IL-1β in the cocultue supernatant of CAR-T cells/tumor cells/macrophages (figure 1G).
Supplemental material
Activation of the AIM2 inflammasome pathway is triggered by CAR-T-induced tumor cell DNA release
AIM2 is a cytosolic double-stranded DNA (dsDNA) sensor that can only be activated by dsDNA in the cytosol, regardless of whether it is derived from pathogens or damaged tissues.25 26 A recent study showed that CAR-T cells induce target cell pyroptosis that is characterized by apoptosis-like chromatin condensation and DNA fragmentation in the early stage, followed by necrosis-like cell membrane pore formation, cellular swelling, and membrane rupture, leading to release of cellular contents, including dsDNA.27–30 We speculated that activation of the AIM2 inflammasome during the CAR-T treatment might be triggered by the dsDNA, which is released from dying tumor cells and engulfed by macrophages.
To test this hypothesis, we analyzed the levels of dsDNA in the coculture supernatant of Raji cells with the T cells that are genetically modified to express a CAR specific for CD19 (CD19 CAR-T cells). As shown in figure 2A, the level of dsDNA was significantly higher in the supernatant of Raji cells cocultured with CD19 CAR-T cells than in the supernatant of Raji cells alone or cocultured with mock T cells. To observe the phagocytosis of tumor cell DNAs by macrophages, the cellular DNAs were prelabeled by EdU in live Raji cells and the macrophages were labeled with CellTracker Deep Red (CTDR) before the coculture. The proportion of CTDR+/EdU+ cells, which represent the macrophages phagocytozing the tumor cell DNAs, was analyzed by flow cytometry. The results showed that the percentage of the CTDR+/EdU+ cells was significantly higher in the coculture of CAR-T cells/tumor cells/macrophages than in the coculture of tumor cells/macrophages (figure 2B). No tumor cell DNA was detected in the macrophages cultured alone. By confocal microscopy, we observed the co-localization of the tumor cell DNAs with the cytosolic DNA sensor AIM2 in the microphages cocultured with CAR-T and Raji cells (figure 2C,D), indicating that the CAR-T treatment induced the phagocytosis of the tumor cell DNAs by the macrophages and the interaction of tumor cell DNA/AIM2.

Activation of the AIM2 inflammasome pathway is triggered by CAR-T-induced tumor cell DNA release. (A) The level of double-stranded DNA (dsDNA) in the supernatant of Raji cells cocultured with CD19 CAR-T cells or mock T cells was evaluated by using the dsDNA assay kit (***p<0.001). (B) The cellular DNAs were prelabeled by 5-ethynyl-2’-deoxyuridine (EdU) in live Raji cells and the macrophages labeled with CellTracker deep red (CTDR) before the coculture. The proportion of CTDR+/EdU+ cells, which represent the macrophages phagocytozing the tumor cell DNAs, was analyzed by flow cytometry (***p<0.001). (C,D) The co-localization of the tumor cell DNAs with the cytosolic DNA sensor AIM2 in the microphages was examined by confocal microscopy and quantified (D, ***p<0.001). (E) The binding of AIM2 to ASC in the macrophages derived from THP-1 cells (M-THP-1) cocultured with CAR-T and Raji cells was evaluated by co-immunoprecipitation. (F) The expression of mature IL-1β and cleaved caspase-1 in the coculture supernatants and pro-caspase-1, pro-IL-1β, and AIM2 in the M-THP-1 cocultured with CAR-T/Raji cells in the presence of the caspase-1-specific inhibitors VX765 or ac-YVAD-cmk was evaluated by western blot. (G) The concentration of IL-1β in supernatants of the coculture of CAR-T/Raji/M-THP-1 in the presence of VX765 or ac-YVAD-cmk was examined by ELISA assay (**p<0.01, ***p<0.001). Data are presented as mean±SEM and analyzed by by two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test or by one-way ANOVA followed by Dunnett’s test. CAR-T, chimericantigen receptor T; CM, culture medium; WCL, whole cell lysate.
Upon binding to dsDNA, AIM2 recruits the adapter protein, which is the speck-like protein associated to apoptosis (ASC) and initiates the assembly of AIM2/ASC, leading to auto-cleavage of procaspase-1 and release of its active form.25 26 By using co-immunoprecipitation, we demonstrated increased binding of AIM2 to ASC in the coculture of CAR-T cells/Raji cells/macrophages (figure 2E), suggesting that the CAR-T treatment induces the formation and activation of the AIM2 inflammasome in the macrophages. Moreover, the caspase-1-specific inhibitors VX765 or AC-YVAD-CMK repressed the production of bioactive IL-1β in the macrophages cocultured with CAR-T/Raji cells (figure 2F,G). These data support that the CAR-T treatment induces the release of tumor cell DNA. The latter is then phagocytozed by macrophages and sensed by AIM2, resulting in AIM2 inflammasome formation, caspase-1 activation, and IL-1β release.
