Article Text

Abstract
Background The efficacy of cancer immunotherapy can be limited by the poor immunogenicity of cancer and the immunosuppressive tumor microenvironment (TME). Immunologically programming the TME and creating an immune-inflamed tumor phenotype is critical for improving the immune-responsiveness of cancers. Here, we interrogate the immune modulator Flagrp170, engineered via incorporation of a pathogen-associated molecular pattern (ie, flagellin) into an immunostimulatory chaperone molecule, in transforming poorly immunogenic tumors and establishing a highly immunostimulatory milieu for immune augmentation.
Methods Multiple murine cancer models were used to evaluate the immunostimulatory activity, antitumor potency, and potential side effects of Flagrp170 on administration into the tumors using a replication impaired adenovirus. Antibody neutralization and mice deficient in pattern recognition receptors, that is, toll-like receptor 5 (TLR5) and NOD like receptor (NLR) family caspase activation and recruitment domain (CARD) domain-containing protein 4 (NLRC4), both of which can recognize flagellin, were employed to understand the immunological mechanism of action of the Flagrp170.
Results Intratumoral delivery of mouse or human version of Flagrp170 resulted in robust inhibition of multiple malignancies including head and neck squamous cell carcinoma and breast cancer, without tissue toxicities. This in situ Flagrp170 treatment induced a set of cytokines in the TME known to support Th1/Tc1-dominant antitumor immunity. Additionally, granulocyte macrophage colony-stimulating factor derived from mobilized CD8+ T cells was involved in the therapeutic activity of Flagrp170. We also made a striking finding that NLRC4, not TLR5, is required for Flagrp170-mediated antitumor immune responses.
Conclusion Our results elucidate a novel immune-potentiating activity of Flagrp170 via engaging the innate pattern recognition receptor NLRC4, and support its potential clinical use to reshape cancer immune phenotype for overcoming therapeutic resistance.
- tumor microenvironment
- immunotherapy
- T-lymphocytes
- dendritic cells
- vaccination
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
It is now well-established that the cooperative interactions between cancer cells, non-transformed stromal cells and immune cells in the tumor microenvironment (TME) promote carcinogenesis, cancer progression and invasion, as well as their responses to treatments.1 The TME is sculpted towards an immunosuppressive state via an “immunoediting” process, which includes impaired antigen cross-presentation, recruitment of immunosuppressive cells or factors, and overexpression of inhibitory immune checkpoint molecules.2 The resulting immune evasion within the TME is thus proposed to be one of the emerging “cancer hallmarks” and also represents a critical factor in limiting the efficacy of cancer immunotherapies.1 3–6 Several clinical studies have shown that cancers responding to immune checkpoint inhibitors (ICIs) often express an inflammatory gene signature or a T-cell “inflamed” phenotype characterized by high tumor infiltration of cytotoxic T lymphocytes (CTLs),7–10 which suggests that the limited success of ICIs in the clinic may be partially attributed to a lack of pre-existing tumor-reactive T cells in the TME.11 12 Thus, strategically using novel approaches or agents for programming tumors to create a highly immunostimulatory milieu or to convert immune-excluded or immune-deserted tumors into “inflamed” ones can help mobilize immune effector cell for tumor destruction and improve outcomes of other cancer treatment modalities.
We recently engineered a multifunctional immunotherapeutic agent termed Flagrp170 by incorporating the microbial protein flagellin-derived, NF-κB-stimulating sequence into the Grp170-based molecular chaperoning platform.13 When compared with either flagellin or unmodified chaperone molecule, integration of this pathogen-associated molecular pattern confers the Flagrp170 with markedly improved therapeutic potency against mouse melanoma, prostate cancer, and colorectal cancer.13 The superior immunostimulatory activity of the Flagrp170 cargo results from its highly efficient antigen-shuttling and presenting property coupled with strong costimulation provided by the embedded pathogen-associated “danger” signal.13–16 As a novel class of immune modulators with unique immune-potentiating activity, Flagrp170 could be used to reshape the immune landscape of TME to favor the immune response against tumor and/or to enhance cancer susceptibility to therapies.
In the present research, we conduct studies to evaluate the impact of intratumoral administration of Flagrp170 on the immune compartment of the TME, systemic immune activation, and antitumor outcomes using multiple preclinical cancer models. We show that Flagrp170 mobilizes a unique cytokine network in the TME that involves cytokines crucial for polarization of type 1 CD4+ (Th1)/CD8+(Tc1)-dominant antitumor immunity. Using transgenic mouse models, we also investigate the molecular basis of immune pattern recognition of the therapeutically delivered Flagrp170. Our studies lead to an unexpected finding of the distinct dependence of in situ Flagrp170 therapy on two innate pattern recognition receptors, that is, toll-like receptor 5 (TLR5) and the NOD like receptor (NLR) family caspase activation and recruitment domain (CARD) domain-containing protein 4 (NLRC4). Our results indicate that the targeted immune programming with Flagrp170 is highly capable of creating an immunologically “hot” TME and driving tumor-reactive immune activation locally and systemically.
Methods
Mice and cell lines
C57BL/6 mice, C3H/HeN mice, BALB/c mice, Tlr5−/− mice, and Pmel transgenic mice carrying T cell receptor (TCR) transgene specific for the mouse homolog (pmel-17) of human gp100 (6–8 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). Nlrc4−/− mice on C57BL/6 background were kindly provided by Dr Richard Flavell (Yale University). Murine mammary tumor 4T1 line was purchased from American Type Culture Collection (ATCC, Manassas, Virginia, USA). Murine head and neck squamous cell carcinoma VII(SCCVII) cells were from Dr Gal Shafirstein (Roswell Park Comprehensive Cancer Center). Human gp100-expressing mouse melanoma B16 line was from Dr Alexander Rakhmilevich (University of Wisconsin-Madison). B16 cells and 4T1 cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (FBS), 10 mM 2-[4-(2-Hydroxyethyl)-1-piperazinyl]-ethanesulfonic acid (HEPES), 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin. SCCVII cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 10 mM HEPES, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin. All cell lines were routinely tested for mycoplasma contamination using a PCR-based mycoplasma detection kit (ATCC).
Reagent and antibodies
Fluorochrome-conjugated anti-mouse monoclonal antibodies, including FITC-CD3 (17A2), APC-CD4 (GK1.5), PerCP/Cy5.5-CD8 (2.43), FITC-granzyme B (GB11), FITC-CD80 (16-10A1), PE-IFN-γ (XMG1.2), APC-NK1.1 (PK136), FITC-CD45 (30-F11), PE-GM-CSF (MP1-22E9), PerCP/Cy5.5-CD11b (M1/70), PE-F4/80 (BM8), APC-CD11c (N418), PE-CD86 (GL-1) as well as CD16/CD32 (2.4G2), isotype control rat IgG2b (RTK4530), and IgG1 (RTK2071) were purchased from BioLegend (San Diego, California, USA). Phycoerythrin (PE) conjugated anti-mouse TLR5 (ACT5) was purchased from BD Biosciences (San Jose, California, USA). Depletion Abs for CD4 T cells (GK1.5), CD8 T cells (2.43), granulocyte macrophage colony-stimulating factor (GM-CSF)-neutralizing antibody (MP1-22E9), and IgG control were purchased from BioXcell (West Lebanon, New Hampshire, USA). Human gp10025–33 (KVPRNQDWL) peptides were from AnaSpec (Fremont, California, USA). Antibodies against phospho-IκBα (Ser32/36), IκBα, phospho-p65, and p65 were from Cell Signaling Biotechnology (Beverly, Massachusetts, USA). Mouse tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-12p40, interferon (IFN)-γ, IL-1α, IL-1β, and IL-2 ELISA kits were purchased from BioLegend. Mouse IL-18 ELISA kit was purchased from ThermoFisher Scientific (Whaltham, Massachusetts, USA). Alanine aminotransferase (ALT) colorimetric activity assay kit was purchased from Cayman (Ann Arbor, Michigan, USA).
