Article Text

Abstract
Background Chimeric antigen receptor (CAR) T-cell therapy is an emerging option for cancer treatment, but its efficacy is limited, especially in solid tumors. This is partly because the CAR T cells become dysfunctional and exhausted in the tumor microenvironment. However, the key pathways responsible for impaired function of exhausted cells remain unclear, which is essential to overcome CAR T-cell exhaustion.
Methods Analysis of RNA-sequencing data from CD8+ tumor-infiltrating lymphocytes (TILs) led to identification of Cbl-b as a potential target. The sequencing data were validated using a syngeneic MC38 colon cancer model. To analyze the in vivo role of Cbl-b in T-cell exhaustion, tumor growth, % PD1+Tim3+ cells, and expression of effector cytokines were analyzed in cbl-b+/+ and cbl-b–/– mice. To evaluate the therapeutic potential of Cbl-b depletion, we generated a new CAR construct, hCEAscFv-CD28-CD3ζ.GFP, that recognizes human carcinoembryonic antigen (CEA). cbl-b+/+ and cbl-b–/– CEA-CAR T cells were generated by retroviral transduction. Rag–/– mice bearing MC38-CEA cells were injected with cbl-b+/+ and cbl-b–/–; CEA-CAR T cells, tumor growth, % PD1+Tim3+ cells and expression of effector cytokines were analyzed.
Results Our results show that the E3 ubiquitin ligase Cbl-b is upregulated in exhausted (PD1+Tim3+) CD8+ TILs. CRISPR-Cas9-mediated inhibition of Cbl-b restores the effector function of exhausted CD8+ TILs. Importantly, the reduced growth of syngeneic MC38 tumors in cbl-b–/– mice was associated with a marked reduction of PD1+Tim3+ CD8+ TILs. Depletion of Cbl-b inhibited CAR T-cell exhaustion, resulting in reduced MC38-CEA tumor growth, reduced PD1+Tim3+ cells and increased expression of interferon gamma, tumor necrosis factor alpha, and increased tumor cell killing.
Conclusion Our studies demonstrate that deficiency of Cbl-b overcomes endogenous CD8+ T-cell exhaustion, and deletion of Cbl-b in CAR T cells renders them resistant to exhaustion. Our results could facilitate the development of efficient CAR T-cell therapy for solid tumors by targeting Cbl-b.
- cell engineering
- CD8-positive T-lymphocytes
- immunotherapy
- immunotherapy
- adoptive
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Adoptive transfer T-cell therapy is an emerging option for cancer treatment,1 but its efficacy is limited, especially in solid tumors, because the effector CD8+ T-cells that promote antitumor immunity become dysfunctional and exhausted in the tumor microenvironment (TME). Exhausted T cells exhibit progressive loss of effector function (expression of interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α) and tumor cell killing) and express inhibitory receptors (PD1, Tim3, and LAG3).2 However, the key pathways responsible for impaired function of exhausted cells remain unclear.
Post-translational modification mediated by ubiquitin conjugation plays an indispensable role in immune cells. Ubiquitination involves a cascade of biochemical reactions through ubiquitin activating (E1) enzymes, ubiquitin-conjugating (E2) enzymes, and ubiquitin ligase (E3) enzymes. The E3 ubiquitin ligases are critical components of this system because they recognize and target specific target proteins for ubiquitination.3
Cbl-b is an E3 ubiquitin ligase that belongs to the Really Interesting New Gene (RING) family. Cbl-b contains an N-terminal tyrosine kinase-binding domain, a RING finger, and a C-terminal proline-rich sequence, and can thus function as both an E3 ligase and a molecular adaptor.3 Here, we demonstrate that Cbl-b is upregulated in exhausted CD8+ tumor-infiltrating lymphocytes (TILs) and plays a crucial role in dysfunction of tumor-reactive TILs.
Materials and methods
Mice
C57BL/6, Rag–/– and cbl-b–/– mice were housed in microisolator cages, and the experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of UT Southwestern Medical Center.
MC38-carcinoembryonic antigen (CEA) tumor and CD8+ TIL isolation
cbl-b+/+ and cbl-b–/– mice were injected subcutaneously with 1×106 MC38-CEA cells and tumor growth was analyzed.4 Kaplan-Meier estimate was used to analyze the survival of mice. Mice were counted as dead when the tumor diameter reached the length limit of 15 mm.5 CD8+ TILs were isolated using CD8 TIL Microbeads (Miltenyi).