Activation of α1-AR augments CAR-T treatment-induced AIM2 inflammasome activation
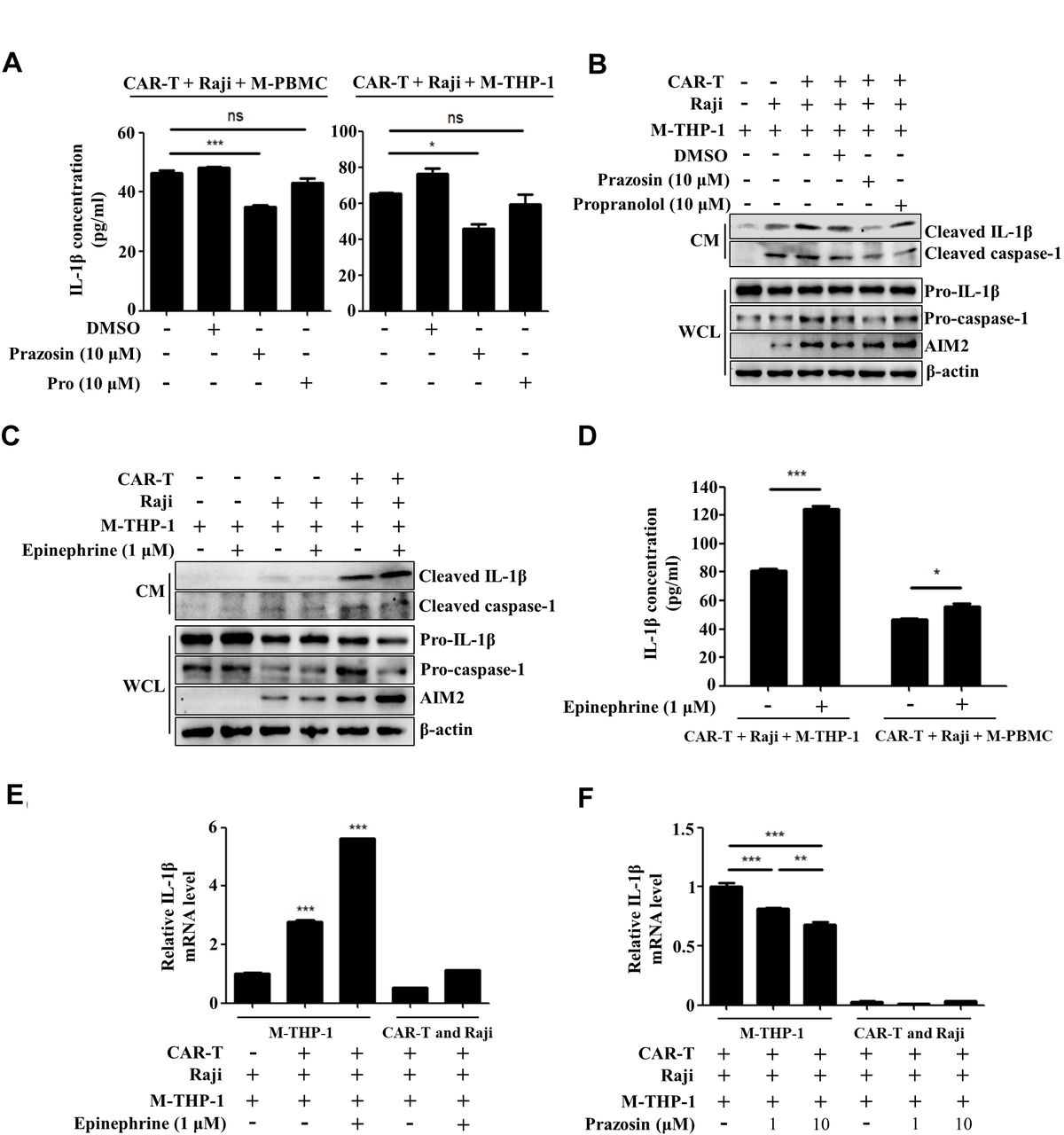
The previous study showed that cytokine release induced by T cell-activating therapeutic agents was accompanied by a catecholamine surge and inhibition of catecholamine synthesis reduced cytokine release.31 Thus, we attempted to determine the role of adrenergic receptors (ARs) in CAR-T treatment-induced IL-1β release. Figure 3A shows that the release of IL-1β was moderately repressed by prazosin, the antagonist of α1-AR but not propranolol, the inhibitor of β-AR when the macrophages were cocultured with CAR-T and tumor cells, as determined by ELISA. In addition, online supplemental figure 5 shows that addition of both prazosin and thalidomide more evidently repressed the upregulation of IL-1β release compared with the antagonist of α1-AR or AIM2 used alone. This indicates that activation of α1-AR adrenergic signaling triggered by CAR-T treatment plays a key role in AIM2 inflammasome activation. Figure 3B confirms the above data, as the expressions of AIM2 and pro-caspase-1 in the macrophages and the release of cleaved IL-1β into the coculture supernatant of CAR-T cells/tumor cells/macrophages could be inhibited by prazosin but not propranolol. The cleaved IL-1β release could be further enhanced by addition of epinephrine into the coculture system (figure 3C,D). We collected the CAR-T cells and Raji cells growing in the suspension and the adherently growing THP-1-derived macrophages after coculture for 24 hours, and examined the transcription of IL-1β mRNA. The upregulation of the IL-1β mRNA level predominantly occurred in the macrophages and epinephrine further promoted the transcription of IL-1β (figure 3E). The increased transcription of IL-1β mRNA in the macrophages could be remarkably inhibited by prazosin (figure 3F). These data prove that α1-AR-mediated adrenergic signaling promotes the activation of the AIM2 inflammasome and production of IL-1β in the macrophages interacting with tumor cells and CAR-T cells. The upregulation of the inflammasome-associated genes including IL-1β and AIM2, as a priming signal, is vital for the activation of the AIM2 inflammasome.32 33 Thus, our findings suggest that α1-adrenergic signaling augments CAR-T treatment-induced AIM2 inflammasome activation at least partially through enhancing the priming signal.