Generation of replication-defective recombinant adenoviruses
To obtain a replication-deficient adenovirus carrying human Grp170 (hGrp170) fused with NF-κB-activating sequence derived from flagellin, the coding sequence of ATP-binding domain-truncated Grp170 was amplified from pMD18-T-hGrp170 (Sino Biological, Wayne, Pennsylvania, USA) and in-frame linked to the flagellin NC fragment via the second flexible linker. Signal peptide for hGrp170 was added and the “KNDEL” ER retention sequence was removed to generate a secretable form of hFlagrp170 as we previously described.13 Recombinant adenoviruses were packaged, amplified and prepared using AdenoPACK Maxi spin columns (Sartorius Stedim Biotech, Bohemia, New York, USA) as we previously described.13 17
Tumor studies
Tumors were established in the dorsal flank by subcutaneous inoculation of 2×105 of SCCVII cells to female C3H/HeN mice, 2×105 of B16 cells to C57BL/6 mice, or 5×105 of 4T1 cells to female BALB/c mice, respectively. Mice were randomized into different treatment groups when tumor size reached 4~5 mm in diameters. Adenovirus (2×108 p.f.u. in 50 µL PBS) was administered intratumorally. Treatment continued every other day for a total of five doses or otherwise as indicated. Tumor sizes were measured in a blinded manner by using a vernier caliper. Tumor volume (mm3) was calculated using the formula V=(shortest diameter2×longest diameter)/2. Mice were euthanized when tumors reached 16 mm in diameter or caused loss of 10% of body weight. Monoclonal antibodies (200 µg) for depletion of CD4+ or CD8+ T cells, or for neutralization of GM-CSF were injected intraperitonially every 3 days starting 1 day prior to viral treatment. Control mice received IgG (200 µg, intraperitonially). Tumor tissues were digested by incubation in collagenase D (1 mg/mL) and DNase I (20 µg/mL) at 37°C for 1 hour. Cell suspensions were filtered through 70 µm cell strainers and subjected to percoll gradient centrifugation to enrich viable mononuclear cells for analysis of tumor-infiltrating immune cells. Hearts, kidneys, livers, and spleens were collected following Flagrp170 treatment and subjected to H&E staining to evaluate the presence of tissue toxicity. Images were taken using an Olympus BX41 fluorescence microscope (Tokyo, Japan). ALT activity in serum samples was assayed using a kit from Cayman.
Bone morrow-derived dendritic cells and T cell priming in vitro
Bone morrow-derived dendritic cells (BMDCs) were generated by culturing mouse bone marrow cells in the presence of GM-CSF as we previously described.13 18 19 For assays of DC activation by Flagrp170, DCs were infected by Flagrp170-encoding or empty adenovirus (multiplicity of infection, MOI=300) for 3 hours. Adenoviruses were removed by extensive washing with complete RPMI 1640 medium. Expression of costimulatory molecules and production of inflammatory cytokines was determined by flow cytometry and ELISA, respectively. For T cell priming, infected BMDCs were pulsed with gp10025-33 peptide (1 µg/mL) for 1 hour. DCs were then serially diluted and incubated with 105 purified Pmel CD8+ T cells for 60 hours. 3H-thymidine (3H-TdR, 0.5 mCi/well) was added to the wells for the last 12–18 hours of culture. T cell proliferation was measured using 3H-TdR incorporation assay. IFN-γ production from T cells were measured by ELISA or intracellular cytokine staining to assess T cell activation.
Intracellular cytokine staining and flow cytometry analysis
Cells were stimulated by phorbol 12′-myristate-13′-acetate (PMA, 10 nM) plus ionomycin (0.5 µM) for 3 hours, followed by treatment with brefeldin A (BFA, 5 µg/mL) to block intracellular protein transport for another 2 hours. Cells were incubated with anti-CD16/CD32 antibodies for 20 min on ice before staining with fluorescently labeled antibodies. Cells were then fixed, permeabilized, and stained with PE-conjugated anti-IFN-γ or FITC-conjugated antigranzyme B antibodies for 30 min at room temperature. For GM-CSF staining of tumor-infiltrating leukocytes, tumor-bearing mice were injected intravenous with BFA (250 µg/mouse) 16 hours prior to collection of tumor tissues for preparing cell suspensions.
Quantitative PCR analysis
Real-time quantitative PCR was performed as previously described.20 Briefly, total RNA was extracted using TRIzol Reagent (ThermoFisher Scientific). Reverse transcription and quantitative PCR were conducted using carboxyfluorescein (FAM)-labeled probe sets from ThermoFisher Scientific. Gene expression was quantified relative to the expression of β-actin, and normalized to that measured in control groups by standard 2(−ΔΔCt) calculation.
Statistical analysis
Data were analyzed using SigmaPlot software and expressed as mean±SD values. Statistical significance between groups within experiments was determined by the two-way repeated measures analysis of variance test or Student’s t-test. Survival of tumor bearing mice between experimental groups was compared using the log-rank test. P values less than 0.05 were considered statistically significant.
Results
Programming the TME with Flagrp170 generates a potent antitumor immune response
We first examined the effect of intratumoral delivery of Flagrp170 on the growth of SCCVII squamous cell carcinoma established in C3H/HeN mice, which is widely used as a model for human oral squamous cell carcinoma.21In situ Flagrp170 therapy resulted in profound inhibition of SCCVII tumors when compared with mock treatment with an empty adenovirus, that is, null (figure 1A). This was accompanied by substantially prolonged life-span of Flagrp170-treated mice (figure 1B). Analysis of tumor tissues showed that immune programming by Flagrp170 caused marked elevation of Th1 immunity-related cytokine genes including ifng and il12a (figure 1C). On stimulation with SCCVII tumor lysates, the splenocytes from Flagrp170-treated mice also produced significantly higher levels of IFN-γ (figure 1D) that was associated with increased frequencies of IFN-γ-expressing CD8+ or CD4+ T cells (figure 1E) compared with those from mock-treated mice, suggesting a systemic antitumor response induced by local Flagrp170 treatment.

