Article Text

Abstract
Background Uveal melanoma (UM) is the most frequent intraocular malignancy and is resistant to immunotherapy. Nearly 50% of patients with UM develop metastatic disease, and the overall survival outcome remains very poor. Therefore, a treatment regimen that simultaneously targets primary UM and prevents metastasis is needed. Here, we suggest an immunotherapeutic strategy for UM involving a combination of local photodynamic therapy (PDT), rho-kinase (ROCK) inhibitor, and PD-1/PD-L1 immune checkpoint blockade.
Methods The antitumor efficacy and immune response of monotreatment or combinational treatment were evaluated in B16F10-bearing syngeneic mouse models. Abscopal antitumor immune responses induced by triple-combinational treatment were validated in syngeneic bilateral B16F10 models. After each treatment, the immune profiles and functional examinations were assessed in tumors and tumor draining lymph nodes by flow cytometry, ELISA, and immunofluorescence assays. In orthotopic intraocular melanoma models, the location of the immune infiltrate in the tumor microenvironment (TME) was evaluated after each treatment by multiplex immunohistochemistry and metastatic nodules were monitored.
Results PDT with Ce6-embedded nanophotosensitizer (FIC-PDT) elicited immunogenic cell death and stimulated antigen-presenting cells. In situ immunogenic clearance induced by a combination of FIC-PDT with ripasudil, a clinically approved ROCK inhibitor, stimulated antigen-presenting cells, which in turn primed tumor-specific cytotoxic T cells. Moreover, local immunogenic clearance sensitized PD-1/PD-L1 immune checkpoint blockade responses to reconstruct the TME immune phenotypes of cold tumors into hot tumors, resulting in recruitment of robust cytotoxic CD8+ T cells in the TME, propagation of systemic antitumor immunity to mediate abscopal effects, and prolonged survival. In an immune-privileged orthotopic intraocular melanoma model, even low-dose FIC-PDT and ripasudil combined with anti-PD-L1 antibody reduced the primary tumor burden and prevented metastasis.
Conclusions A combination of localized FIC-PDT and a ROCK inhibitor exerted a cancer vaccine-like function. Immunogenic clearance led to the trafficking of CD8+ T cells into the primary tumor site and sensitized the immune checkpoint blockade response to evoke systemic antitumor immunity to inhibit metastasis, one of the major challenges in UM therapy. Thus, immunogenic clearance induced by FIC-PDT and ROCK inhibitor combined with anti-PD-L1 antibody could be a potent immunotherapeutic strategy for UM.
- immunity
- innate
- immunogenicity
- vaccine
- immunotherapy
- phagocytosis
- programmed cell death 1 receptor
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Uveal melanoma (UM) accounts for only <5% of all melanomas1 and first-line UM treatment options currently include surgical enucleation and plaque brachytherapy, which show effective local tumor control.2 3 Therefore, there is renewed interest in treating UM with a focus on functional outcomes, such as retaining visual function and preserving the eye without causing adverse side effects. Moreover, although UM is a rare disease and treatment options are successful in removing early-stage UM, the prognosis of patients with UM is poor, with nearly 50% of patients with UM developing metastatic disease.4 Therefore, it is crucial to target primary UM and prevent further metastasis simultaneously. Here, we suggest a novel and optimal therapeutic treatment regimen for patients with UM, lowering the burden on patients, and curing or preventing lethal metastatic disease by inducing systemic anti-tumor immunity. To achieve whole antitumor immunity, we previously proposed intrinsic cancer vaccination5 induced by combination of immunogenic cell death (ICD) and phagocytosis enhancement of antigen presenting cells (APCs) which initiates innate immune reactions leading to the production of tumor-specific cytotoxic T cells that can be further activated by immune checkpoint blockade (ICB).6
Photodynamic therapy (PDT) is a minimally invasive, topical treatment for superficial cancer. Although PDT has been approved and used for various cancer types, including amelanotic melanoma, lung and bladder tumors,7 8 numerous current photosensitizers show limited therapeutic efficacy against melanoma because of its malignancy and melanin pigmentation, which generally limits the efficacy by absorbing light across broad wavelengths.9 However, some photosensitizers, such as chlorins, including chlorin e6 (Ce6) and verteporfin, are effective and well-tolerated antimelanoma agents that are unaffected by optical interference caused by melanin.10 11 In clinical trial studies, PDT employing these photosensitizing agents and their derivatives exhibited excellent antitumor effects and safety in patients with various cancer types.12 13 In our previous study, a Ce6-embedded nanophotosensitizer (FIC) was formulated with Pluronic F-127 and iodine-rich diatrizoic acid, which are biocompatible and used clinically to increase photodynamic efficacy.14 Recently, PDT with certain photosensitizing agents has been studied for its applicability in immunotherapeutic approaches because of its potential to induce ICD in cancer cells.15 We hypothesized that PDT with Ce6-embedded nanophotosensitizer (FIC-PDT) is an excellent candidate as an effective ICD agent targeting melanoma cells and subsequently stimulating APCs.
Rho-kinase (ROCK) is a downstream effector of small GTPase and regulates cellular functions and structures by organizing the actin cytoskeleton.16 ROCK signaling is also involved in various tumorigenic pathways.17 Ripasudil, a ROCK inhibitor, has been clinically approved for treating glaucoma and ocular hypertension in the form of an eye drop solution. Based on the immune-stimulatory effects of ROCK inhibitors, including enhancing the phagocytic activity of APCs and their ability to processing tumor antigens, and immediate cross-priming of naïve T cells,18 ripasudil can be repurposed as an antitumor compound.
In the present study, we evaluated the potency of FIC-PDT as an efficient ICD inducer and the phagocytic activity of APCs against immunogenically dying tumor cells on treatment with ripasudil. We also observed potent antitumor immune responses in a syngeneic melanoma model by promoting the antigenicity and adjuvanticity of dying tumor cells and inducing immunogenic phagocytosis after combined-treatment with FIC-PDT and ripasudil. Moreover, combination treatment with FIC-PDT and ripasudil enhanced the therapeutic efficacy of α-PD-L1 antibody treatment of mice bearing highly aggressive cold B16F10 tumors resistant to ICB therapy.19 Furthermore, we validated local immunogenic clearance induced by FIC-PDT and ROCK inhibition in combination with PD-1/PD-L1 blockade therapy to evoke systemic therapeutic efficacy and convert the tumor microenvironment (TME) immune phenotypes of cold tumors into hot tumors in both an abscopal model and immune-privileged orthotopic model. Overall, these findings suggest a new immunotherapeutic strategy for UM that employs clinically applicable therapeutic tools and components that target the primary tumors and induce systemic antitumor immunity to inhibit metastasis, the present major problem in UM therapy.
Methods
Cell culture
B16F10 melanoma cells and CT26 colon cancer cells were obtained from the American Type Culture Collection and 92.1 UM cells were kindly provided by Dr Hyun Woo Park, Yonsei University, Seoul. Cells were cultured in DMEM and RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% antibiotics under 5% CO2 at 37°C.
Bone marrow (BM) cells were obtained from 6 to 8 week-old C57BL/6 and BALB/c mice. Bone marrow-derived macrophages (BMDMs) were differentiated in the presence of 20 ng/mL M-CSF (PeproTech) for 7 days, and bone marrow-derived dendritic cells (BMDCs) were differentiated using 200 ng/mL Flt-3 ligand (R&D Systems) and 0.1% β-mercaptoethanol for 10 days.
In vitro PDT and immunogenic cell death
Cells at a 60%–80% confluence were treated with various concentrations of FIC for 4 hours. After replacing the media containing FIC with conditioned media, B16F10 and CT26 cells were irradiated using an LED (633 nm, 65 mW/cm2) for 3 min, and 92.1 cell was irradiated for 30 s.