Supplemental material
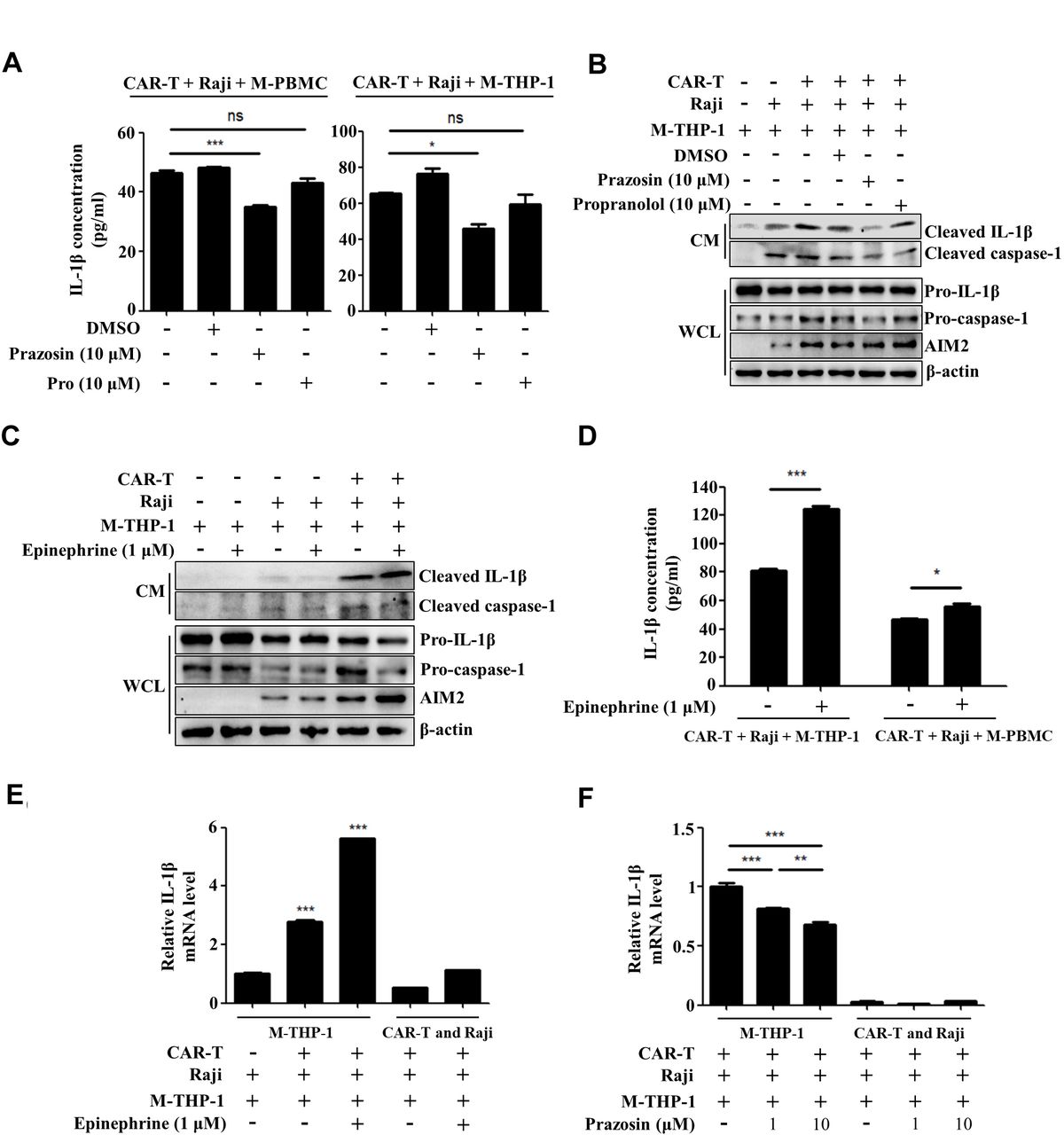
Activation of α1-adrenergic receptor augments CAR-T treatment-induced AIM2 inflammasome activation. (A) The concentration of IL-1β in supernatants of the coculture of CAR-T/Raji/M-PBMC or macrophages derived from THP-1 cells (M-THP-1) in the presence of prazosin or propranolol was examined by ELISA assay (*p<0.05, ***p<0.001). (B,C) The expression of mature IL-1β and cleaved caspase-1 in the coculture supernatants and pro-caspase-1, pro-IL-1β, and AIM2 in M-THP-1 in response to prazosin, propranolol or epinephrine treatment was examined by western blot. (D) In response to epinephrine treatment, the concentration of IL-1β in supernatants of the coculture of CAR-T/Raji/M-PBMC or M-THP-1 was examined by ELISA assay (*p<0.05, ***p<0.001). (E,F) After coculture for 24 hours, the CAR-T cells and Raji cells growing in suspension and the adherently growing M-THP-1 were collected. The expression of the IL-1β mRNA in response to epinephrine or prazosin treatment was examined by quantitative real-time PCR (qRT-PCR) (**p<0.05, ***p<0.001). Data are presented as mean±SEM and analyzed by one-way ANOVA followed by Dunnett’s test or by two-way ANOVA followed by Bonferroni’s post hoc test. ANOVA, analysis of variance; CAR-T, chimericantigen receptor T; CM, culture medium; M-PBMC, macrophages derived from PBMCs; PBMCs, peripheral blood mononuclear cells; WCL, whole cell lysate.