Construction of retroviral vector (hCEA scFV-GFP) containing chimeric antigen receptor (CAR)
DNA sequences encoding scFv anti-CEA chimeric receptors were synthesized by Genescript (New Jersey, USA and cloned into the m1928z-GFP6 retroviral construct by replacing the CD19scFv region between CD8 leader (CD8L) sequence and CD8 hinge region (CD8TM) at the NotI restriction site. The chimeric gene constructs were composed of the mouse CD8 signal peptide (CD8L), scFv-anti-CEA mAb, a membrane proximal hinge region along with the transmembrane domain, and mouse stimulatory domain, which included mouse CD28 and mouse CD3ζ.
CRISPR-Cas9 targeting
Three top-ranking guide sequences against Cbl-b exons were designed and cloned into lenti-CRISPR v2 plasmid (Addgene 52961). HEK293FT cells (Thermo Fisher) were transfected with lenti-CRISPR v2 plasmid, along with packaging plasmid pMD2.G (Addgene 12259) and psPAX2 (Addgene 12260). Lentivirus was harvested after 48–72 hours of incubation, passed through 0.45 µm filter and stored at −80°C. The details of the guide RNAs targeting Cbl-b gene are as follows:
Cbl-b guide RNA1: GATGGGATGTCGCATCCTCG.
Cbl-b guide RNA2: GCACTTCGTGCCTTACCGCG.
Cbl-b guide RNA3: GGCGAGTGTTCGAAAGTGCA.
Retroviral transduction, CAR T-cell production and adoptive transfer
CD8+ T-cells were stimulated for 24 hours with anti-CD3 clone (1 µg/mL) and anti-CD28 (1 µg/mL) antibodies and were cultured with mIL-2 (100 U/mL) and transduced with the retrovirus by spinoculation method.7 On the day of adoptive transfer, GFP+ cells were sorted by flow cytometry and (GFP+CD8+ CAR T cells) adoptively transferred via tail–vein injections.
Statistical analysis
Statistical analysis was performed with the Prism V.8.0 software, and all results were summarized as mean±SD. Differences between groups were evaluated by two-tailed Student’s t-test. Log-rank test was used for survival analysis. A p value of <0.05 was considered as statistically significant.
Results
