Article Text

Abstract
Background Metastatic breast cancer is a leading cause of cancer-related death in women worldwide. Infusion of natural killer (NK) cells is an emerging immunotherapy for such malignant tumors, although elimination of the immunosuppressive tumor environment is required to improve its efficacy. The effects of this “metastatic” tumor environment on NK cells, however, remain largely unknown. Previous studies, including our own, have demonstrated that metastasis-associated macrophages (MAMs) are one of the most abundant immune cell types in the metastatic tumor niche in mouse models of metastatic breast cancer. We thus investigated the effects of MAMs on antitumor functions of NK cells in the metastatic tumor microenvironment.
Methods MAMs were isolated from the tumor-bearing lung of C57BL/6 mice intravenously injected with E0771-LG mouse mammary tumor cells. The effects of MAMs on NK cell cytotoxicity towards E0771-LG cells were evaluated in vitro by real-time fluorescence microscopy. The effects of MAM depletion on NK cell activation, maturation, and accumulation in the metastatic lung were evaluated by flow cytometry (CD69, CD11b, CD27) and in situ hybridization (Ncr1) using colony-stimulating factor 1 (CSF-1) receptor conditional knockout (Csf1r-cKO) mice. Finally, metastatic tumor loads in the chest region of mice were determined by bioluminescence imaging in order to evaluate the effect of MAM depletion on therapeutic efficacy of endogenous and adoptively transferred NK cells in suppressing metastatic tumor growth.
Results MAMs isolated from the metastatic lung suppressed NK cell-induced tumor cell apoptosis in vitro via membrane-bound transforming growth factor β (TGF-β) dependent mechanisms. In the tumor-challenged mice, depletion of MAMs increased the percentage of activated (CD69+) and mature (CD11b+CD27–) NK cells and the number of Ncr1+ NK cells as well as NK cell-mediated tumor rejection in the metastatic site. Moreover, MAM depletion or TGF-β receptor antagonist treatment significantly enhanced the therapeutic efficacy of NK cell infusion in suppressing early metastatic tumor outgrowth.
Conclusion This study demonstrates that MAMs are a main negative regulator of NK cell function within the metastatic tumor niche, and MAM targeting is an attractive strategy to improve NK cell-based immunotherapy for metastatic breast cancer.
- breast neoplasms
- tumor microenvironment
- macrophages
- killer cells
- natural
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Metastatic breast cancer is largely refractory to current therapies and thus the most common cause of cancer-related death in women worldwide.1 Since up to 10% of patients with breast cancer already have metastatic tumors at diagnosis,2 novel therapies that suppress metastatic tumor outgrowth are needed to improve patients’ survival. Given the recent success with checkpoint inhibitors that enhance CD8+ T cell-mediated tumor elimination, immunotherapy is now considered one of the most promising strategies to treat metastatic tumors.3 However, most advanced breast cancers downregulate major histocompatibility complex class-I (MHC-I) that is essential for effective killing by CD8+ T cells,4 and only a minority (1/25) of metastatic patients with breast cancer respond to checkpoint inhibitors.5 Adoptive transfer of natural killer (NK) cells is proposed as a novel immunotherapy to treat such types of cancer, since NK cells do not require MHC-I dependent priming to exert their cytotoxicity.6 Instead, loss of MHC-I on target cells drives activation of NK cells.7 The infusion of ex vivo expanded NK cells has been evaluated in a clinical context and found to exert modest clinical responses without any side effects in patients with metastatic breast cancer.6 8 Conversely, it has been suggested that the cytotoxicity of NK cells is restricted within the tumor microenvironment and elimination of this immune suppressive environment is needed to improve efficacy of NK cell infusion therapy.8 9
In this study, we identified that NK cell cytotoxicity is suppressed by metastasis-associated macrophages (MAMs) via transforming growth factor β (TGF-β) and that depletion of MAMs or blockade of TGF-β signaling enhances efficacy of infused NK cells in suppressing early metastatic tumor outgrowth (online supplemental file 1).