CAR-T therapy induces macrophage phenotype switch by upregulating PD-L1 and IDO
Macrophages are highly plastic and undergo phenotypic transformation in response to microenvironmental stimuli. Previous studies showed that phagocytosis of apoptotic cells by macrophages in damaged tissues induced immunosuppressive phenotypes, leading to immune tolerance.34 To explore the effects of the interaction between CAR-T cells, macrophages and tumor cells on the cytotoxicity of CAR-T cells, CD19 CAR-T cells, Raji cells and macrophages derived from PBMCs are cocultured for 24 hours, and then the Raji/Luc cells were added into the coculture system for 48 hours. The luciferase activity was measured to exhibit the cytotoxicity of CAR-T cells. Online supplemental figure 6 shows that the coculture of macrophages with both CAR-T and Raji cells critically inhibits the cytotoxicity of CD19 CAR-T cells. To further investigate whether the phenotype of the macrophages is altered after contacting with CAR-T and tumor cells, the macrophages derived from human PBMCs were preincubated with CAR-T and Raji cells for 24 hours and then co-incubated with CAR-T cells for 36 hours. The CAR-T cells were then isolated and incubated with Raji/Luc cells. Figure 4A,B shows that the macrophages contacting with both CAR-T and Raji cells remarkably inhibit the cytotoxicity of CD19 CAR-T (figure 4A) but not mock T cells (figure 4B), indicating that the phenotype of macrophages is altered after encountering CAR-T and tumor cells. A recent study reported that induction of bioactive IL-1β mediated by caspase-1 triggers upregulation of PD-L1 and IDO, which exert immunosuppressive effects, in macrophages.35 To explore the mechanisms of the phenotype switch of the macrophages, we examined the expression of PD-L1 and IDO in the macrophages. Western blot analysis demonstrates that the expressions of PD-L1 and IDO are markedly upregulated in the macrophages derived from human PBMCs after coculturing with CAR-T and Raji cells (figure 4C). The macrophage phenotype switch is also confirmed by the T cell proliferation assay. The macrophages isolated from the coculture were then co-incubated with the activated autologous T cells labeled with CFSE. Flow cytometry analysis of cell division by dilution of CFSE was used to assess the effect of the macrophages on T cell proliferation. The data in figure 4D,E shows that the macrophages contacting with both CAR-T and Raji cells remarkably inhibited the proliferation of the activated T cells. This phenomenon was also confirmed by using macrophages derived from THP-1 cells (figure 4F,G). To further confirm the upregulation of PD-L1 and IDO, the macrophages derived from THP-1 cells were labeled with CTDR and then cocultured with CAR-T and tumor cells. The expressions of PD-L1 and IDO were analyzed by flow cytometry and confocal microscopy. The results show that the increased expressions of PD-L1 and IDO are clearly detected on the surface or in the cytoplasm of the macrophages after encountering CAR-T and tumor cells (figure 4H,I). Online supplemental figure 6 shows that blockade of PD-L1 or IDO with anti-PD-L1 antibody or IDO inhibitor (1-MT) reverses the inhibition of CAR-T cells cytotoxicity induced by the coculture of CAR-T cells/tumor cells/macrophages. To further illustrate whether PD-L1 and IDO mediate the immunosuppressive effects of the macrophages, PD-L1 antibody or 1-MT was added to the coculture of the preactivated T cells and the macrophages from the coculture system with CAR-T and Raji cells. Figure 4J,K demonstrated that PD-L1 neutralization or IDO inhibition effectively alleviated the repressive effects of the macrophages on the proliferation of T cells. Noticeably, addition of both anti-PD-L1 antibody and IDO inhibitor completely reversed the inhibitory effects of the macrophages (figure 4J,K). These data suggest that CAR-T therapy induces the phenotype switch of the macrophages by upregulating the expression of PD-L1 and IDO.
Supplemental material

CAR-T therapy induces macrophage phenotype switch by upregulating PD-L1 and IDO. (A,B) The macrophages derived from human PBMCs were preincubated with CAR-T and Raji cells for 24 hours and then co-incubated with CAR-T or mock T cells for 36 hours. The CAR-T cells (A) or mock T cells (B) were then isolated and incubated with Raji/Luc cells and then luciferase activity was measured to exhibit the cytotoxicity of CAR-T cells. (C) The expression of PD-L1 and IDO in the M-PBMCs after coculturing with CAR-T and Raji cells was examined by western blot. (D–G) M-PBMCs (D) or macrophages derived from THP-1 cells (M-THP-1) (F) were preincubated with CAR-T and Raji cells for 24 hours and then cocultured with the activated autologous T cells labeled with CFSE. Flow cytometry analysis of cell division by dilution of CFSE was used to assess the effect of the macrophages on T cell proliferation. The divided T cells were quantified (*p<0.05, ***p<0.001) (E,G). (H) Flow cytometry was employed to evaluate the level of PD-L1 and IDO in M-THP-1 after coculturing with CAR-T and Raji cells (***p<0.001). (I) M-THP-1 cells were labeled with CellTracker Deep Red (CTDR) and then cocultured with CAR-T and Raji cells. The expressions of PD-L1 and IDO were analyzed by confocal microscopy. (J) The effects of the macrophages on T cell proliferation in the presence of PD-L1 antibody or IDO inhibitor (1-MT) were examined by flow cytometry analysis of cell division by dilution of CFSE. (K) The divided T cells were quantified (***p<0.001). Data are presented as mean±SEM and analyzed by one-way ANOVA followed by Dunnett’s test. ANOVA, analysis of variance; CAR-T, chimericantigen receptor T; CFSE, carboxyfluorescein succinimidyl ester; IDO, indoleamine 2,3-dioxygenase; M-PBMCs, macrophages derived from PBMCs; PBMCs, peripheral blood mononuclear cells; PD-L1, programmed cell death-ligand 1.