Cbl-b is upregulated in PD1+Tim3+ exhausted TILs
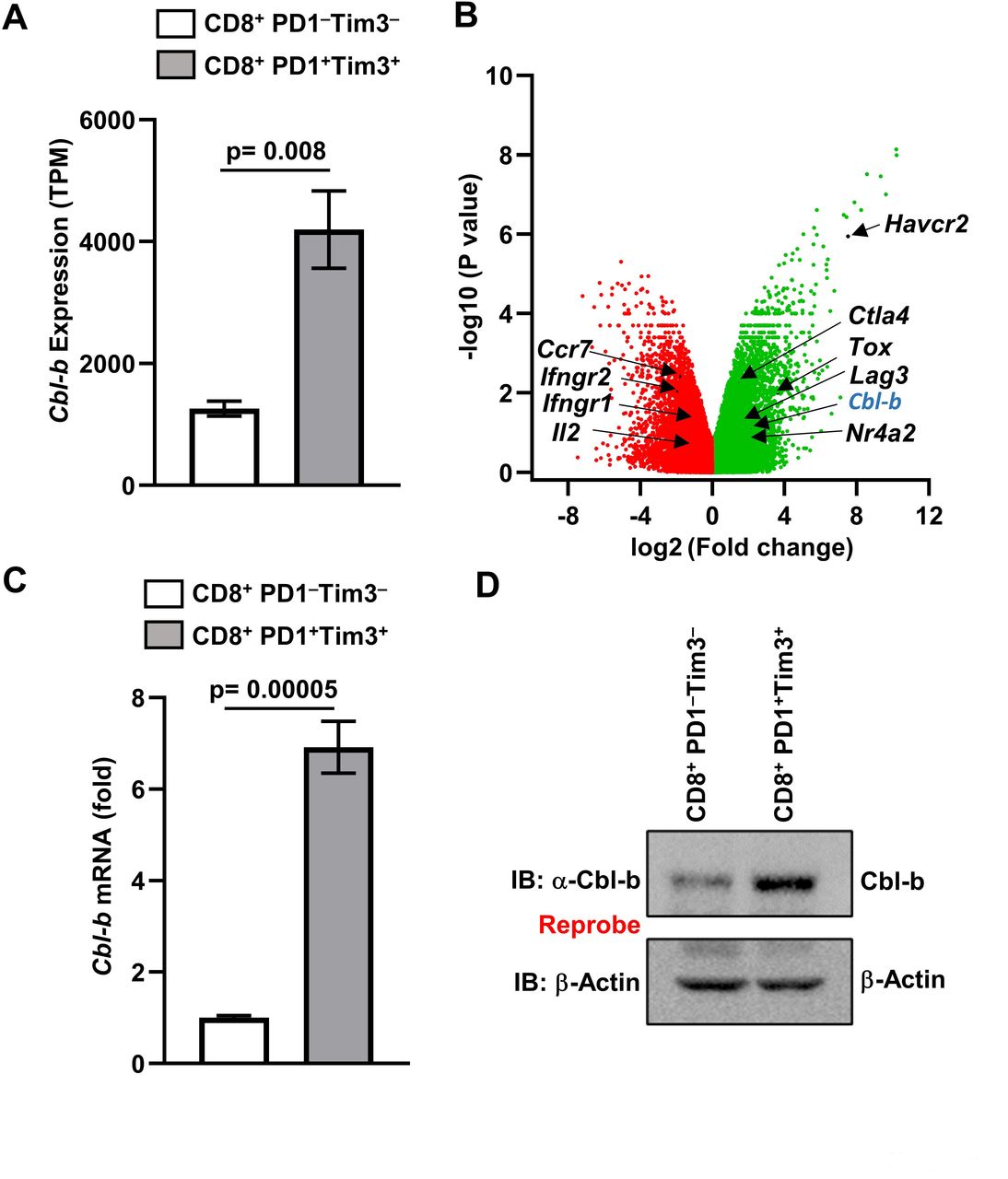
Cbl-b has been shown to be an important negative regulator of T cells,3 and deletion of Cbl-b in T cells results in rejection of established tumors.8 Analysis of published RNA-sequencing data (GSE85947)9 showed that messenger RNA (mRNA) encoding the Cbl-b gene was preferentially upregulated in PD1+Tim3+ exhausted CD8+ TILs in the CT26 colon carcinoma model (figure 1A,B). Further, analysis of the published sequencing results showed that Cbl-b expression was upregulated in exhausted CD8+ TILs in multiple tumor models (GSE123738 and GSE85947),7 as well as in a lymphocytic choriomeningitis virus infection model (GSE88987).10 To confirm these sequencing results, we adopted the syngeneic MC38 colon cancer model. As shown in figure 1C, real-time PCR analysis of RNA isolated from PD1+Tim3+ TILs showed increased expression of Cbl-b compared with PD1–Tim3– cells. Increased Cbl-b expression in PD1+Tim3+ TILs was further confirmed by immunoblotting (figure 1D). These data collectively suggest that Cbl-b expression is preferentially upregulated in exhausted CD8+ TILs in the TME.

Cbl-b is upregulated in exhausted CD8+PD1+Tim3+ TILs. (A) Geo dataset (GSE85947) was analyzed and normalized read counts (TPM) for Cbl-b are shown. (B) Volcano plot of differentially expressed genes (−log10 p value vs log2 fold change) in PD1+Tim3+ versus PD1–Tim3– TILs. (C) MC38 tumors were implanted subcutaneously into C57BL6 mice; PD1–Tim3– and PD1+Tim3+ TILs were sorted; and Cbl-b messenger RNA levels were analyzed by real-time PCR. (D) Immunoblot of Cbl-b protein using Fluorescence-activated cell sorted (FACS) PD1–Tim3– versus PD1+Tim3+ TILs. The data are representative of three independent experiments. Statistics are mean±SD, calculated by Student’s t-test (two-tailed). TIL, tumor-infiltrating lymphocyte. TPM, transcript per million; IB, Immunoblotting, FACS, Fluorescence-activated cell sorting.