Apoptosis was examined by flow cytometric analysis. Briefly, PDT-treated cells were incubated for 24 hours. The media and adherent cells were collected and resuspended in 1×binding buffer. Cells were incubated with a mixture of 1.5 µL of fluorochrome-conjugated Annexin V (AdipoGen) and 1 µL of propidium iodide (PI) staining solution (Thermo Fisher Scientific) for 15 min at room temperature (RT) in the dark. Finally, 400 µL 1× binding buffer was added to each sample, and the samples were analyzed by flow cytometry.
For CRT detection, following 1 hour of incubation after PDT treatment, the cells were collected, washed with Phosphate buffered saline (PBS), and fixed with 0.25% paraformaldehyde for 5 min. The cells were washed with PBS and incubated in 3% bovine serum albumin containing PBS blocking buffer for 30 min at 4°C; this was followed by adding anticalreticulin primary antibody (ab2907, Abcam) to each sample. The samples were stained with Alexa Fluor 488-conjugated antibody (711-545-152, Jackson ImmunoResearch) and analyzed by flow cytometry with gating on PI-negative cells.
To detect high-mobility group box 1 (HMGB1) and heat shock protein 70 (HSP70) release, after PDT treatment, the supernatants were collected. Equal amounts of protein were measured using the Bradford protein assay, and samples were resolved by SDS-PAGE. Following gel electrophoresis, the proteins were transferred to nitrocellulose membranes (Bio-Rad) and preblocked in 5% skim milk in Tris-buffered saline containing Tween 20. The membranes were incubated with HMGB1 or HSP70 primary antibodies (ab18256 and ab181606, Abcam) overnight, washed, and incubated with secondary antibody (a0545, Sigma-Aldrich) for 1 hour at RT. Finally, the chemiluminescent substrate was added to visualize the blots.
ATP was evaluated in the medium at 4 hour post-PDT treatment using a luciferin-based ENLITEN ATP assay (Promega) according to the manufacturer’s manual.
Phagocytosis assay
APC phagocytosis assay was evaluated by flow cytometry and immunofluorescence as described previously.20 Briefly, BMDMs or BMDCs were stained with green 5-chloromethylfluorescein diacetate (CMFDA) (1 μM; Thermo Fisher Scientific) before coculture with cancer cells, and incubated with or without ripasudil (30 μM; Selleckchem) for 1 hour and washed. For flow cytometric analysis, FIC-PDT treated or non-treated cancer cells were labeled with CellTracker Deep Red (2 μM; Thermo Fisher Scientific) and for immunofluorescence, cancer cells were stained with pH-sensitive red pHrodo succinimidyl ester (pHrodo SE) (100 μM for PDT-treated cancer cells; 60 μM for non-treated cancer cells; Invitrogen). Cancer cells were added to BMDMs or BMDCs at a ratio of 1:2 and coincubated for 30 min.
Animals and in vivo tumor models
Male C57BL/6 mice (6–8 weeks old) were purchased from Orient Bio and maintained at a specific-pathogen free animal facility of the Korea Institute of Science and Technology (KIST). Animal studies were conducted following the guidelines of the Institutional Animal Care and Use Committee of the KIST. A total of 5×105 B16F10 cells were subcutaneously inoculated in the left flanks of the mice. To establish a bilateral tumor model for evaluating abscopal effects, at 3 days postprimary tumor inoculation, 3×105 B16F10 cells were inoculated into the contralateral flank of each mouse. On day 6 after tumor inoculation, when the size had reached 50–80 mm3 and on day 8, FIC (0.6 µg) was injected intratumorally, and after 1 hour, the mice were irradiated using an LED for 30 min. From day 6, ripasudil was administered into tumors six times daily by intratumoral injection. For combination therapy, at 1 hour post-PDT treatment, ripasudil was administered to the mice. In some experiments, mice were injected with 10 mg/kg anti-PD-L1 monoclonal antibody (10F.9G2; BioXCell) intraperitoneally, 4 times at 2-day intervals. Tumor volumes were calculated using calipers and the formula volume (mm3)=d2×D/2. Tumor volume was measured every 3 days, and the mice were euthanized when the tumor dimension exceeded 20 mm or signs of impaired health were observed in the mice.
For orthotopic intraocular melanoma model, 1×104 B16F10 cells/2 μL PBS were inoculated into the subretinal space of the right eye of each C57BL/6 mouse. To prepare a needle track, a 30-gage needle was introduced behind the limbus to access the choroid, and the cell suspension was inoculated using a Hamilton microliter syringe (87900, Reno) through the needle track. On day 10 after inoculation, FIC (0.06 μg) was injected intratumorally, and after 1 hour, the mice were irradiated using a laser (655 nm, 300 mW/cm2) for 10 min. From day 10, ripasudil was administered into the tumors three times daily by intratumoral injection. Anti-PD-L1 antibody was intraperitoneally injected into the mice every other day for a total of 4 times starting on day 12. On day 18, the mice were sacrificed, and their tumors and lungs were excised.
Immunofluorescence assays
Mice were sacrificed and their tumors were harvested. Sectioned and blocked samples were probed with the following antibodies at 4°C overnight: for ICD analysis, anticalreticulin, anti-HMGB1, and anti-HSP70 (ab2907, ab18256, and ab181606, Abcam); for assessment of CD8 infiltration, anti-CD8a (550281, BD Pharmingen); and for evaluation of PD-L1 expression in TME, anti-PD-L1 (AF1019, R&D Systems). Samples were washed with PBS three times and incubated with Alexa Fluor 488-conjugated antibody for 1 hour at RT. After three washes, mounting medium with DAPI was used to stain the nuclei. Images were visualized using an immunofluorescence microscope.
Single-cell preparation and flow cytometric analysis
Tumors and tumor draining lymph nodes (TDLNs) were harvested from B16F10 bearing mice. Tumor tissues were enzymatically dissociated using a tumor dissociation kit (Miltenyi Biotec) and GentleMACS dissociator (Miltenyi Biotec), and TDLNs were gently mashed. The digested tumors and TDLNs were filtered through a 40 µm strainer. Red blood cells were eliminated using red blood cell lysis buffer (BioLegend). For tumor cell analysis, dead cells were removed using a dead cell removal kit (Miltenyi Biotec).
For flow cytometric analysis, the following antibodies were purchased from BioLegend: anti-CD16/CD32 (2.4G2), anti-F4/80 (BM8), anti-CD11c (N418), anti-CD40 (3/23), anti-CD86 (GL-1), anti-CD45.2 (104), anti-CD3 (17A2), anti-CD8a (53–6.7), anti-CD274 (10F.9G2), anti-CD103 (2E7), anti-CD44 (IM7), anti-Granzyme B Recombinant (QA16A02), anti-CD4 (GK1.5), anti-CD25 (3C7), anti-Ki-67 (16A8), and anti-IFN-γ (XMG1.2). The CytoFix/CytoPerm kit (BD Biosciences) was used for intracellular staining. Flow cytometric analysis was performed using an Accuri C6 platform (BD Biosciences), and data were analyzed with FlowJo (v10) software (TreeStar).
Ex vivo IFN-γ detection
TDLNs were isolated from B16F10 cell-bearing mice at 15 days post-tumor inoculation and dissociated cells were seeded into 96-well round-bottom plates at 5×105 cells/well. 5×103 UV-irradiated B16F10 cells and IL-2 (100 ng/mL) were added to the seeded TDLN cells for 24 hours for stimulation. The supernatants were collected, and a mouse IFN-γ Quantikine ELISA Kit (R&D Systems) was used to detect IFN-γ production according to the manufacturer’s instructions.