Activation of the AIM2 inflammasome triggered by CAR-T treatment induces macrophage phenotype switch
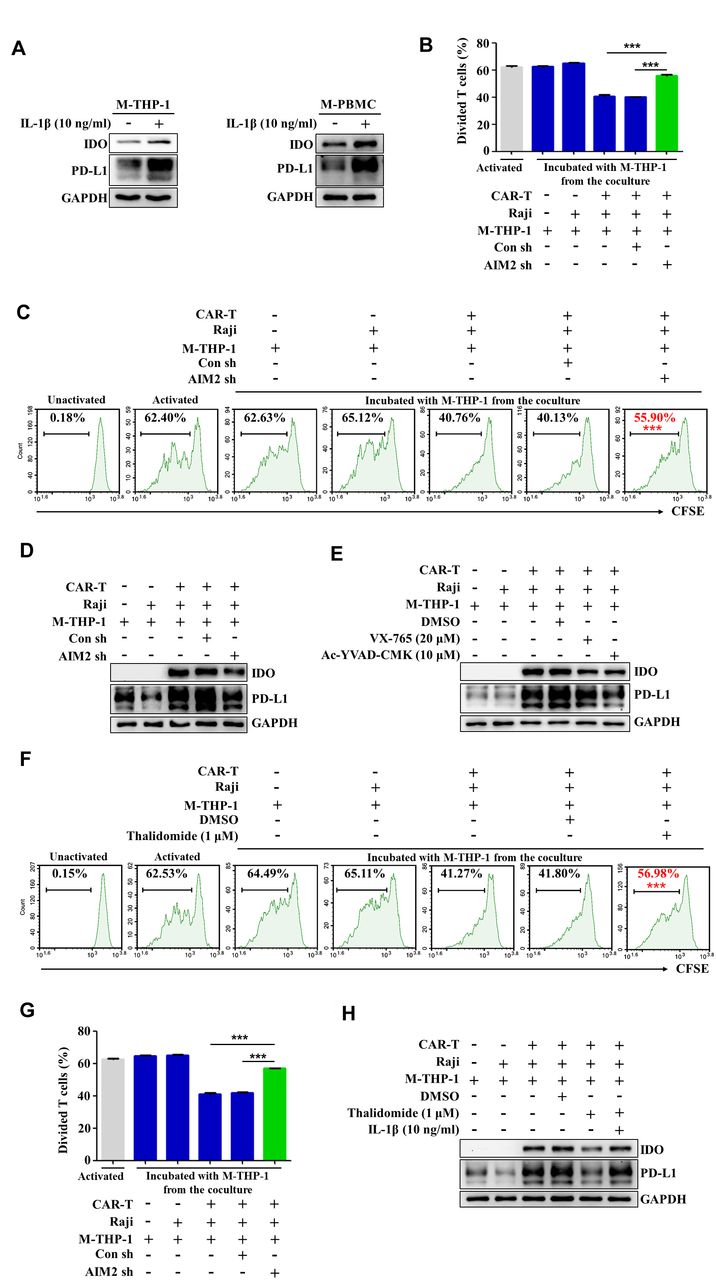
It is considered that IFN-γ (Interferon γ) released by activated T cells is a major inducer of PD-L1 expression by cancer cells. Recent studies demonstrated that IL-1β released by macrophages is also an important inducer of PD-L1 expression by cancer cells.36 To further explore the mechanisms about the phenotype switch of macrophages in the presence of CAR-T and tumor cells, we treated the macrophages with recombinant human IL-1β. As shown in figure 5A, human IL-1β evidently enhanced the expression of PD-L1 and IDO in the macrophages derived from THP-1 cells or human PBMCs. We therefore speculated that activation of the AIM2 inflammasome, which is responsible for IL-1β production (figures 1 and 2), might be involved in the induction of immunosuppressive phenotypes in the macrophages. Blockade of AIM2 with thalidomide evidently relieved the repression on CAR-T cell cytotoxicity induced by coculture of CAR-T cells/tumor cells/macrophages (online supplemental figure 6). To further determine the role of the AIM2 inflammasome in inducing the immunosuppressive phenotype, AIM2 expression in the macrophages was knocked down by the specific shRNAs. Figure 5B,C shows that the inhibitory effect of the macrophages on the activated T cell proliferation was effectively reversed by knockdown of AIM2. The expressions of PD-L1 and IDO in the macrophages induced by coculture with CAR-T and Raji cells were also repressed (figure 5D). The specific inhibitors of caspase-1, VX765 or AC-YVAD-CMK, had similar effects on the expression of PD-L1 and IDO (figure 5E). The growth inhibitory effect on preactivated T cells could be antagonized by addition of the inhibitor of the AIM2 inflammasome, thalidomide, into the coculture of CAR-T cells/tumor cells/macrophages. The data in figure 5F–H demonstrate that blocking AIM2 inflammasome activation alleviated the suppressive effects of the macrophages on T cells and reversed the upregulation of PD-L1 and IDO of the macrophages induced by contacting with CAR-T and tumor cells. Importantly, treatment with recombinant human IL-1β upregulated PD-L1 and IDO in the presence of thalidomide to the level comparable to CAR-T treatment-induced macrophages (figure 5H). Collectively, our data indicate that activation of the AIM2 inflammasome governs the phenotype switch of macrophages during CAR-T therapy.