Programming tumor microenvironment with Flagrp170 induces a potent antitumor immunity. (A, B) C3H/HeN mice (n=5) bearing SCCVII tumors (4~5 mm in diameter) were treated intratumoral with an empty adenovirus (ie, null) or an adenovirus encoding Flagrp170 every other day for a total of five doses. Tumor growth (A) and animal survival (B) were followed. (C) Transcription of ifng and il12a genes in tumor tissues (n=3) following treatments was assayed by quantitative PCR. (D, E) Systemic T cell activation by Flagrp170-mediated immune programming of tumor environment. Splenocytes from treated mice (n=3) were stimulated with SCCVII tumor cell lysates at a ratio of 3:1 for 96 hours. IFN-γ level in the culture media was examined using ELISA (D) and the frequency of IFN-γ-producing CD8+ or CD4+ T cells were determined using intracellular cytokine staining (E). (F–H) Comparable antitumor potency of human and mouse versions of Flagrp170. Mice bearing B16 tumors of 4–5 mm sizes (n=5) received treatment with mouse version of Flagrp170 (mFlagrp170) or its human counterpart, that is, hFlagrp170 (F). Immune activation in the tumor tissues (n=3) was evaluated by analyzing the transcription of ifng and il12a (G). Splenocytes (left) or lymph node cells (right) from mice treated with or without mFlagrp170/hFlagrp170 (n=3) were stimulated with MHC I-restricted gp10025-33 peptide, followed by assessment of IFN-γ production using ELISA (H). Data represent three different experiments with similar results. *p<0.05, **p<0.01, ***p<0.001, NS, not significant, using two-way repeated measures analysis of variance test (A, F), log-rank test (B) and Student’s t-test (C, D, G, H). IFN, interferon; MHC I, major histocompatibility complex class I; SCC, squamous cellcarcinoma.
To prepare for clinical testing of this immunotherapeutic agent in the treatment of human malignancies, we engineered a human version of Flagrp170, in which mouse Grp170 sequence was replaced with the corresponding human sequence (online supplemental figure 1). Grp170 is an evolutionally highly conserved chaperone molecule.22 Sequence alignment showed that murine version (mFlagrp170) and human version (hFlagrp170) share 93.5% similarity (online supplemental figure 2). Comparison of these two molecules in the treatment of established tumors showed that hFlagrp170 was as therapeutically effective as mFlagrp170 in controlling B16 melanomas (figure 1F), which correlated with strong immune augmentation in the TME and lymphoid organs (figure 1G,H). No evident tissue toxicity associated with in situ Flagrp170 immunotherapy was detected, as measured by changes in body weight and ALT levels as well as histological analysis of major organs (online supplemental figure 3). Study of different treatment regimens in SCCVII tumor model also showed a dose-dependent antitumor activity of hFlagrp170 without apparent adverse effects, evidenced by no significant elevation of hepatic ALT or decrease in body weight (online supplemental figure 4).
Supplemental material
Supplemental material
Supplemental material
Supplemental material
GM-CSF is required for antitumor immunity induced by Flagrp170-based tumor programming
Given a critical role of Th1 immunity in eradication of cancer, we performed a time-course analysis of B16 tumor tissues during in situ Flagrp170 therapy. Examination of the changes in cytokines known to be involved in Th1/Tc1 immunity showed that gene transcription of il12a, ifng, and gmcsf was strongly upregulated in a time or dose dependent manner (figure 2A), suggesting that Flagrp170 treatment skews the tumor milieu towards a Th1/Tc1-polarized, immunostimulatory status.

GM-CSF production induced by Flagrp170 immune programming is critical for its antitumor efficacy. (A) B16 tumor-bearing C57BL/6 mice (n=3) received in situ Flagrp170 therapy for a total of five doses. Tumor tissues were collected 24 hours after each treatment and subjected to analyzes of Th1 immunity-related genes. (B and C) Production of GM-CSF by tumor-infiltrating immune cell subsets was examined by intracellular cytokine staining and flow cytometry. (D) B16 tumor-bearing mice (n=3) were depleted of CD4+ or CD8+ T cells prior to Flagrp170 therapy by administration of the corresponding antibodies. Transcription of gmcsf gene in tumor tissues was assayed by quantitative PCR. Flagrp170-treated mice receiving normal IgG and mice receiving empty adenovirus serve as controls. (E) Neutralization of GM-CSF inhibits antitumor activity of Flagrp170. B16 tumor-bearing mice (n=5) were injected with GM-CSF neutralizing antibodies during in situ Flagrp170 treatment. (F) Tumor infiltration by CD11b+CD11c+ dendritic cells was determined by flow cytometry. (G) Transcription of ifng, il12a, and gmcsf in tumor tissues (n=3) was assayed by quantitative PCR. Data shown are representative of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, using Student’s t-test (A, D, F, G) and two-way repeated measures analysis of variance test (E). GM-CSF, granulocyte macrophage colony-stimulating factor.
Given the substantial induction of GM-CSF in tumors, we next attempted to define the GM-CSF sources within the tumors using intracellular cytokine staining followed by flow cytometric analysis. We showed that GM-CSF expression was mainly increased in CD45+ leukocytes but not in CD11b+ myeloid cells (figure 2B). Direct infection of T cells with Ad.Flagrp170 failed to induce any detectable GM-CSF production (online supplemental figure 5A), suggesting that production of GM-CSF required immunostimulatory events (eg, DC activation) prior to T cell activation. Since both CD8+ and CD4+ T cells expressed GM-CSF (figure 2C), we further performed antibody depletion of either CD8+ or CD4+ T cells to determine their effects on the GM-CSF levels in the TME. Removal of CD8+ T cells, not CD4+ T cells, substantially decreased the levels of GM-CSF in the TME (figure 2D). Although tumor-infiltrating CD4+ T cells similarly produced GM-CSF on Flagrp170 therapy, depleting CD8+ T cells significantly reduced GM-CSF expression in these CD4+ T cells (online supplemental figure 5B), suggesting that CD8+ T cells are essential for GM-CSF induction in the TME, even though both T cell subsets are the source of GM-CSF.
Supplemental material
To examine the involvement of GM-CSF in Flagrp170-elicited antitumor immunity, we neutralized GM-CSF by injection of anti-GM-CSF antibodies. The therapeutic activity of Flagrp170 against established B16 melanoma was partially abrogated on neutralization of GM-CSF (figure 2E). Absence of GM-CSF also blocked the Flagrp170-induced recruitment/expansion of CD11c+ DCs (figure 2F) and F4/80+ macrophages (online supplemental figure 6), which is consistent with the notion that GM-CSF is a connecting conduit between myeloid cells and T cells for immune activation.23 The elevation of cytokine genes ifng and il12a, previously shown to be critical for Flagrp170-induced antitumor immunity,13 was also diminished after administration of anti-GM-CSF antibodies. However, the transcriptional levels of gmcsf gene was not altered (figure 2G).
Supplemental material
We next validated the role of GM-CSF using murine 4T1 mammary tumor model. Similarly, immune programming of established 4T1 tumors with Flagrp170 induced a potent antitumor response, which was compromised by neutralization of GM-CSF (figure 3A). GM-CSF blockade also interfered with elevation of ifng and il12a gene transcription in the TME (figure 3B). The loss of GM-CSF functionally impaired tumor-infiltrating immune effector cells, as indicated by a substantial decrease in IFN-γ or granzyme B expression in both CD8+ T cells and NK cells (figure 3C). In addition to reduced immune activation in the tumor sites, analyzes of splenocytes (figure 3D) and lymph node cells (figure 3E) from GM-CSF-neutralized mice showed significantly decreased production of IFN-γ and IL-2 on stimulation with 4T1 tumor lysates.