Multiplex immunohistochemistry, image acquisition, and analysis
Formalin-fixed paraffin-embedded tissue blocks were obtained from the orthotopic mouse model and cut into 4-μm-thick sections. Prepared slides were stained with primary antibodies for Ki-67 (ab16667, Abcam), F4/80 (NB600-404, Novus), PD-L1, CD4, CD8 (25 229S, 98 941S, 13 684S, Cell Signaling Technology), FOXP3 (14-5773-82, Invitrogen), for 30 min and then each protein was detected using HRP-polymer conjugated secondary antibodies (ab214880, Abcam; MP-7444, Vector Laboratories). Tyramide signal amplification was performed to visualize immunofluorescence signals using Opal seven-color immunohistochemistry (IHC) Kit dyes (FP1502, PerkinElmer). DAPI-stained nuclei were continuously visualized, and the sections were cover-slipped.
Statistical analysis
Student’s unpaired two-tailed t-test was performed to compare two experimental groups, and other statistical analyses were performed using GraphPad Prism 5.0 (GraphPad). Data were analyzed by one-way analysis of variance, followed by Tukey’s posthoc test. Statistical significance is denoted by *, **, and, *** for p<0.05,<0.01, and <0.001, respectively. In vitro data are presented as the mean±SD, and in vivo data are presented as the mean±SE of the mean.
Results
FIC-PDT elicits immunogenic cancer cell death
We evaluated the in vitro photocytotoxicity of FIC-PDT towards mouse melanoma B16F10, human UM 92.1 and mouse colon cancer CT26 cells. Cells were treated with various concentrations of FIC and irradiated with LED. The cytotoxic effects of FIC-PDT increased with increasing doses of FIC; however, cells incubated with FIC but without LED irradiation showed no obvious signs of cell death (figure 1A, online supplemental figures S1A, S2A, and S3A).
Supplemental material

Analysis of cell death induction and detection of hallmarks of immunogenic cell death triggered by FIC-PDT in B16F10 cells. (A) Dying or dead B16F10 cells after FIC-PDT treatment with varying concentrations of FIC. The percentage represents early (PI-negative) and late (PI-positive) apoptosis. (B) Flow cytometric analyses of CRT expression on the surface of cell membrane-gated PI- intact cells. Data are presented as the relative mean fluorescence intensity. (C) Immunoblotting of HMGB1 and HSP70 released in conditioned media after FIC-PDT treatment or not. (D) Quantification of secreted ATP from FIC-PDT treated B16F10 cells. One-way ANOVA followed by Tukey’s posthoc test was used for statistical analysis; *p<0.05, **p<0.01, ***p<0.001. Data are presented as the mean±SD. ANOVA, analysis of variance; CRT, Calreticulin; FIC-PDT, PDT with Ce6-embedded nanophotosensitizer; PDT, photodynamic therapy; PI, propidium iodide.
Next, we examined whether FIC-PDT-induced cell death is immunogenic. We investigated essential Damage associated molecular patterns (DAMPs) after treatment with FIC-PDT. Figure 1B and online supplemental figures S2B and S3B show that Calreticulin (CRT) expression was increased by PDT in an FIC concentration-dependent manner; without LED irradiation, there were no fold-changes in CRT expression in cells treated with different doses of FIC. To further identify CRT exposure on the cell surface, confocal microscopy examination was performed. The images revealed that FIC-PDT treatment promoted CRT translocation to the cell membrane (online supplemental figure S1B). We also evaluated the release of HMGB1 and HSP70, which are APC-stimulating proteins, from FIC-PDT treated cells by immunoblotting. Clear bands corresponding to each protein were observed for FIC-PDT-treated supernatants (figure 1C, online supplemental figures S2C and S3C). Moreover, cells treated with FIC-PDT secreted higher levels of ATP than basal and non-irradiated cells (figure 1D, online supplemental figure S2D). Collectively, these results demonstrate that FIC-based PDT represents a candidate therapeutic modality involving ICD induction.
Combination of FIC-PDT with ripasudil enhances engulfment of cancer cells by APCs
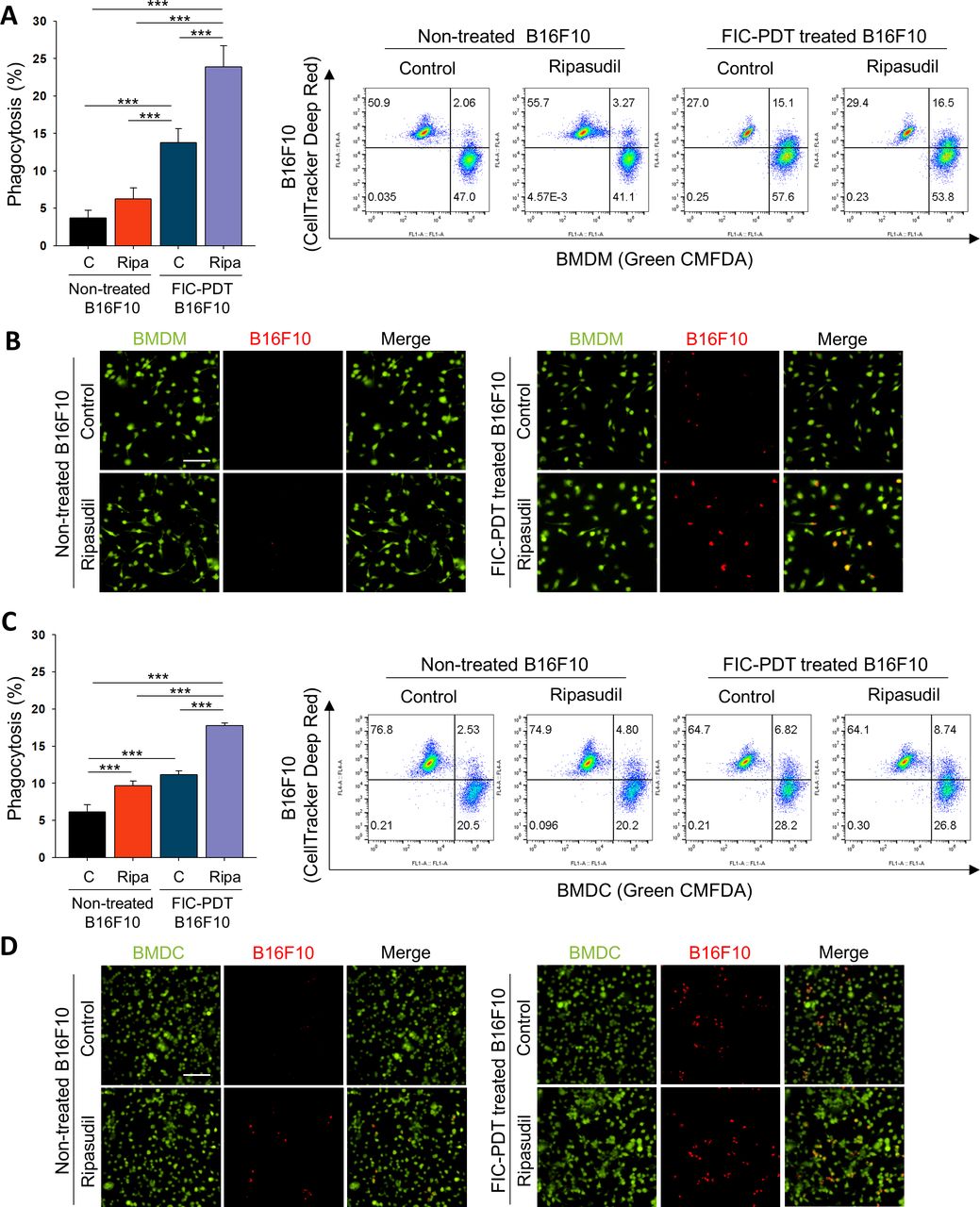
We next evaluated the phagocytic activity of APCs on treatment with ripasudil. We performed phagocytosis assays with BMDMs and BMDCs obtained from C57BL/6 and BALB/c mice (online supplemental figure S4A). CRT expression on dying cell surfaces acts as an “eat-me” signal, which guides phagocytes to clear dying cells.21 We further treated BMDMs and BMDCs with ripasudil to block Rho/ROCK signaling, which is a negative regulator of phagocytosis.22 FIC-PDT-treated or non-treated B16F10 cells were co-incubated with BMDMs or BMDCs non-treated or pretreated with ripasudil, after which phagocytosis assays were performed. Flow cytometry revealed that FIC-PDT-treated cancer cells were engulfed by BMDMs and BMDCs more frequently than cancer cells not undergoing ICD, respectively. Furthermore, in the presence of ripasudil, the phagocytic percentages of BMDMs and BMDCs were elevated (figure 2A,C). To confirm the significant engulfment of cancer cells by phagocytes, cancer cells with or without ICD induction were labeled with acid-sensitive pHrodo red dye, and phagocytosis was analyzed by fluorescence microscopy. In accordance with the results obtained by flow cytometry, treatment with both FIC-PDT and ripasudil promoted the phagocytosis of cancer cells by BMDMs and BMDCs (figure 2B,D, online supplemental figures S4B and S5). These results demonstrate that combining ICD induction by FIC-PDT and ROCK inhibition significantly augments the phagocytic activity of APCs, suggesting that the innate immune response against tumor cells can be efficiently induced by combining PDT-mediated ICD with ROCK inhibition.