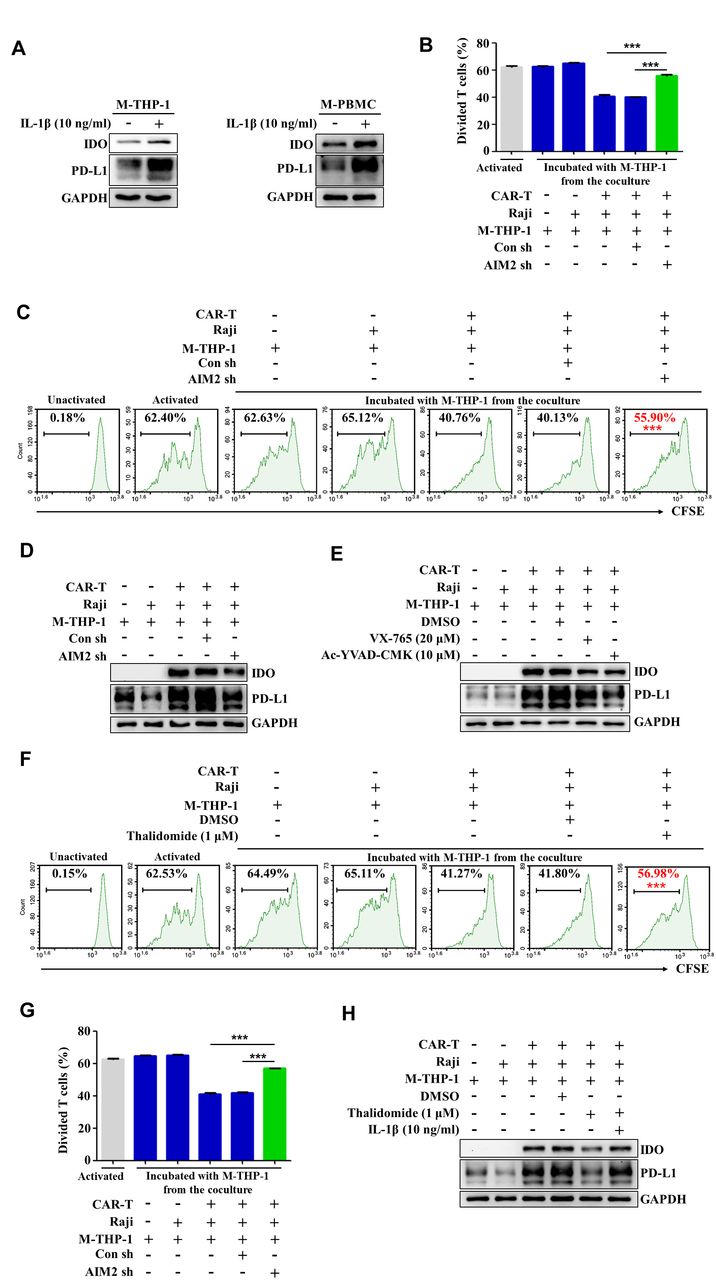
Activation of the AIM2 inflammasome triggered by CAR-T treatment induces macrophage phenotype switch. (A) Macrophages derived from THP-1 cells (M-THP-1) or M-PBMCs were treated with recombinant human IL-1β. The expression of PD-L1 and IDO in the macrophages was detected by western blot. (B,C) M-THP-1 infected with AIM2-shRNA expressing lentivirals were preincubated with CAR-T and Raji cells for 24 hours and then cocultured with the activated autologous T cells labeled with CFSE. T cell division was evaluated by flow cytometry (***p<0.001). (D,E) The expression of PD-L1 and IDO in M-THP-1 after coculturing with CAR-T and Raji cells in response to AIM2 knocking down or caspase-1 inhibitor (VX765 or ac-YVAD-cmk) treatment was detected by western blot. (F,G) M-THP-1 were preincubated with CAR-T and Raji cells for 24 hours in the presence of the AIM2 inhibitor thalidomide and then cocultured with the activated autologous T cells labeled with CFSE. The effects of the macrophages on T cell proliferation were examined by flow cytometry. The divided T cells were quantified (***p<0.001). (H) In response to thalidomide and IL-1β treatment, the expression of PD-L1 and IDO in M-THP-1 cocultured with CAR-T and Raji cells detected by western blot. Data are presented as mean±SEM and analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s test. CAR-T, chimericantigen receptor T; CFSE, carboxyfluorescein succinimidyl ester; IDO, indoleamine 2,3-dioxygenase; M-PBMCs, macrophages derived from PBMCs; PBMCs, peripheral blood mononuclear cells; PD-L1, programmed cell death-ligand 1.
Activation of α1-AR is involved in phenotype switch of macrophages induced by contacting with CAR-T and tumor cells
Previous data demonstrated that α1-AR signaling was involved in the activation of the AIM2 inflammasome and release of bioactive IL-1β in the macrophages in the coculture system (figure 3). Therefore, α1-AR-mediated adrenergic signaling may also participate in inducing the phenotype switch of the macrophages. Online supplemental figure 6 showed that blockade of α1-AR by using prazosin alleviated the repression of CAR-T cell cytotoxicity induced by coculture of CAR-T cells/tumor cells/macrophages. To confirm the role of α1-AR in the phenotype manipulation of macrophages, prazosin was added to the coculture of CAR-T cells/tumor cells/macrophages and then the isolated macrophages were cocultured with preactivated T cells. The data demonstrated that blockade of α1-AR relieved the suppressive effects of the macrophages on T cell proliferation (figure 6A,B). Upregulation of PD-L1 and IDO in macrophages was also inhibited by prazosin but not propranolol (figure 6C). Addition of recombinant human IL-1β into the coculture system restored the expression of PD-L1 and IDO in the presence of prazosin (figure 6D). The agonist of the adrenergic signaling, epinephrine, significantly enhanced the expression of PD-L1 and IDO (figure 6E). These data suggest that α1-AR-mediated adrenergic signaling may modulate the phenotype switch of macrophages in the setting of CAR-T therapy through enhancing IL-1β production.