Inhibition of Cbl-b restores the effector function of exhausted CD8+ TILs
To test the hypothesis that upregulated Cbl-b in PD1+Tim3+ cells plays an essential role in CD8+ T-cell exhaustion, we knocked out Cbl-b using CRISPR-Cas9. We transduced the PD1+Tim3+ CD8+ TILs with guide RNA targeting exons of the Cbl-b gene or non-targeted control guide RNA, along with CRISPR-Cas9-expression lentiviruses. The efficiency of CRISPR-Cas9-mediated deletion of Cbl-b was confirmed by immunoblotting (online supplemental figure 1A). Deleting Cbl-b restored expression of IFN-γ, TNF-α, granzyme B and IL-2 in PD1+Tim3+ CD8+ TILs (figure 2A–D).
Supplemental material

Inhibition of Cbl-b restores effector function of CD8+PD1+Tim3+ TILs. Cbl-b was knocked out using Cbl-b-guide RNA-pLenti-CRISPR-v2 in sorted CD8+PD1+Tim3+cells from MC38 tumor-bearing C57BL6 mice. (A) Expression of IFN-γ, (B) TNF-α, (C) granzyme B and (D) IL-2 was measured by real-time PCR. The data are representative of three independent experiments. Statistics are mean±SD, calculated by Student’s t-test (two tailed). IFN-γ, intergeron gamma; IL, interleukin; TIL, tumor-infiltrating lymphocyte; TNF-α, tumor necrosis factor alpha, gRNA. guide RNAs.
Cbl-b deficiency leads to reduced PD1+Tim3+ TILs
To examine the in vivo role of Cbl-b in regulation of T-cell exhaustion and tumor growth, we inoculated cbl-b+/+ and cbl-b–/– mice with MC38-CEA cells. A markedly reduced growth of MC38 tumors was observed in cbl-b–/– mice compared with cbl-b+/+ mice (figure 3A,B). Our results are consistent with previous reports that cbl-b–/– mice mount a robust antitumor response against transplanted (TC-1, EL4, EG7, and B16 melanoma), chemically induced and spontaneous tumors.8 11 The increased antitumor activity in cbl-b–/– mice was attributed primarily to CD8+ T-cells.8 11 Similarly, we found an increased number of CD8+ T-cell infiltration into MC38 tumors in cbl-b–/– mice (online supplemental figure 1B).

Reduced MC38 tumor growth in cbl-b–/– mice. cbl-b+/+ and cbl-b–/– mice (n=5) were subcutaneously injected with MC38-CEA cells. (A) Tumor volume. (B) Representative images of tumors. (C) Percentage of CD8+PD1+Tim3+ TILs. (D) Representative image showing percentage of CD8+PD1+Tim3+ TILs. (E) CD8+PD1+Tim3+ TILs were sorted and expression of IFN-γ, TNF-α and granzyme B was analyzed by real-time PCR. The data are representative of three independent experiments. Statistics are mean±SD, calculated by Student’s t-test (two-tailed). CEA, carcinoembryonic antigen; IFN-γ, intergeron gamma; TIL, tumor-infiltrating lymphocyte; TNF-α, tumor necrosis factor alpha.
To test if Cbl-b deficiency effected CD8+ T-cell exhaustion, we isolated CD8+ TILs and stained the cells with anti-PD1 and anti-Tim3 antibodies. Flow cytometry analysis showed a significantly reduced number of PD1+Tim3+ cells in cbl-b–/– mice compared with cbl-b+/+ mice (figure 3C,D). Further, we sorted the CD8+PD1+Tim3+ TILs from tumors grown in cbl-b+/+ and cbl-b–/– mice, and real-time PCR analysis showed that cbl-b–/– CD8+PD1+Tim3+ TILs but not cbl-b+/+ TILs expressed IFN-γ, TNF-α and granzyme B (figure 3E). These data collectively suggest that Cbl-b plays an important role in CD8+ T-cell exhaustion.