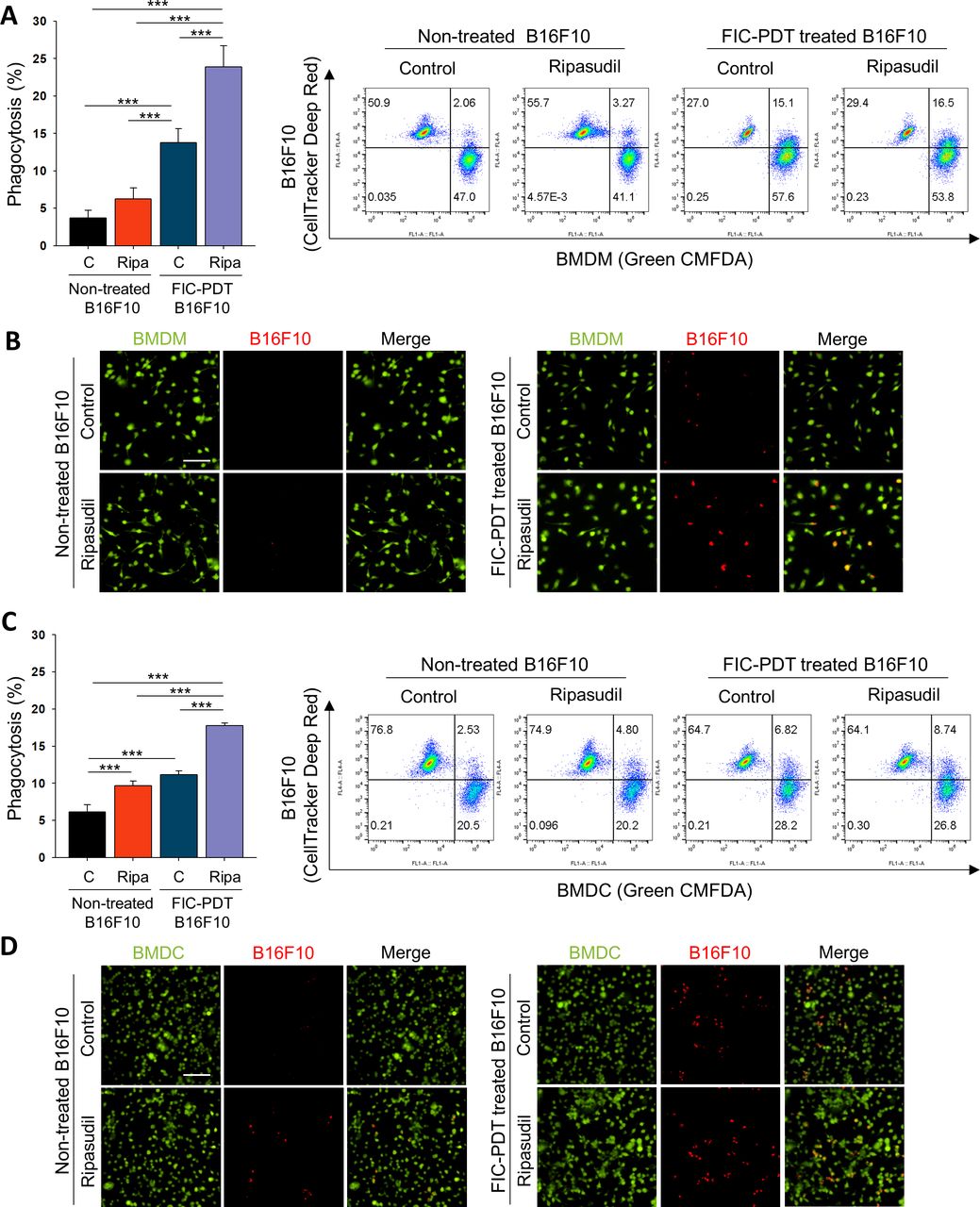
Tumor cell clearance activity of phagocytes was enhanced after ROCK signaling inhibition of BMDMs and BMDCs. (A–D) Phagocytic activity of (A,B) BMDMs and (C,D) BMDCs after ripasudil (30 μm) treatment or non-treatment for 1 hour and further cocultured with non-treated or FIC-PDT pretreated B16F10 cells for 30 min. Engulfed B16F10 cells by BMDMs and BMDCs were assessed by flow cytometry (A,C) and CLSM (B,D). One-way ANOVA followed by Tukey’s posthoc test was used for statistical analysis; *p<0.05, **p<0.01, ***p<0.001. Data are presented as the mean±SD. ANOVA, analysis of variance; BMDC, bone marrow-derived dendritic cell; BMDM, bone marrow-derived macrophage; CLSM, Confocal laser scanning microsope; FIC-PDT, PDT with Ce6-embedded nanophotosensitizer; PDT, photodynamic therapy; ROCK, rho-kinase.