Activation of α1-adrenergic receptor (α1-AR) is involved in the phenotype switch of macrophages induced by contacting with CAR-T and tumor cells. (A,B) The macrophages derived from THP-1 cells (M-THP-1) were preincubated with CAR-T and Raji cells in the presence of prazosin for 24 hours and then cocultured with the activated autologous T cells labeled with CFSE. T cell division was evaluated by flow cytometry (***p<0.001). (C–E) Western blot was employed to detect the expression of PD-L1 and IDO in M-THP-1 after coculturing with CAR-T and Raji cells in the presence of prazosin, propranolol, IL-1β or epinephrine. Data are presented as mean±SEM and analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s test. CAR-T, chimericantigen receptor T; CFSE, carboxyfluorescein succinimidyl ester; IDO, indoleamine 2,3-dioxygenase; PD-L1, programmed cell death-ligand 1.
Discussion
Previous studies demonstrated that CAR-T therapy-induced CRS is mediated by proinflammatory mediators (such as IL-6, IL-1, etc) produced by recipient macrophages but not by CAR-T cell-derived cytokines.10 11 The side effects of CAR-T therapy could be abated by the IL-1 receptor antagonist anakinra, indicating that IL-1 is one primary cause of the adverse events associated with CAR-T therapy. It was reported that the release of IL-1 preceded the production of IL-6. Moreover, IL-1 could trigger the production of IL-6.11 37 Therefore, the induction of IL-1 may be the upstream event of the catastrophic cascade of CRS and neurotoxicity. Unveiling the mechanisms for IL-1 production in CAR-T therapy will help to establish effective prevention and control strategies to minimize the risk of serious adverse events.
The maturation and secretion of IL-1β is strictly controlled by the inflammasome, which activates caspase-1 that cleaves pro–IL-1β into the bioactive IL-1β.38 Our study demonstrated that the expressions of AIM2 and pro-IL-1β were dramatically increased in the macrophages, whereas mature IL-1β and cleaved caspase-1 were released into the coculture supernatant of CAR-T cells, Raji cells, and macrophages. Noticeably, the release of bioactive IL-1β was predominantly from the macrophages and maximal release of IL-1β only occurred in the presence of both CAR-T and tumor cells. Inhibition of the AIM2 inflammasome pathway by thalidomide abolished the effects on the activation of caspase-1 and cleavage of pro-IL-1β in the macrophages. Suppression of caspase-1 activation by VX765 or AC-YVAD-CMK also effectively abated the production of mature IL-1β. The data suggest that the interactions of macrophages with CAR-T and tumor cells trigger the release of bioactive IL-1β through activation of the AIM2 inflammasome pathway.
AIM2 recognizes cytosolic dsDNA and then forms an active inflammasome complex with ASC, inducing activation of downstream caspase-1.39 A recent study showed that tumor cell pyroptosis, a form of lytic programmed cell death, which is initiated by inflammasomes, triggers CRS during CAR-T therapy.27 During pyrotosis of host cells, caspase-1-dependent membrane pore formation causes cell swelling and osmotic lysis, leading to the release of intracellular contents and proinflammatory cytokines.40 Our study indicated that the level of tumor cell DNA in the coculture supernatant of macrophages/CAR-T cells/Raji cells was markedly elevated. The phagocytosis of the tumor cell DNA by the macrophages and obvious co-localization of tumor cell DNA and AIM2 were clearly observed. Concurrent with the release of tumor cell DNA, the AIM2/ASC complex formation was detected by co-immunoprecipitation. The production of bioactive IL-1β could be repressed by the caspase-1 inhibitor. It is reasonable to believe that the cellular DNA released by pyroptotic tumor cells during CAR-T therapy was phagocytozed by macrophages, thereby triggering the activation of the AIM2 inflammasome and release of the bioactive IL-1β, which drives the production of inflammatory cytokines in CRS.
A previous study demonstrated that cytokine release induced by T cell-activating therapeutic agents was accompanied by a catecholamine surge. Catecholamine produced by macrophages promoted the secretion of diverse cytokines in the setting of immunotherapy.31 However, how catecholamines boost cytokine production is unknown. The types of adrenergic receptors in human cells for the effects of catecholamines on cytokine production remain to be identified.41 Our study shows that epinephrine significantly enhanced AIM2 expression and bioactive IL-1β release in the macrophages induced by coculturing with CAR-T cells and Raji cells. The effect of epinephrine could be blocked by the α1-AR antagonist prazosin, suggesting that catecholamine-mediated α1-AR signaling promotes the maturation of IL-1β through augmenting the AIM2 inflammasome activation. The activation of inflammasome is tightly controlled by a priming signal, which upregulates the expression of inflammasome-associated genes. On engagement of dsDNA, AIM2 initiates the downstream signaling and assembly of an inflammasome.32 We observed that epinephrine markedly promoted the transcription of IL-1β in the macrophages triggered by contacting with CAR-T/Raji cells. The effect resulted from the interactions of the macrophages/CAR-T/Raji cells on the transcription of IL-1β could be antagonized by prazosin, implicating that catecholamines boost proinflammatory cytokine release at least through triggering the priming signal of the AIM2 inflammasome.