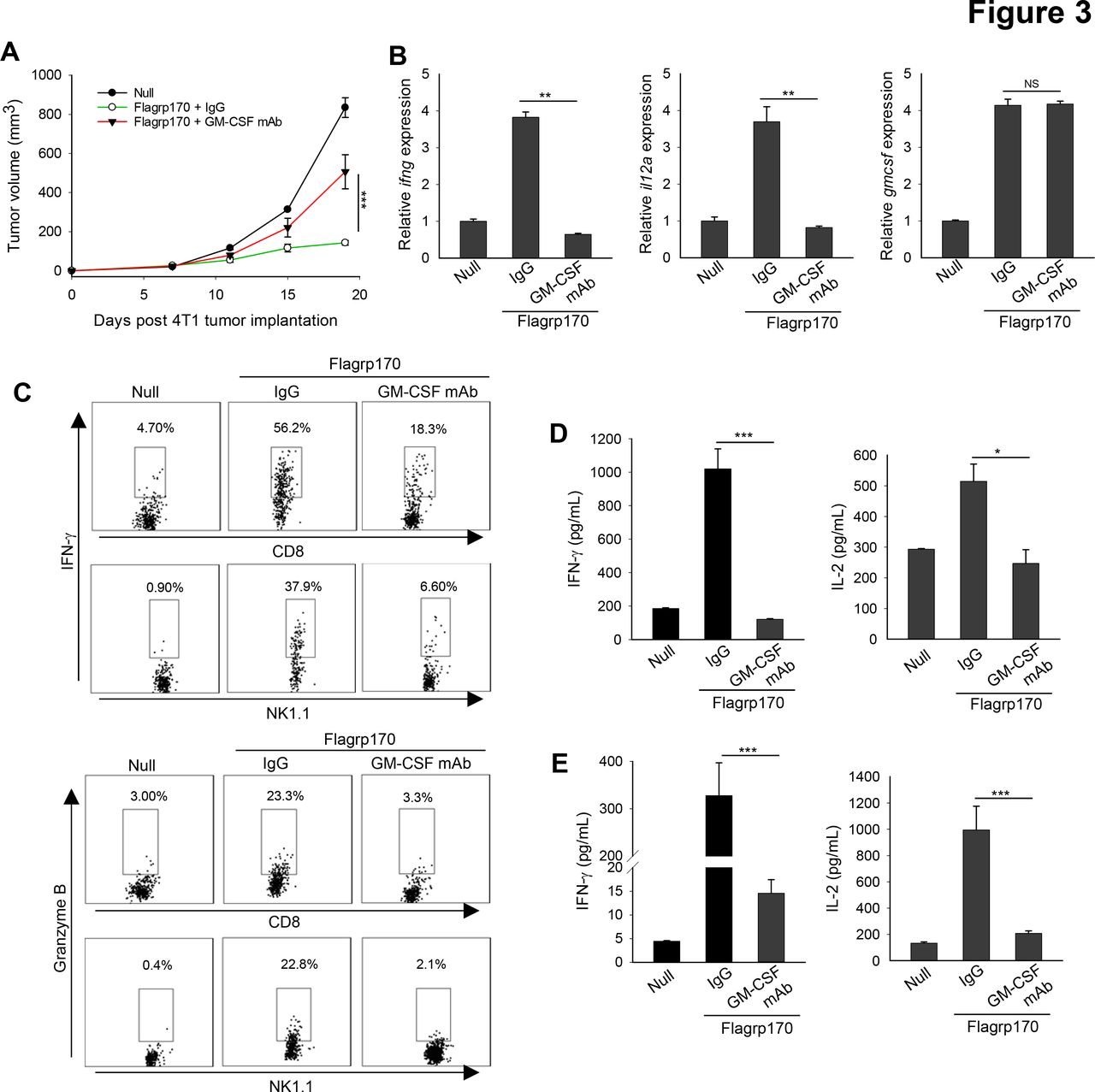
In situ Flagrp170 immunotherapy induces GM-CSF-dependent antitumor response against mouse mammary tumor. (A) BALB/c mice (n=5) with established 4T1 tumors of 4~5 mm sizes were treated with an empty or Flagrp170-encoding adenovirus as indicated. GM-CSF antibodies (200 µg) was administrated intraperitonially 1 day before the first treatment and continued at 3-day intervals for four doses. (B) Gene transcription of ifng, il12a, and gmcsf in tumor tissues (n=3) was assayed 2 weeks after the Flagrp170 treatment. (C) Expression of IFN-γ and granzyme B in tumor-infiltrating CD8+ T cells or NK cells were examined by intracellular cytokine staining. Splenocytes (D) or draining lymph node cells (E) from treated mice (n=3) were stimulated with 4T1 cell lysates. Production of cytokines IFN-γ and IL-2 was assessed using ELISA. Data shown are representative of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, using two-way repeated measures analysis of variance test (A) and Student’s t-test (B, D, E). GM-CSF, granulocyte macrophage colony-stimulating factor; IFN, interferon; IL, interleukin.
Consistent with these observations, neutralization of GM-CSF also diminished the antitumor activity of Flagrp170 in the treatment of SCCVII squamous cell carcinoma and reduced the transcriptional level of ifng gene in the tumor tissues (online supplemental figure 7A,B). Similarly, GM-CSF was shown to be necessary for Flagrp170 immunotherapy induced tumor infiltration by activated CD8+/CD4+ T cells (online supplemental figure 7C,D).
Supplemental material
Flagrp170 enhanced DC activation does not require TLR5
Given that the flagellin-derived NF-κB-stimulating sequence was integrated into the backbone of Flagrp170 and that DCs are crucial for the Flagrp170-mediated antitumor immune response,13 we next examined the potential involvement of TLR5 in activation of DCs by Flagrp170. We showed that infection with the Flagrp170-encoding adenovirus induced activation of NF-κB efficiently in both wild-type (WT) and Tlr5−/− DCs, indicated by similar levels of phosphorylation of IκB or p65 (figure 4A). WT or Tlr5−/− DCs also showed comparable elevation of co-stimulatory molecules CD86 and CD80 (figure 4B). Flagrp170-induced production of inflammatory cytokines including TNF-α, IL-6, and IL-12p40 was not affected in the absence of TLR5 (figure 4C). Additionally, Flagrp170-stimulated Tlr5−/− DCs were as effective as WT counterparts in stimulating tumor antigen (ie, gp100)-specific Pmel-1 T cells in vitro, as determined by T-cell proliferation and IL-2 production (figure 4D), suggesting a dispensable role of TLR5.