Combined therapy with FIC-PDT and ripasudil synergistically promotes antitumor T cell responses by dendritic cell stimulation
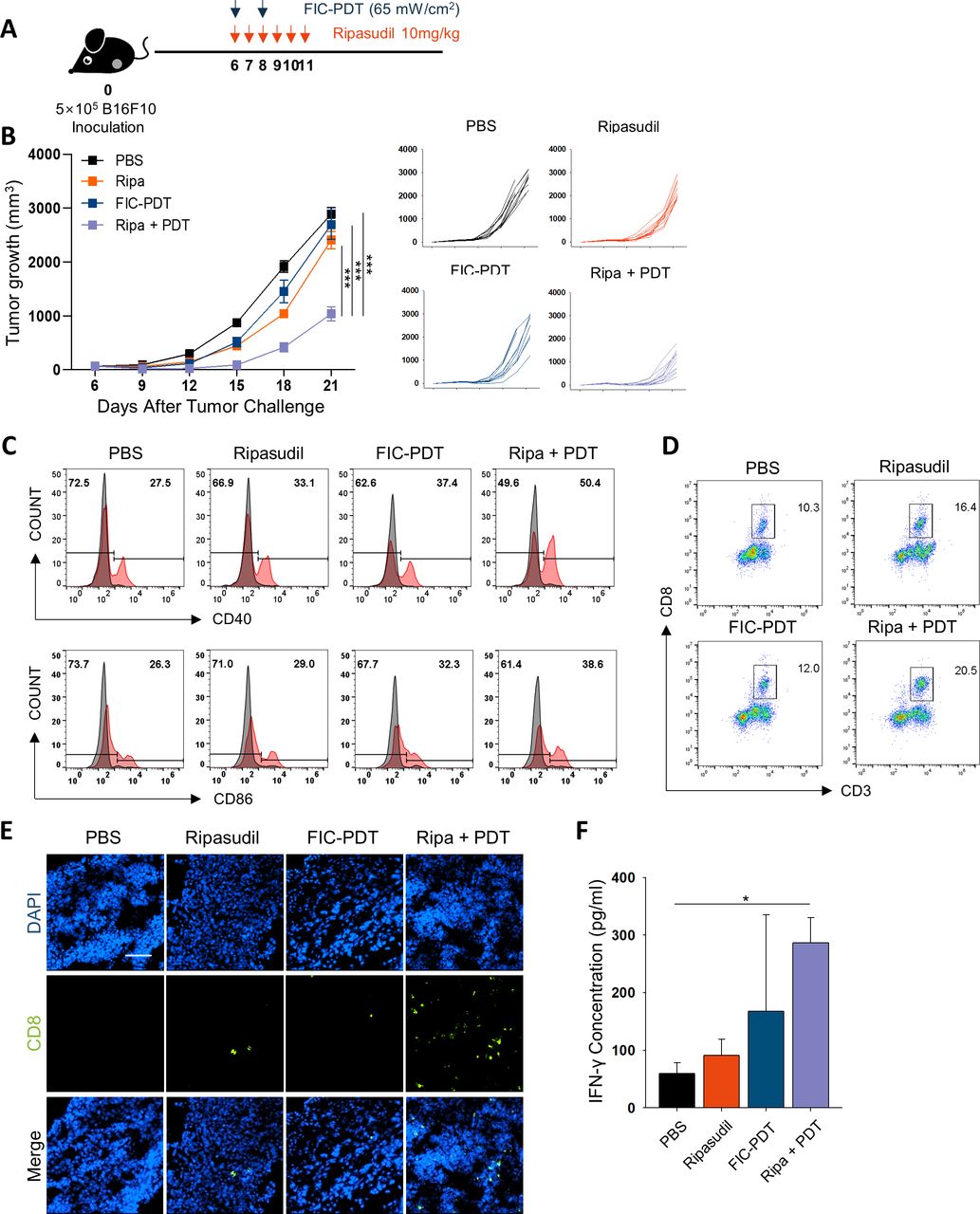
To evaluate the antitumor efficacy of combined FIC-PDT and ripasudil therapy in terms of the induction of in situ immunogenicity, we established a B16F10 tumor-bearing C57BL/6 mouse model. FIC-PDT and ripasudil treatment was administered following the schedule presented in figure 3A. Only slight beneficial therapeutic outcomes were observed when either FIC-PDT or ripasudil alone was used for treatment. Interestingly, combination therapy of FIC-PDT and ripasudil achieved significant synergistic antitumor effects compared with those of individual treatments, inhibiting tumor growth by >65% (figure 3B). In addition, body weight changes were not observed in any group, indicating that combination treatment had minimal cytotoxic effects (online supplemental figure S6A). Neither FIC nor LED irradiation alone affected tumor growth (online supplemental figure S6B, C).

Evaluation of antitumor efficacy and immune response in vivo after combined-treatment with FIC-PDT and ripasudil. (A) Treatment schedule of FIC-PDT and ripasudil monotherapy and combination therapy. (B) Tumor growth in B16F10 tumor-bearing mice after various treatments (n=9–12 per group). (C) Flow cytometry analyses of the percentage of costimulatory molecules CD40 and CD86 in CD11c+ cells from TDLNs (n=3 per group). (D, E) Tumor-infiltrating CD8+ T cells in tumor site analyzed by flow cytometry (D) or under fluorescent microscope (E) (n=3–6 per group). (F) T cells from TDLNs of B16F10 tumor-bearing mice were stimulated with irradiated B16F10 tumor lysates and IFN-γ production was examined by ELISA. (n=3–4 per group). One-way ANOVA followed by Tukey’s posthoc test was used for statistical analysis; *p<0.05, **p<0.01, ***p<0.001. Data are presented as the mean±SEM. ANOVA, analysis of variance; FIC-PDT, PDT with Ce6-embedded nanophotosensitizer; PDT, photodynamic therapy; TDLN, tumor draining lymph node.
To verify FIC-PDT-induced ICD in vivo, we excised tumors from the mice 1 hour after the last FIC-PDT treatment and examined hallmarks of ICD. We found that DAMP signals, such as CRT, HMGB1, and HSP70 proteins, were significantly upregulated in FIC-PDT-treated tumors (online supplemental figure S7A). As increased immunogenic danger signals are expected to promote dendritic cell (DC) maturation, we analyzed CD11c+ DCs in TDLNs by flow cytometry. Whereas FIC-PDT monotreatment only slightly upregulated the expression of T cell costimulatory molecules such as CD40 and CD86, combination therapy notably upregulated the percentage and relative mean fluorescence intensity of the markers (figure 3C, online supplemental figure S7B).
To investigate the infiltration of CD8+ T cells into the TME mediated by activated APCs 23 we isolated tumors and evaluated tumor-infiltrating CD8+ T cells by flow cytometry. Combination treatment with ripasudil and FIC-PDT markedly increased the proportion of CD8+ T cells (figure 3D, online supplemental figure S7C). To visually estimate the number of CD8+ T cells, we performed immunofluorescent staining of tumor tissue samples. The images revealed similar results to those obtained by flow cytometric analysis in that the number of CD8+ T cells infiltrated into tumor tissues was highest in the combined-treatment group (figure 3E, online supplemental figure S7D). We next assessed the cytotoxic function of CD8+ T cells against B16F10 cells by evaluating inflammatory IFN-γ production. Significantly higher IFN-γ secretion was observed in the combined-treatment group (figure 3F). These results demonstrate that immunogenic clearance induced by combined-treatment with FIC-PDT and ripasudil generates effective anti-tumor T cell immune responses.