Depletion of Cbl-b prevents CAR T-cell exhaustion
Exhaustion is a major barrier in the success of CAR T cells.2 Similar to endogenously exhausted CD8+ T-cells, Cbl-b was also upregulated in exhausted CAR T cells.7 Therefore, we investigated if depletion of Cbl-b would rescue CAR T cells from exhaustion resulting in enhanced antitumor activity. We replaced CD19scFv with CEA-scFv (MFE23), which recognizes human carcinoembryonic antigen (hCEA)12 of a CAR construct against CD19, CD19scFv-CD28-CD3ζ.GFP,13 generating a new CAR construct, hCEAscFv-CD28-CD3ζ.GFP (figure 4A). We chose CEA as a model antigen because CEA is overexpressed in many human cancers, most notably in colorectal adenocarcinomas.14

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Depletion of Cbl-b inhibits CAR T-cell exhaustion and promotes tumor regression. (A) Schematic representation of the CEA-reactive CAR construct. (B) 6×106 cbl-b+/+ and cbl-b–/– CAR T cells were adoptively transferred into Rag–/– mice (n=6) on days 3 and 8 after tumor inoculation. Percentage survival of Rag–/– mice that received cbl-b+/+ and cbl-b–/– CAR T cells. The mice were considered dead when tumor size became 15 mm. Log-rank test was used for statistical analysis. (C) Tumor volume is shown; (D) sorted cbl-b+/+ and cbl-b–/– CD8+PD1+Tim3+ TILs were cocultured with MC38-CEA cells (target) labeled with calcein-AM. Specific lysis and expression of IFN-γ, TNF-α and granzyme B are shown. (E) Representative images of spleen and H&E-stained sections of the colon, lung and liver. The data are representative of three independent experiments. Statistics are mean±SD, calculated by Student’s t-test (two-tailed). AM, acetoxymethyl; CAR, chimeric antigen receptor; CEA, carcinoembryonic antigen; hCEA, human carcinoembryonic antigen; IFN-γ, intergeron gamma; scFv, single-chain variable fragment; TIL, tumor-infiltrating lymphocyte; TNF-α, tumor necrosis factor alpha.
We retrovirally transduced naïve CD8+ T-cells with either control (mock) (without hCEAscFv) or hCEAscFv-CD28-CD3ζ.GFP. The transduction efficiency varied between 60% and 65% (online supplemental figure 2A). We cocultured fluorescence-activated cell sorted GFP+ cells (online supplemental figure 2B) with MC38-CEA cells, a mouse colon cancer cell line expressing hCEA.15 Real-time PCR analysis showed that CEA-CAR T cells, but not mock transduced cells, expressed IFN-γ, TNF-α, granzyme B, and IL-2 (online supplemental figure 2C).
Supplemental material
Next, we injected MC38-CEA cells into the right flank of Rag–/– mice, and on days 3 and 8, we inoculated with cbl-b+/+ or cbl-b–/– CEA-CAR T cells. The use of cbl-b–/– CAR T cells considerably enhanced the survival of Rag–/– mice as shown in figure 4B. In addition, we observed significantly reduced tumor growth in Rag–/– mice that received cbl-b–/– CAR T cells compared with the mice that received cbl-b+/+ CAR T cells (figure 4C).
To investigate if Cbl-b deficiency effected CAR T-cell exhaustion, tumors were collected and CD8+ TILs were enriched. These cells were stained with anti-PD1 and anti-Tim3 antibodies and were analyzed by flow cytometry. The percentage of PD1+Tim3+ cells were markedly reduced in the tumors in mice that received cbl-b–/– CAR T cells (between 30% and 35% in cbl-b+/+ and 4%–6% in cbl-b–/–. Next, we sorted the PD1–Tim3– and PD1+Tim3+ cells that were gated on GFP+ cells and cocultured with MC38-CEA cells to analyze their tumor killing ability using calcein-acetoxymethyl.16 As shown in figure 4D, coculture of MC38-CEA cells with cbl-b+/+ PD1+Tim3+ CEA-CAR T cells exhibited markedly reduced tumor cell killing ability. However, the cbl-b–/– cells retained their tumor killing ability. Further, cbl-b–/– CEA-CAR T cells cocultured with MC38-CEA cells expressed IFN-γ, TNF-α and granzyme B but not the cbl-b+/+ cells (figure 4D). These data collectively suggest that deletion of cbl-b–/– potentiates the antitumor activity of CAR T cells. Since Cbl-b deficiency in T cells is linked to autoimmunity,3 we looked for signs of toxicity in mice that received cbl-b–/– cells. No significant change in spleen size was observed between Rag−/− mice that received either cbl-b+/+ or cbl-b–/– cells. Similarly, histological analysis of H&E-stained sections of colon, lung and liver tissues of mice that received cbl-b–/– CAR T cells did not show any signs of toxicity (figure 4E).