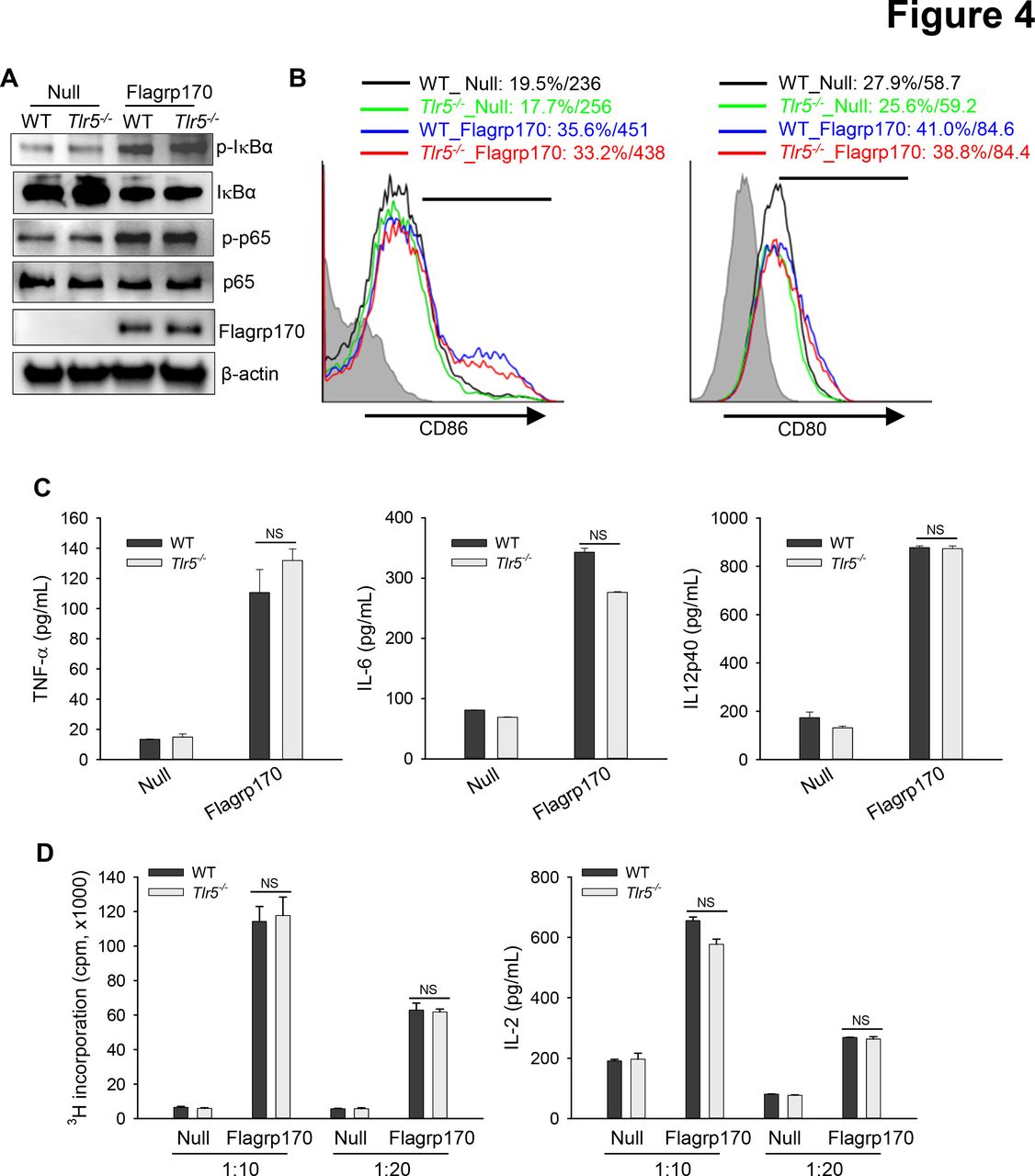
TLR5 is not required for Flagrp170-enhanced dendritic cell (DC) activation in vitro. (A) WT or Tlr5−/− bone marrow-derived DCs were infected with an empty or Flagrp170-encoding virus at a multiplicity of infection of 300. 24 hours later, protein lysates were prepared and analyzed for NF-κB activation by immunoblotting. (B) The expression of costimulatory molecules CD86 and CD80 was also assessed using flow cytometry. (C) Production of TNF-α, IL-6, and IL-12p40 by DCs were measured using ELISA. (D) Virus-infected WT or Tlr5−/− bone morrow-derived dendritic cells were pulsed with gp10025-33 peptide and cocultured with gp100-specific Pmel cells for 3 days. T-cell proliferation was assessed by 3H-TdR incorporation assays (left) and ELISA of the IL-2 levels in culture medium (right). The experiments were repeated three times with similar results. NS, not significant, using Student’s t-test (C and D). IL, interleukin; TNF, tumor necrosis factor; WT, wild-type.
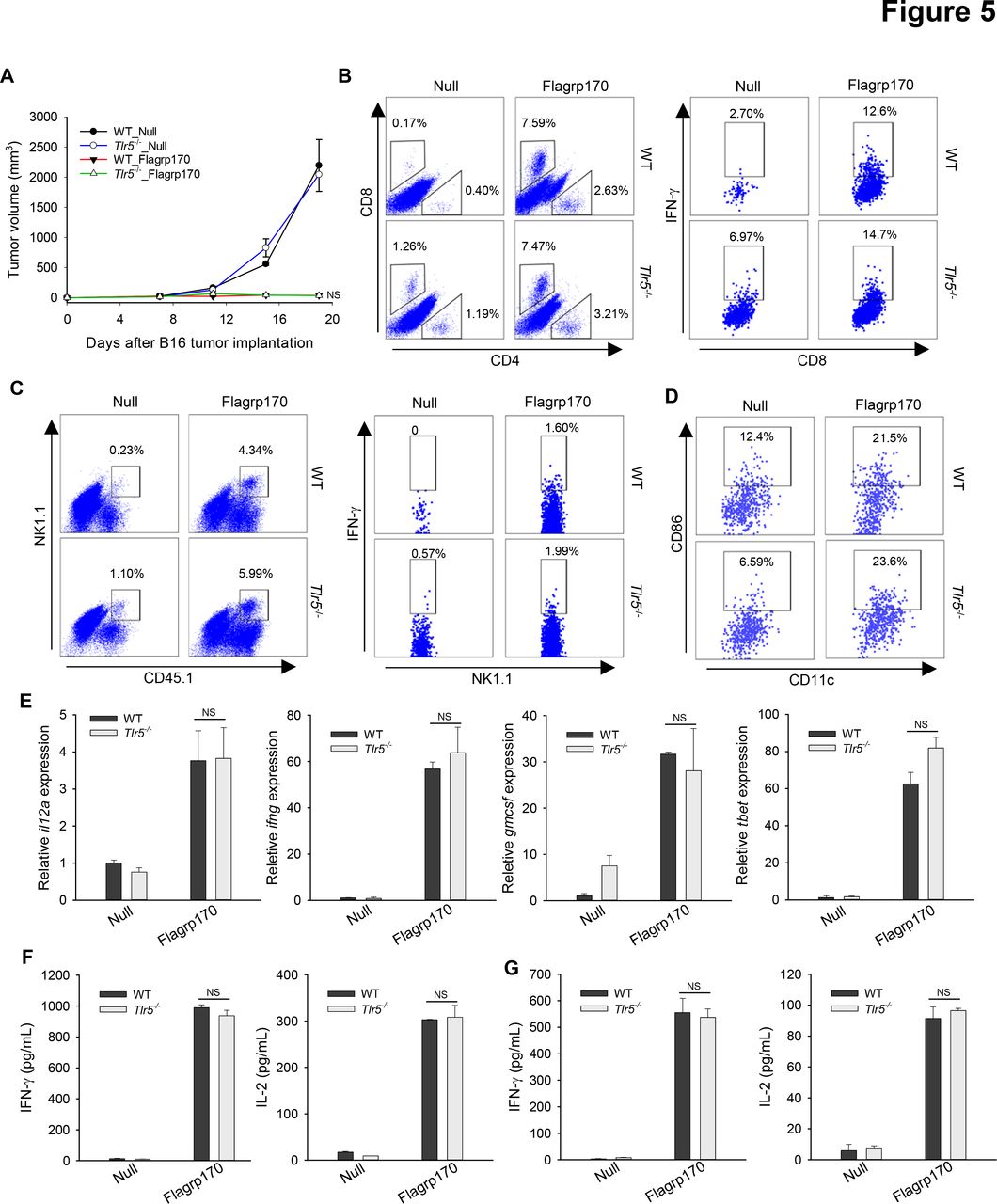
TLR5 is not involved in antitumor immunity generated by in situ Flagrp170 therapy
We sought to address the question whether TLR5 is required for therapeutic activity of Flagrp170 in vivo using TLR5 knockout mice. Strikingly, the antitumor potency of Flagrp170 was not affected by the loss of TLR5, as shown by similar tumor inhibition in WT or Tlr5−/− mice (figure 5A). Lack of TLR5 did not appear to alter the recruitment or activation of immune effector cells (figure 5B,C). We also showed that TLR5 was mainly expressed on DCs, not on B16 tumor cells (online supplemental figure 8). Consistent with in vitro study, tumor-infiltrating DCs from Flagrp170-treated WT or Tlr5−/− mice showed similar CD86 expression (figure 5D). Flagrp170 treatment-induced Th1 immunity-related genes within the tumors, such as il12a, ifng, gmcsf, and tbet, were also comparable in WT or Tlr5−/− mice (figure 5E). Finally, Flagrp170-augmented systemic activation of CD8+ T cells remained intact, indicated by similar production of IFN-γ or IL-2 by splenocytes (figure 5F) or lymph node cells (figure 5G) from WT and Tlr5−/− mice on stimulation with MHC I-restricted gp10025-33 peptide.