Combined-treatment sensitizes the therapeutic efficacy of ICB
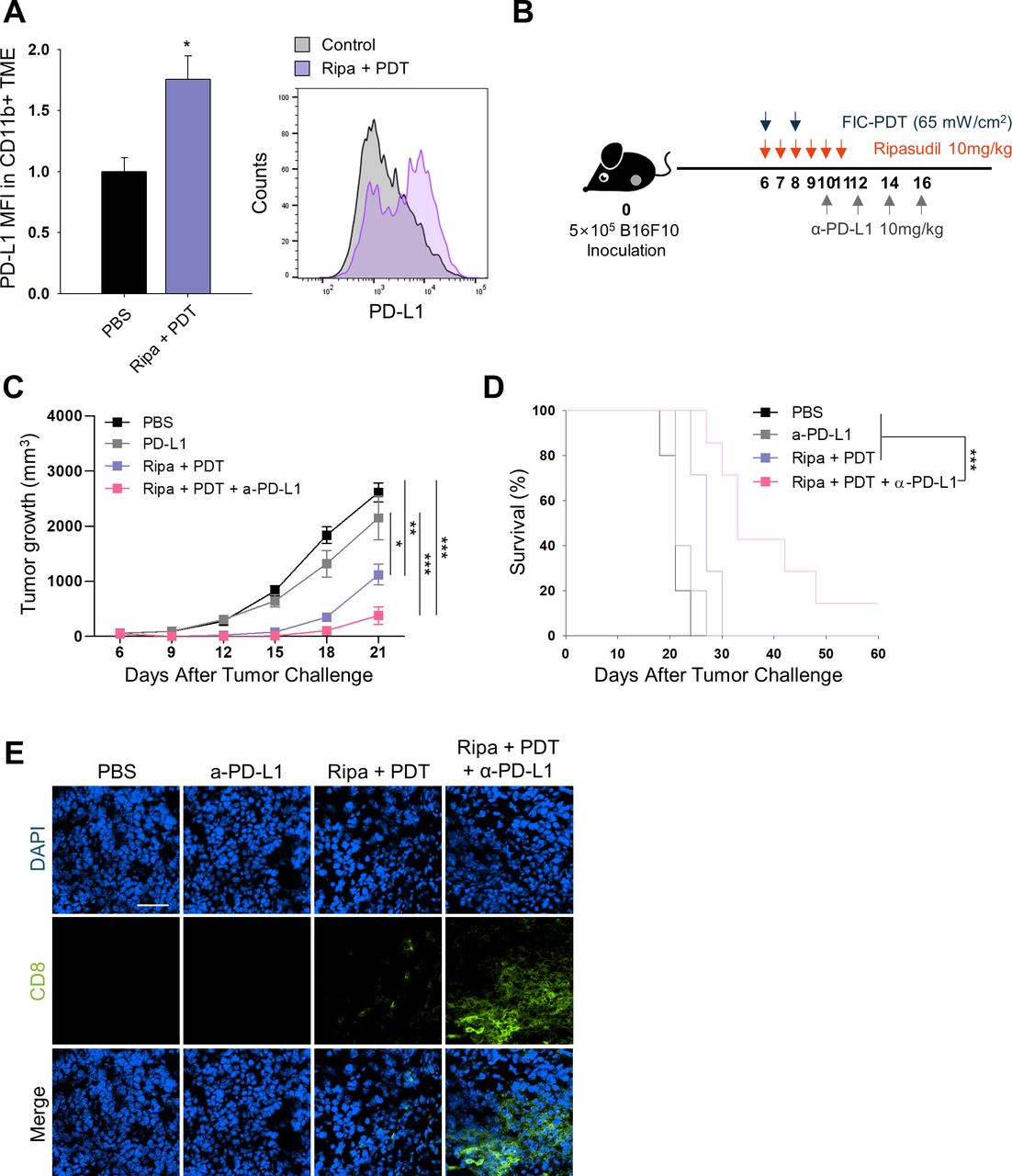
After demonstrating increased CD8+ T cell numbers in the TME and their activation on combined-treatment with ripasudil and FIC-PDT, we next evaluated whether the treatment could enhance tumor responsiveness to ICB. We assessed PD-L1 expression in tumors in the PBS group and Ripa +PDT group. Flow cytometry and immunofluorescent analysis demonstrated that overall PD-L1 expression was not significantly altered on combination treatment (online supplemental figure S8A,B); however, when defined for myeloid cells, combined-treatment upregulated the levels of PD-L1 by 1.7-fold in myeloid cells (figure 4A). PD-L1 expression in the host adaptive immune system has been reported to predict therapeutic outcomes in PD-1/PD-L1 blockade.24 25 Therefore, our combination treatment may sensitize tumors to respond to ICB therapy. To examine antitumor responses to combined FIC-PDT and ripasudil treatment with α-PD-L1 antibody as per the schedule indicated in figure 4B. Triple-combination therapy remarkably amplified the therapeutic potential of ICB by inhibiting tumor growth (figure 4C). Moreover, triple-combination therapy prolonged survival rates, and one mouse treated with this therapy remained completely tumor-free until day 60 (figure 4D). This result was impressive, considering that the B16F10 melanoma model is highly resistant to immunotherapy.19 Improved tumor growth inhibition was accompanied by higher CD8+ T cell infiltration into the TME in the group treated with triple-combination therapy compared with that in the group treated with α-PD-L1 antibody alone, or treated with Ripa +PDT (figure 4E). Taken together, these findings indicate that triple-combination therapy can facilitate T cell infiltration into the TME and extend T cell activation and prevent T cell exhaustion.
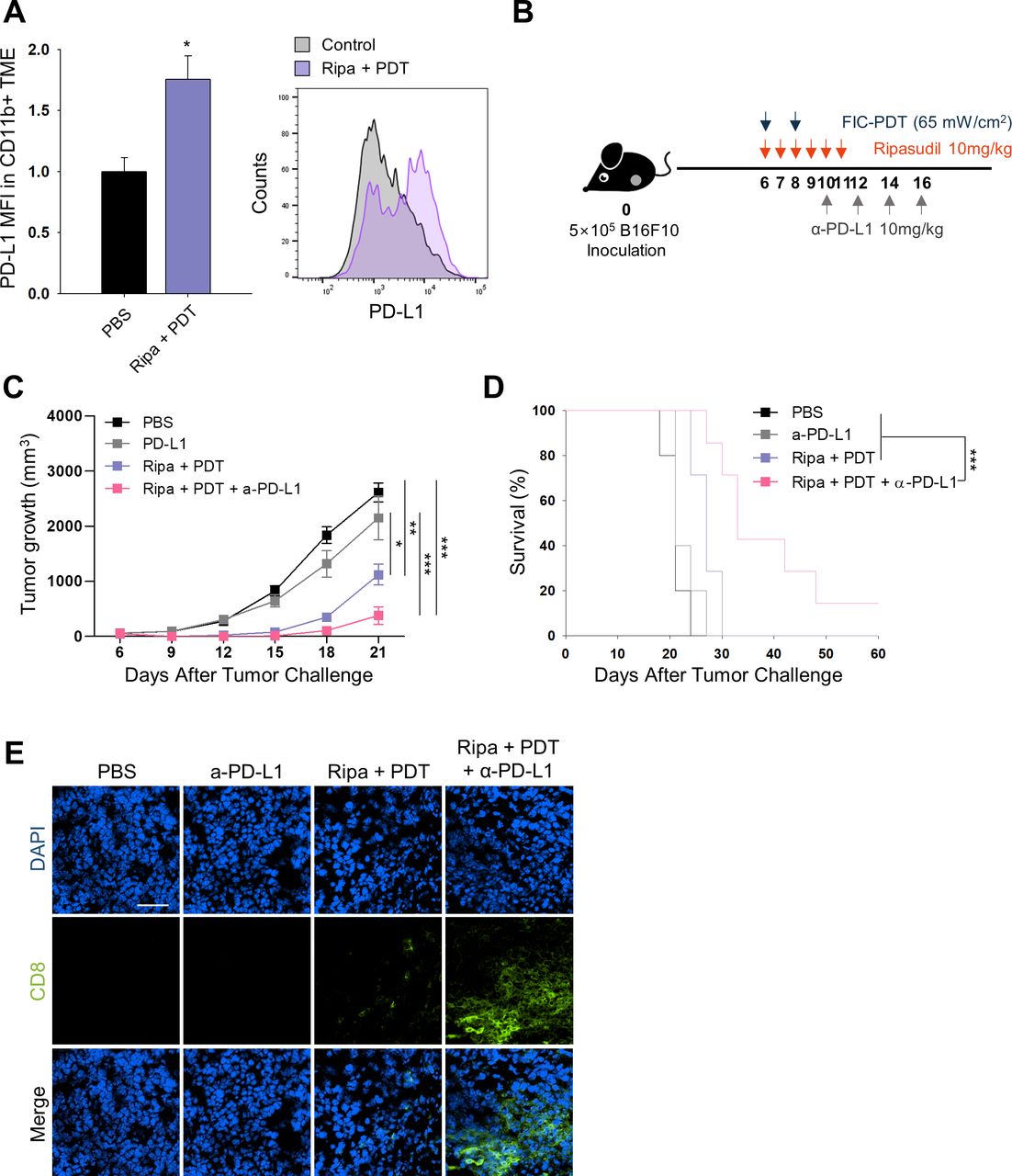
In vivo PD-L1 expression and combined therapy with FIC-PDT, ripasudil, and anti-PD-L1 antibody. (A) PD-L1 expression of CD11b+ cells in TME assessed by flow cytometry (n=3 per group) (B) Treatment schedule of FIC-PDT, ripasudil and α-PD-L1 antibody. (C) Tumor growth curves (n=8–10 per group) and (D) survival curves (n=5–7 per group) in B16F10 tumor-bearing mice after various treatments. (E) Tumor-infiltrating CD8+ T cells in tumor under fluorescent microscope (n=4 per group). One-way ANOVA followed by Tukey’s posthoc test was used for statistical analysis; *p<0.05, **p<0.01, ***p<0.001. Data are presented as the mean±SEM. Survival was determined using the log‐rank test. ANOVA, analysis of variance; FIC-PDT, PDT with Ce6-embedded nanophotosensitizer; PDT, photodynamic therapy; TME, tumor microenvironment.