Discussion
Immunotherapy for cancer via checkpoint blockade and adoptive transfer of tumor antigen-specific CAR T cells have shown promising results.1 However, only a small subset of patients achieves complete remission with checkpoint blockade. Similarly, CAR T cells have been effective against hematopoietic malignancies but not against solid tumors. Several lines of evidence implicate exhaustion in limiting the potency of CAR T cells.2 Here, we demonstrate that deletion of Cbl-b inhibits CAR T-cell exhaustion, which can be exploited therapeutically.
The mechanism by which Cbl-b deficiency leads to such a remarkable effect on CD8+ T-cell exhaustion currently remains unclear. However, recent studies suggest that exhaustion results due to activation of nuclear factor of activated T-cells (NFAT) in the absence of AP-117 and several studies have implicated the NFAT-driven transcription factors NR4A7 and TOX in T-cell exhaustion.18–20 Interestingly, Cbl-b expression was induced by constitutively active NFAT,17 suggesting the potential role of Cbl-b in NFAT driven mechanisms.
Previous studies have shown that Cbl-b deficiency leads to tumor rejection, which is predominantly mediated by CD8+ T-cells.8 11 Further, the antitumor activity of cbl-b–/– CD8+ T-cells was shown to be independent of CD4+ T-cell help.8 11 In line with these studies, we found reduced MC38 tumor growth, which was associated with increased infiltration of CD8+ T-cells in cbl-b–/– mice. However, Cbl-b has also been shown to regulate natural killer cells and dendritic cells.21 22 Therefore, we do not completely exclude the function of these immune cells in Cbl-b-mediated antitumor response.
It was demonstrated that Cbl-b deficiency uncouples the requirement for costimulation for T cells and hence heightened antitumor activity.3 Our results show that Cbl-b is upregulated in PD1+Tim3+ exhausted CD8+ T-cells, and depletion of Cbl-b restores their effector function, suggesting a novel role for Cbl-b in CD8+ T-cells. Further, adoptive transfer of CEA-specific CD8+ CAR T cells resulted in reduced tumor growth and substantial survival benefit in Rag−/− mice, suggesting the potential of targeting Cbl-b in CAR T cells. However, a significant difference exists between spontaneous tumors that arise in humans and the transplanted tumors. Therefore, further investigation in spontaneous tumor models and humanized mice is essential before the translation into clinical trials.
CAR T cells specific to CEA have shown clinical efficacy in gastrointestinal adenocarcinomas.23 However, another trial failed due to preconditioning-dependent respiratory toxicity.24 While we did not observe any significant toxicity in mice that received cbl-b–/– CAR T cells, further studies using mice transgenically expressing human CEA in gastrointestinal or lung epithelia cells are essential to fully assess the potential toxicity. Nevertheless, our studies clearly suggest that Cbl-b could be potentially targeted to overcome CAR T-cell exhaustion.
Conclusion
Our studies demonstrate that deficiency of Cbl-b overcomes endogenous CD8+ T-cell exhaustion, and deletion of Cbl-b in CAR T cells render them resistant to exhaustion. Our results could facilitate the development of efficient CAR T-cell therapy for solid tumors by targeting Cbl-b.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @marcoldavila
Contributors J.K, R.K, A.S, E.L.T, M.K performed the experiments, analyzed the data and helped to prepare manuscript. M.J.R, A.T, M.D helped to prepare manuscript. K.V conceived the project, designed the experiment and wrote the manuscript.
Funding This work was supported by grants from the National Institutes of Health (R01-DK115668) and Cancer Prevention Research Institute of Texas (RP160577 and RP190527) to VP and R01- DK117001 to VP and ALT.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study protocol has been approved by the institutional review board of UT Southwestern Medical Center.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All the data are available and will be shared upon request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.