Supplemental material
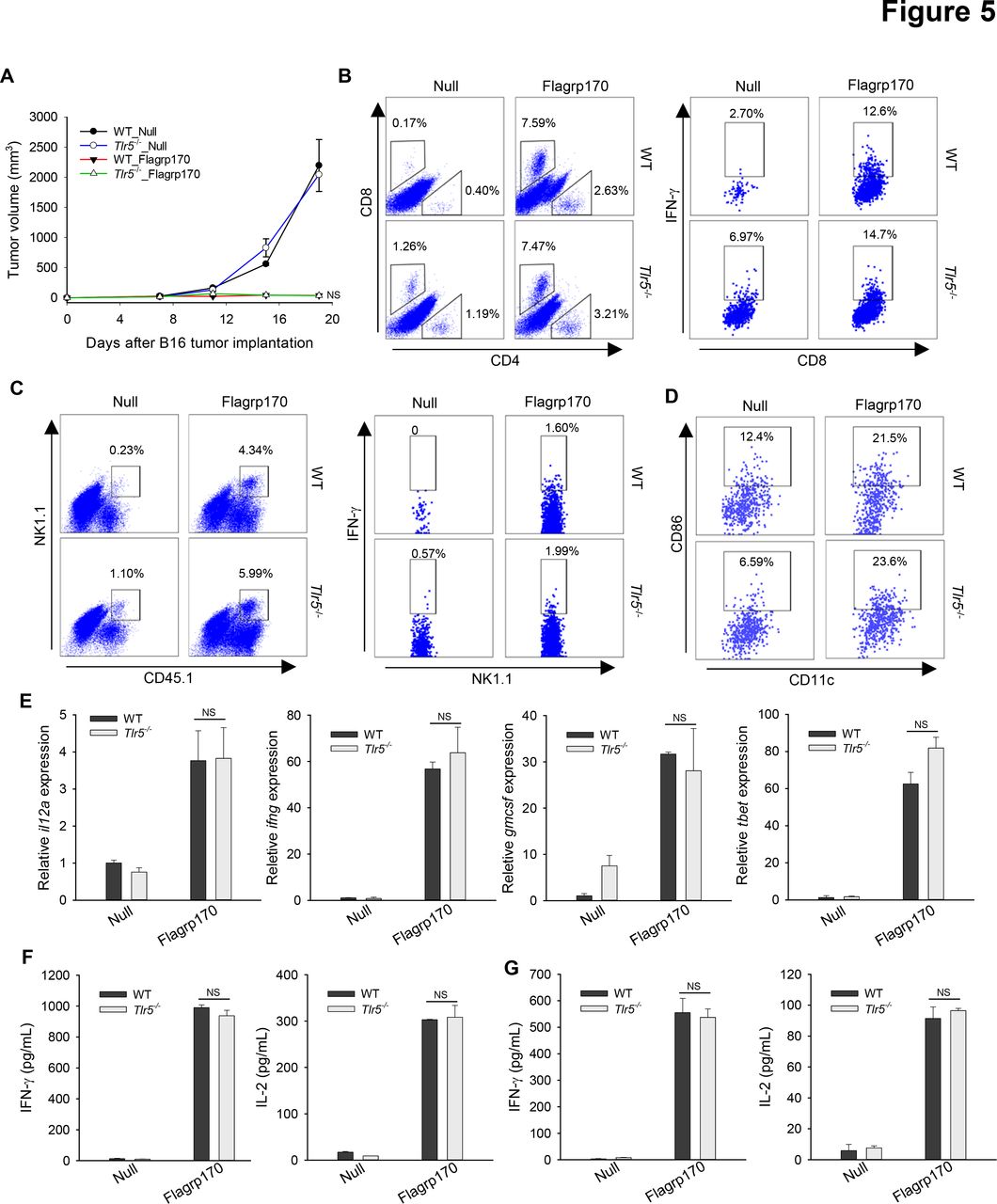
Toll-like receptor 5 is dispensable for Flagrp170-elicited antitumor immunity in vivo. (A) WT or Tlr5−/− mice (n=5) with B16 tumors of 4~5 mm sizes were treated with an empty or Flagrp170-encoding adenovirus. Tumor infiltration by T cells (B), NK cells (C), and dendritic cells (DCs) (D) and their activation status were assayed by intracellular cytokine staining and flow cytometry. (E) Transcription of il12a, ifng, gmcsf, and tbet in tumors (n=3) was examined 2 weeks after treatment. Splenocytes from mock or Flagrp170 treated mice (n=3) were stimulated with B16 tumor lysates (F) or gp10025-33 peptide (G), followed by analyzes of IFN-γ and IL-2 levels in the culture media using ELISA. Data shown are representative of three independent experiments. NS, not significant, using two-way repeated measures analysis of variance test (A) and Student’s t-test (E–G). IFN, interferon; IL, interleukin; WT, wild-type.