The phenotypes and functions of macrophages are critically regulated by the surrounding microenvironment. In response to growth factors and cytokines released in the local tissue microenvironment macrophages undergo marked phenotypic and functional changes.12 42 It has been reported that cancer treatment, such as chemotherapy or targeted therapy, may induce phenotypic switch of macrophages.35 43 During CAR-T therapy numerous tumor cells were killed, accompanied by a massive and abrupt release of cellular constituents. The phagocytozed tumor cell DNAs were sensed by AIM2, leading to the activation of the AIM2 inflammasome pathway and subsequent release of proinflammatory cytokines, which may influence the phenotypes and functions of macrophages. In the present study, we demonstrated that an apparent phenotypic switch in macrophages occurred after interacting with CAR-T/tumor cells. The immunosuppressive molecules PD-L1 and IDO were greatly upregulated in the macrophages. The anti-PD-L1 antibody and IDO inhibitor effectively abrogated the repressive effects of the macrophages on the proliferation of activated T cells, suggesting that CAR-T therapy induces the phenotype switch of the macrophages by upregulating the expression of PD-L1 and IDO. Blocking the AIM2 inflammasome or α1-AR inhibited the upregulation of PD-L1 and IDO and the phenotypic switch of the macrophages induced by CAR-T/tumor cells, indicating that the activation of the AIM2 inflammasome and adrenergic signaling synergistically promoted the immunosuppressive phenotype of macrophages during CAR-T therapy.
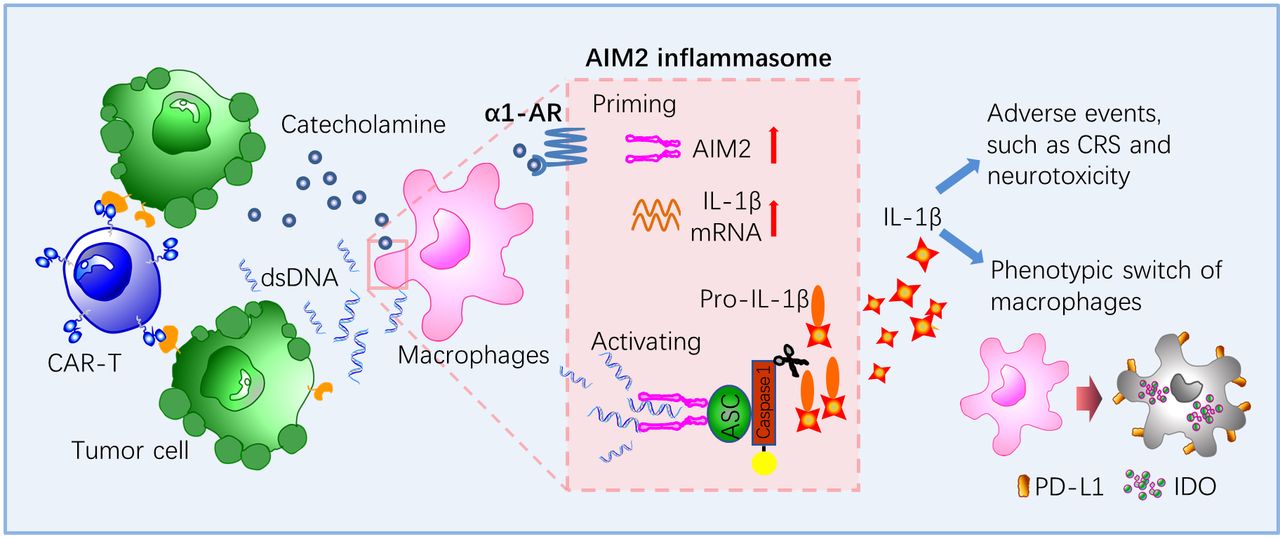
Taken together, our findings reveal that CAR-T therapy induces the production of bioactive IL-1β and immunosuppresive phenotypes of macrophages mainly through activating the AIM2 inflammasome and α1-AR-mediated adrenergic signaling pathway. Our study implicates that CAR-T therapy combined with blockade of the AIM2 inflammasome and α1-AR may ensure the antitumor effects of CAR-T therapy and relieve the toxic side effects associated with the treatment (figure 7). In addition, prazosin and thalidomide are capable of penetrating the blood-brain barrier, and thus may block the inflammatory cascade within the central nervous system. These two drugs have been approved by the Food and Drug Administration for the treatment of hypertension and multiple myeloma, respectively. Experiences have been accumulated regarding the dosage, side effect profile, and toxicity management of these drugs. The findings in our study may be helpful in guiding the design and implementation of future clinical trials.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram showing the role of the AIM2 inflammasome and α1-adrenergic receptor (α1-AR) in the release of IL-1β and macrophage-mediated immunosuppression triggered by CAR-T treatment. CAR-T, chimericantigen receptor T; CRS, cytokine releasesyndrome; ds DNA, double-stranded DNA; IDO, indoleamine 2,3-dioxygenase; PD-L1, programmed cell death-ligand 1.
Acknowledgments
The authors thank Dr Jiang Cao for providing technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
DL and XX contributed equally.
Contributors MS, JZ and DL initiated, designed and supervised the study. XX, DL, YD, XZ, SB, LZ and SL designed and performed the experiments. WM, YL and SB analyzed the data. DL and MS wrote the paper. All authors read and approved the final manuscript.
Funding This work is supported by the National Natural Science Foundation of China (No. 81972719, 81773258, 81 773 086 and 82003164), Jiangsu Natural Science Foundation (No. BK20171161 and BK20201012), Key University Science Research Project of Jiangsu Province (No. 17KJA320009), Key Research & Development Plan of Jiangsu Province (No. BE2018634), key young talents in medicine of Jiangsu Province (No. QNRC2016803), Jiangsu Province Innovation and Entrepreneurship Talents Project, and Key Research & Developement Plan of Xuzhou (No. KC18102).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The human tissue study was approved by the Medical Ethics Committee of Affiliated Hospital of Xuzhou Medical University (approval number XYFY2016-KL002-01).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.