Systemic antitumor immune response-mediated abscopal effect is induced by the triple-combination therapy
We next predicted that local administration of combined FIC-PDT and ripasudil therapy could accomplish in situ intrinsic cancer vaccination and that combining this therapy with α-PD-L1 may induce systemic antitumor immune responses. To verify this hypothesis, we established a bilateral B16F10 syngeneic model and tested the abscopal effects of the triple-combination therapy. As presented in figure 5A, FIC-PDT and ripasudil were locally administered at the primary tumor site and α-PD-L1 antibody was then systemically injected. The triple-combination group showed a significantly higher capability to delay the growth of both primary and secondary tumors compared with those of other groups (figure 5B, online supplemental figure S9A), demonstrating that systemic antitumor responses against B16F10 were induced. Next, we evaluated the recruitment of CD103+ DCs in TDLN, a key mediator of CD8+ T cell priming and trafficking to tumor sites.26 27 As expected, the percentage of CD103+ DCs was higher in the triple-combination group than in the other groups, suggesting increased priming of CD8+ T cells in TDLN (figure 5C). Consistent with these results, we observed an increased frequency of antigen-experienced CD44+CD8+ T cells in TDLN after triple-combination treatment (figure 5D).

Abscopal effect of local administration of FIC-PDT and ripasudil combined with α-PD-L1 antibody. (A) Treatment schedule for abscopal animal model after various treatments. (B) Tumor growth curves of primary and secondary tumors (n=8–12 per group). (C,D) Percentage of CD103-expressing DCs (C) and CD44+CD8+ T cells (D) in TDLNs. (E,F) Percentage of CD8+ T cells (E) and granzyme B+ T cells (F) in primary and secondary tumors was quantified by flow cytometry. (G) Ratio of CD8+ T cells over regulatory (CD25+CD4+) T cells in primary and secondary tumors. One-way ANOVA followed by Tukey’s posthoc test was used for statistical analysis; *p<0.05, **p<0.01, ***p<0.001. Data are presented as the mean±SEM. ANOVA, analysis of variance; FIC-PDT, PDT with Ce6-embedded nanophotosensitizer; PDT, photodynamic therapy; TDLN, tumor draining lymph node.
Furthermore, we explored the infiltration of T cells into primary and secondary tumor sites and tested their cytotoxic effects and proliferation capacity. Consistent with previous results, an enlarged population of CD8+ T cells was identified in both primary and secondary tumors in the triple-combination treatment group (figure 5E). There was a larger granzyme B+ population of CD8+ T cells after Ripa+PDT treatment compared with after PBS and α-PD-L1 treatment; moreover, after triple-combination therapy, the population was further expanded at both the primary and secondary tumor sites (figure 5F). Additionally, both IFN-γ+ and Ki-67+ populations of CD8+ T cells were identified in primary tumors after combination treatment (online supplemental figure S9B,C), indicating that cytotoxic and proliferating CD8+ T cells were more abundant in tumor tissues treated with double-combination and triple-combination therapy. Finally, we analyzed regulatory T (Treg) cells from both types of tumor sites in mice. There was a decreasing tendency of the numbers of Treg cells in both types of tumor sites after Ripa+PDT treatment and Ripa+PDT+α-PD-L1 treatment (online supplemental figure S9D). In addition, the ratio of tumor-infiltrating CD8+ T cells over Treg cells was considerably increased using the triple-combination approach (figure 5G), suggesting that it improved the immune contexture of the TME to favor inhibition of tumor growth. Thus, our results demonstrate that triple-combination treatment successfully evoked systemic anti-tumor immunity and converted the cold TME into a hot TME.
Triple-combination therapy reconstructs the immunosuppressive TME and prevents metastasis in the orthotopic intraocular melanoma model
As the therapeutic efficacy of immunotherapy is highly dependent on the properties of the host organ site, we employed an orthotopic intraocular melanoma model to verify the therapeutic efficacy of our triple-combination therapy. To specifically target focused ocular tumor regions, laser-based FIC-PDT treatment was applied and showed similar cytotoxic effects and aspects of ICD induction to those of LED-based FIC-PDT treatment (online supplemental figure S10A,B). In this model, FIC-PDT treatment and ripasudil injection were applied to tumor sites as indicated in figure 6A. Additionally, because both treatment modalities were directly injected into tumor sites inside the eye, FIC and ripasudil at a 10% dose were used. Although the Ripa+PDT group treated with low doses showed slight therapeutic effect, triple-combination treatment inhibited tumor growth in the orthotopic model, similar to previous results obtained using a syngeneic model (figure 6B, online supplemental figure S11A). H&E staining revealed impaired tumor structures after Ripa+PDT and Ripa+PDT+α-PD-L1 treatments (online supplemental figure S11B).

Antitumor effect, immune context conversion, and metastasis inhibition after each treatment in orthotopic intraocular melanoma model. (A) Treatment schedule for orthotopic intraocular melanoma model. (B) Tumor weights excised from orthotopic mouse model on day 20 after tumor inoculation. (C,D) Observable metastatic nodules in the lung. Representative images of lungs in each group (C) and number of metastatic nodules in the lung after various treatments (D). One-way ANOVA followed by Tukey’s posthoc test was used for statistical analysis; *p<0.05, **p<0.01, ***p<0.001. Data are presented as the mean±SEM. ANOVA, analysis of variance; FIC-PDT, PDT with Ce6-embedded nanophotosensitizer; PDT, photodynamic therapy.
Furthermore, because the occurrence of tumor metastases is the main problem in UM and accounts for low survival rates,4 we investigated whether our combinational treatment strategy could inhibit metastasis and build systemic immunity against tumors. On day 20, we excised the lungs, the primary metastatic site of the B16F10 cell line, from mice treated with PBS, α-PD-L1, Ripa +PDT, and Ripa+PDT+α-PD-L1 to monitor the development of metastatic nodules. Although other groups displayed visible metastatic nodules in the lung, the triple-combination treatment group presented inhibition of metastasis (figure 6C,D). Moreover, the immune expression profiles of numerous markers were investigated by multiplexed IHC, which enables accurate identification of the immune contexture. The tumor site was nearly non-accessible to CD8+ T cells in the PBS group; however, in the α-PD-L1 antibody-treated group, the tumor phenotypes were restored to immune-excluded types, allowing T cells to approach to tumor invasive margins and also forming PD-L1 barriers preventing infiltration into intratumoral regions. Additionally, Ripa+PDT treatment elicited immune cells to traffic into tumors, the numbers of CD8+ T cells were insufficient to combat tumor cells. However, consistent with the syngeneic model, triple-combination treatment afforded tumor infiltration of robust CD8+ T cells (figure 7A). Further, multiplexed IHC images revealed that proliferation of CD8+ T cells and the ratio of CD8+ T cells to FOXP3+ cells were upregulated in tumor beds after triple-combination treatment (figure 7B,C, online supplemental figure S12 A,B). Taken together, these data indicate that the triple-combination treatment strategy provides guidance for its future clinical use in simultaneous targeting of primary UM and metastasis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Population and location of immune cell subsets in tumor using multiplex IHC. (A) Exact location and density of CD8+ cells (green) and PD-L1+ cells (red) in the TME were investigated using multiplex IHC. (B) Tumor tissues were stained with Ki67 (green), PD-L1 (red), FOXP3 (orange), CD8 (white) and nuclei was costained with DAPI (blue). Yellow arrows represents the proliferative CD8+ T cells. (C) Percentage of proliferative CD8+ (Ki67+CD8+) cells. IHC, immunohistochemistry; TME, tumor microenvironment.