Flagrp170-induced protective antitumor immunity depends on NLRC4
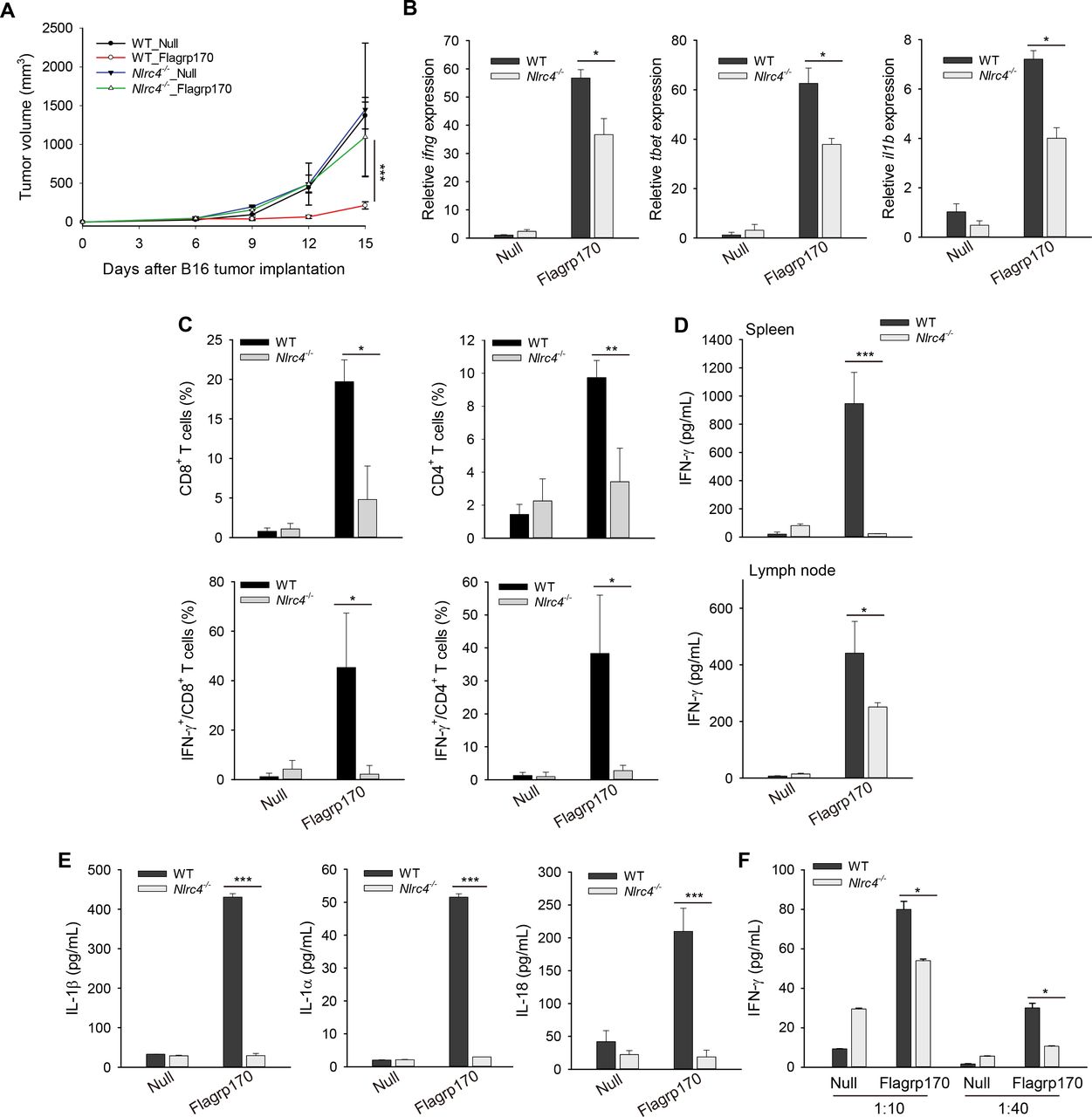
In addition to TLR5 on cell surface, bacterial flagellin can also be recognized by NLRC4, an innate pattern recognition receptor in the cytosolic compartment that plays an important role in innate immune host defense.24–26 To understand the molecular mechanism underlying Flagrp170-induced immune activation, we performed in situ Flagrp170 therapy using WT and Nlrc4−/− mice. We showed that B16 melanoma grew similarly in WT and Nlrc4−/− mice treated with mock virus, suggesting that NLRC4 deficiency did not influence tumor growth per se. However, absence of NLRC4 abrogated the therapeutic activity of Flagrp170 (figure 6A). Flagrp170-induced upregulation of ifng, tbet, and il1b in the TME were significantly inhibited in Nlrc4−/− mice (figure 6B). The loss of NLRC4 also impaired the recruitment and/or activation of CD8+ or CD4+ T cells (figure 6C and online supplemental figure 9A). The levels of tumor-associated NK cells previously shown to contribute to therapeutic activity of Flagrp170 was similarly reduced in Nlrc4−/− mice (online supplemental figure 9B). Additionally, absence of NLRC4 significantly diminished production of IFN-γ by CD8+ T cells in lymphoid organs on stimulation with gp10025-33 peptide (figure 6D).
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The NLR family CARD domain-containing protein (NLRC4) inflammasome is required for antitumor efficacy of in situ Flagrp170 therapy. (A) Abrogation of therapeutic activity of Flagrp170 in the absence of NLRC4. B16 tumor-bearing WT and Nlrc4−/− mice (n=5) were treated with an empty or Flagrp170-encoding virus. Tumor growth was monitored by measuring tumor sizes. Transcription of ifng, tbet, and il1b in tumor tissues (B) and the frequency or activation of tumor-infiltrating T cells (C) were examined (n=3). (D) Splenocytes (top) or lymph node cells (bottom) from mock or Flagrp170 treated mice (n=3) were stimulated with gp10025-33 peptide for 72 hours, followed by ELISA analysis of IFN-γ production. (E) 48 hours after infection, WT or Nlrc4−/− dendritic cells (DCs) was examined for their production of IL-1β, IL-1α, and IL-18. (F) Reduced T-cell priming activity of DCs in the absence of NLRC4. Infected WT or Nlrc4−/− DCs were pulsed with gp10025-33 peptide and cocultured with Pmel cells for 3 days, followed by ELISA analysis of IFN-γ production. Data shown are representative of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, using two-way repeated measures analysis of variance test (A) and Student’s t-test (B–F). CARD, caspase activation and recruitment domain; IFN, interferon; IL, interleukin; NLR, NOD like receptor; WT, wild-type.
As a member of the NLR family, activation of cytosolic NLRC4 by pathogen-associated molecular patterns such as flagellin is required for maturation of cytokine IL-1 and IL-18.25 On infection with the Flagrp170-encoding adenovirus DCs were seen to express Flagrp170 protein in the cytosol and produced high levels of IL-1β, IL-1α, and IL-18, which was abolished in Nlrc4−/− DCs (figure 6E and online supplemental figure 10). Finally, lack of NLRC4 reduced the capacity of Flagrp170-stimulated DCs to prime antigen-specific T cells in vitro (figure 6F), suggesting that engagement of DC-intrinsic NLRC4 contributes to the immunostimulatory and antitumor activity of Flagrp170.