Discussion
Although several treatment options are currently available for UM, vision loss and lethal metastatic disease remain the major challenges.4 Particularly, metastasis is directly related to the survival in patients with UM.28 Because metastatic UM shows similarities to cutaneous melanoma (CM), several immunotherapeutic approaches such as ICB have been applied in clinical trials. However, the clinical benefit of ICB is relatively restricted to “hot tumors” with high immunogenicity29 whereas metastatic UM is considered as “cold tumor” with relatively low mutational load.30 This low genetic instability may also cause low PD-L1 positivity in TME;31 thus, immunotherapies for patients with UM has yielded disappointing results to date.32 33 Moreover, no systemic treatment options are available for targeting primary malignancies and metastatic lesions. Therefore, new systemic therapeutic approaches for generating an in situ tumor-vaccine to elicit a systemic and long-lasting antitumor immune response to prevent metastatic disease are urgently needed.
PDT has been suggested as a treatment modality for UM because of its effectiveness, non-invasiveness, and safety in terms of the low incidence of side effects.34–36 Moreover, PDT treatment exhibits tumor-killing effects and induces dying tumor cells to become immunogenic by releasing DAMPs, which are natural immune adjuvants. Therefore, we hypothesized that localized PDT treatment can exert an adjuvant-like function and thereby inhibit metastasis. In our previous study, we developed a heavy-atom-concentrated nanophotosensitizer, FIC, to intensify the photodynamic efficacy of Ce6.14 In the present study, we validated that FIC-PDT induces crucial ICD hallmarks, including CRT, HSP70, HMGB-1, and ATP suggesting that FIC-PDT is a successful ICD inducer. Certain clinical experiences with PDT have revealed effective tumor control in patients with UM,36 37 but their immunological effects in the systemic antitumor effect remained unclear. We first demonstrated that PDT exerts an in situ vaccine-like function in UM to mediate the systemic antitumor response. Additionally, ripasudil, a clinically used drug for glaucoma and ocular hypertension, was repurposed as an enhancer of phagocyte function. Combined strategies for ICD and phagocytosis enhancement, which are the initial stages of cancer immunity cycle, have been reported to produce and proliferate tumor-specific T cells to propagate antitumor immunity.6 18 38 Consistent with earlier studies,6 18 this immunogenic clearance39 induced by FIC-PDT and ripasudil launched adaptive immune responses to initiate and amplify the cancer-immunity cycle and facilitated CD8+ T cells access to immunologically cold TME. Interferon signaling controls PD-L1 expression levels in TME40 and thereby increases in CD8+ cytotoxic T cells induced by immunogenic clearance led to upregulated PD-L1 expression in the TME, particularly on myeloid cells, the predictive biomarker of responsiveness to anti-PD-L1 treatment.25 Therefore, mice treated with immunogenic clearance in combination with anti-PD-L1 antibody showed elevated therapeutic efficacy and prolonged survival rates.
Recently, some immunotherapeutic approaches for metastatic UM, including DC vaccination loaded with tumor-specific peptides,41 adoptive transfer of tumor-infiltrating lymphocytes,42 and a bispecific protein that recognizes tumor-specific antigen and TCR,43 have been explored. These trials are suggested for metastatic UM, which occurs after the treatment of the primary UM; therefore, a systemic treatment option for targeting UM and metastatic UM remains to be identified. In this study, we evaluated tumor-specific systemic antitumor immune responses by establishing an abscopal model. Local administration of FIC-PDT and ripasudil combined with anti-PD-L1 antibody augmented the recruitment of migratory CD103+ DCs, specific subsets of DCs, which are specialized in CD8+ T cell priming and trafficking of tumor-specific antigens.26 44 Consistent with the functions of CD103+ DCs, triple-combination therapy increased the number of antigen-experienced CD8+ T cells in TDLNs and channeled abundant tumor-specific CD8+ T cells to both primary and secondary tumor sites to drive the potent antitumor therapeutic efficacy. Additionally, immune cell function was improved as evident by the enhanced levels of CD8+ T cells, including those of cytotoxic granzyme B+ CD8+ T cells, and altered immune cell composition with increased number of effector cells after triple-combination therapy. Collectively, triple-combination therapy affected the likelihood of a successful clinical response and evoked a systemic antitumor response to prevent or cure metastatic disease.
It is important to select appropriate model considering the immune-privileged nature of the eye. Therefore, we established an orthotopic intraocular melanoma model to estimate the antitumor efficacy of triple-combination therapy. Unfortunately, there is no optimal animal model for UM available to date, particularly for immunological studies. Therefore, we used UM mouse models intraocularly injected with B16F10 CM cells. Although there are limitations related to the different genetic background and metastatic behavior between CM and UM cell lines, UM mouse models injected with B16F10 remain one of the most commonly used in vivo models for evaluating therapeutic candidates.45 To assess our immunotherapeutic strategy, the immune desert phenotype B16F10 cell line with a low mutation burden was used to represent the immune privileged microenvironment.19 46 47 In highly sensitive organs, we used FIC and ripasudil at a 10% dose. Interestingly, despite the immune privilege, tumor growth was successfully regressed by low-dose FIC-PDT and ripasudil in combination with anti-PD-L1 antibody. Low-dose PDT and ripasudil may exert minimal toxicities and maintain vision acuity, which were reported as clinical outcomes of PDT,37 whereas conventional UM treatments are associated with vision-related complications.48
Further, although understanding the immune profile in preclinical animal models is essential, it has not been clarified in orthotopic model. Therefore, we performed multiplex immunofluorescence analysis for immune profiling with multiplexing antibody staining49 to explore the population, density, and distribution of immune cells in the immune privileged intraocular TME after each monotherapy or combination therapy. The results demonstrate that triple-combination treatment altered the indices indicating the success of antitumor immunotherapy: composition, localization, and activation of immune cells in the TME.50 51 Importantly, localized combination treatment with FIC-PDT and ripasudil combined with anti-PD-L1 resolved occurrence of metastatic disease, the most important challenge in achieving effective UM treatment.
Some limitations still remain that the therapeutic efficacy of our strategy for UM was evaluated through CM cells that were inoculated in the ocular site of mice and majority of immunological studies in this paper were evaluated using CM syngeneic models. Since CM cannot represent the pathology of UM due to differences in genetic level to the activity of some signaling pathways, the results in this study should be cautiously interpreted. While CM has common gene mutations in BRAF, NRAS, or NF1 that deregulates ERK pathway, UM carries GNAQ/GNA11, EIF1AX and, BAP1 mutations.52 53 They may drive different tumor outgrowth, organ tropism of metastases, and immune environment. However, since the syngeneic mice model bearing UM is not available, we had to use B16 CM cell line as the next best one, which is commonly used immunocompetent model to investigate tumor progression and immunological study.54 55
Taken together, our findings provide a preclinical background for a promising immunotherapy for UM using clinically available therapeutic tools and components. Our results indicate that local immunogenic clearance induced by FIC-PDT and ripasudil combined with ICB can achieve successful clinical outcome by modulating all hallmarks of tumor-infiltrating immune cells, accompanied by a systemic antitumor immune response to prevent or cure metastatic diseases. With various therapeutic modalities, our combination strategy to induce immunogenic clearance may provide the evidence to overcome various types of cold tumors, including UM to sensitize ICB therapies.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
All animal studies were conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) of the KIST.
Acknowledgments
The authors would like to thank the animal facility of the Korea Institute of Science and Technology (KIST) for their assistance. Also, we are grateful to Dr Hyun Woo Park for providing 92.1 uveal melanoma cell line.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it first published. The provenance and peer review statement has been included.
Contributors SK conceived the study, designed experiment, conducted experiments, analyzed data, and wrote the manuscript. SK and I-SK directed the study, designed experiments and wrote the manuscript. SAK, G-HN, GBK, and YC supported animal experiment and assisted with writing manuscript. SK and YC assisted experiments and provided feedback. MK and CJ provided expertise and feedback.
Funding This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (2017R1A3B1023418), KU-KIST Graduate School of Converging Science and Technology Program, and KIST Institutional Program.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.