Supplemental material
Discussion
Despite the recent major breakthroughs in the field of immune-oncology, tumor resistance to immunotherapies, including ICIs, remains a major challenge, which has been believed to be caused at least partially by the immunosuppressive or non-immune inflamed TME.27 28 Our study demonstrates that Flagrp170, a new class of immune modulator, can effectively program the tumor milieu by mobilizing a highly immunostimulatory cytokine network critical for induction of Th1 cellular immunity and for profound growth inhibition of tumors. We also provide new molecular insights into the action of Flagrp170 by identifying the NLRC4 as the innate pattern recognition receptor that can sense the Flagrp170-associated microbial signal and orchestrate Flagrp170-induced antitumor immune responses.
The Flagrp170 possesses distinct features for immunologically modifying the tumor antigen-abundant TME and mounting a robust CTL response: promoting cross-presentation of weakly immunogenic tumor antigens with concurrent delivery of crucial costimulation signals.13 Our extensive studies in the current work have further validated the therapeutic potency of Flagrp170 in the treatment of head and neck cancer, melanoma, and breast cancer, supported by strong inhibition of tumor growth and prolonged survival of treated animals. Both mouse and human versions of Flagrp170 constructs display comparable anticancer activities without induction of apparent adverse effects, indicted by no significant changes of body weight, liver injury, and pathologies of major organs in treated animals. Together with our previous data from preclinical models of melanoma, prostate cancer, and colorectal cancer, we have provided compelling evidence supporting the clinical translation of in situ Flagrp170 immunotherapy.
In the present study, we demonstrate that immune programming by Flagrp170 skews the immunosuppressive TME toward one that strongly favors induction of Th1/Tc1 dominant antitumor immunity. This is evidenced by marked elevation of a set of cytokines within the tumors including IL-12, IFN-γ, and GM-CSF that are well documented for their critical roles in driving Th1/Tc1 immune responses against cancers.29–32 While IL-12 and IFN-γ were previously shown to be necessary for the therapeutic activity of Flagrp170,13 the substantial induction of GM-CSF, a hematopoietic growth factor, by in situ Flagrp170 treatment, as reported in this study, is quite intriguing. GM-CSF is believed to be produced mainly by Th cells33 34 and functions at the interface of T-myeloid cells.23 35 We have made a surprising observation that tumor-infiltrating CD8+ T cells recruited by Flagrp170 are critical for GM-CSF upregulation in the TME. Despite its little effect on tumor infiltration by CD4+ T cells, depletion of CD8+ T cells significantly decreases the overall levels of GM-CSF in the TME. Induction of GM-CSF represents a key event involved in immune programming by Flagrp170, in that GM-CSF blockade compromises its therapeutic activity. Consistent with an established role of GM-CSF as a DC recruiter and activator, neutralization of GM-CSF impaired recruitment of DCs and activation of immune effector cells in the TME. Furthermore, the neutralization of GM-CSF also compromises the induction of IL-12 and IFN-γ by Flagrp170, indicating the coordinated and interactive actions of these cytokines within the Flagrp170-primed tumors. Talimogene laherparecvec, an oncolytic viruses encoding GM-CSF for intralesional administration, has been approved by US Food and Drug Administration for treatment of recurrent unresectable melanoma.36 37 Given the distinct action of Flagrp170 in mobilizing an immunostimulatory cytokine network including GM-CSF, it might be advantageous to use Flagrp170 for bridging the innate and adaptive immune compartments in the TME.
While flagellin is commonly known to be a ligand for TLR5 on cell surface, it can also be recognized by the cytosolic NLRC4 inflammasome, another innate pattern recognition receptor involved in the host defense against invading pathogens.26 38 39 We made an unexpected finding that the host TLR5 is dispensable for antitumor immunity generated by Flagrp170. However, the therapeutic efficacy of in situ Flagrp170 treatment is largely dependent on the presence of NLRC4, evidenced by dampened antitumor immune response in Nlrc4−/− mice as well as impaired function of Nlrc4−/− DCs in T cell priming. Reliance on NLRC4 for mediating the therapeutic activity of Flagrp170 likely reflects the fact that Flagrp170 is administrated using a viral delivery platform. Infection of immune cells (eg, DCs) results in expression of Flagrp170 protein in the cytosol, which is then sensed by the intracellular pattern recognition receptor NLRC4 rather than the TLR5 on cell surface. Although activation of inflammasomes such as the nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) is widely known to control maturation and production of IL-1β, the potential role of IL-1β in immunotherapy generated antitumor immune responses is less understood. The IL-1 signaling has been previously reported to facilitate T cell priming and expansion in a non-cancer setting.40 41 Indeed, our discovery of NLRC4-mediated antitumor immunity is in line with the study of Ghiringhelli and colleagues reporting that IL-1β release from DCs on activation of NLRP3 inflammasome can facilitate the antitumor immune responses during chemotherapy.42
The similar growth rate of B16 tumors in WT and Nlrc4−/− mice seen in our study, consistent with the report by Tenthorey et al,43 suggests that NLRC4 may be not involved in cancer immune surveillance in a non-therapeutic setting. Interestingly, Janowski et al44 recently showed that NLRC4 expression in macrophages is required to generate antitumor immunity that is independent of activation of the NLRC4 inflammasome, implicating a possible non-inflammasomal function of NLRC4. However, we conducted our studies in an active immunotherapeutic context where a novel immune-stimulating agent was employed to deliberately activate the NLRC4 inflammasome in immune sentinel cells, that is, DCs. Nonetheless, additional work is necessary to decipher how the NLRC4-IL-1 axis in the myeloid cell compartment governs immunological programming of the TME by Flagrp170 and activation of Th1/Tc1-dominant antitumor immunity.
In summary, targeting the TME with Flagrp170 represents a novel therapeutic strategy to transform the immunosuppressive or non-inflamed tumor milieu into immunologically “hot” tumors, which can achieve immune-mediated tumor destruction and may also help promote tumor response to other cancer treatments such as ICIs. With unique features of Flagrp170 in bridging both innate and adaptive arms of the immune system, immune programming by Flagrp170 not only engages a Th1/Tc1-polarized cytokine network, but also confers the tumors a T-cell “inflamed” phenotype. Moreover, our work advances our understanding of the mechanism of action of Flagrp170 by identifying a previously unrecognized role of the pathogen-sensing NLRC4 inflammasome in dictating Flagrp170-induced antitumor immunity. The robust antitumor response generated by in situ Flagrp170 therapy in the treatment of a wide range of cancer types,13 including head and neck cancer and breast cancer demonstrated in the current study, further supports its potential clinic use as “personalized” vaccination against the entire tumor antigenic repertoire, which can be capitalized together with other treatment modalities for optimized cancer eradication.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
All experimental procedures involving mice were conducted according to the protocols approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee (IACUC), protocol number AD20158 (approval date: February 27, 2018).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @HarryBearMDPhD
XY, WL and CG contributed equally.
Correction notice This article has been corrected since it first published. The provenance and peer review statement has been included.
Contributors CG, X-YW designed and supervised research; XY, WL, CG, SC, XC, PAP, TS, FY, CW performed experiments and collected data; XY, WL, CG, JWL, HDB, ASP, JRS, ER, X-YW analyzed data. CG and X-YW wrote the manuscript.
Funding This work was supported in part by Department of Defense Grant W81XWH-11-0481, National Cancer Institute Grants CA099326, CA154708, and CA175033. Services in support of the research project were provided by the VCU Massey Cancer Center Flow Cytometry Core, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.