Supplemental material
Materials and methods
Mice
C57BL/6JCrl mice were obtained from Charles River and housed in a specific pathogen free room. Colony-stimulating factor 1 (CSF-1) receptor conditional knockout (Csf1r-cKO) mice (ie, rtTA:tetO-Cre:Csf1rF/F) on a C57BL/6 background were obtained by crossing the B6.Cg-Csf1rtm1Jwp/J (ie, Csf1rF/F) mice with ROSA-rtTA and tetO-Cre mice (The Jackson Laboratory).10 11 In some experiments, C57BL/6JCrl mice (4-week-old females) were irradiated with 9 Gy γ-rays and transplanted with 4×106 bone marrow cells isolated from Csf1r-cKO mice. The bone marrow transplanted mice (Csf1r-cKO:BMT) were used for experiments 6 weeks later.
Cell culture
E0771-LG cells, a highly metastatic derivative of E0771 mouse mammary adenocarcinoma cells that originated from a medullary cancer in C57BL/6 mice,12 were manipulated to express firefly luciferase (E0771-LG:Fluc) and a nuclear localized red fluorescent protein mKate2 (E0771-LG:Fluc_NLR) as previously described.11 13 These tumor cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) including 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, 5% CO2. Bone marrow-derived macrophages (BMMs) were prepared from the femur and tibia of C57BL/6JCrl mice,14 and were cultured in αMEM (Gibco) including 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and recombinant murine 20 ng/mL macrophage colony-stimulating factor (M-CSF; PeproTech) or 25 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech). NK cells were isolated from the spleen of C57BL/6JCrl mice (female, 8 weeks of age) using EasySep Mouse NK Cell Isolation Kit (StemCell Technologies) and cultured in αMEM including 20% FBS, 2 mM L-glutamine, 1% non-essential amino acid, 1 mM sodium pyruvate, 50 nM 2-mercaptoethanol, 100 U/mL penicillin, 100 μg/mL streptomycin (all from Gibco), and 1000 U/mL murine interleukin (IL)-2 (PeproTech).
Mouse model of metastatic breast cancer
1×106 of E0771-LG:Fluc cells were injected into the tail vein of syngeneic C57BL/6JCrl or Csf1r-cKO mice (7–12-week-old females). To detect tumor cells in the chest region, mice received intra-peritoneal injection of D-luciferin (1.5 mg/20 g mouse, Gold Biotechnologies) and were imaged by Photon IMAGER Optima (Biospace Lab) every 3–4 days. The metastatic tumor loads indicated by photon count (photon/s/cm2/sr) were quantified by M3 Vision Software (Biospace Lab). At day 4 after tumor injection, animals with micro-metastases (suggested by photon count 2.5×102–1.0×103) were randomly divided into groups and received following treatments. To deplete macrophages, Csf1r-cKO mice were given doxycycline in the drinking water (Sigma, 2 mg/mL in 5% sucrose water) from day 4 after tumor injection until the end of the experiment. To deplete NK cells, anti-NK1.1 neutralizing antibody (BioXCell, 200 mg/20 g mouse) was injected into the peritoneum on days 4 and 7 after tumor injection. In some experiments, 1×106 NK cells were injected via the tail vein on day 7 and day 10 after tumor injection. NK cells were preactivated with 1000 U/mL IL-2 (PeproTech) for 24 hours before injection as described earlier. To block TGF-β signaling, a selective TGF-β receptor antagonist LY346947 (Seleckchem, 0.2 mg/20 g mouse) was injected into the peritoneum every 2 days from day 4 after tumor injection.
Flow cytometry and sorting
Lungs were perfused with phosphate-buffered saline (PBS) and isolated from mice at day 14 after tumor injection. Single-cell suspensions were prepared via enzymatic digestion using the Lung Dissociation Kit (Miltenyi) and subsequent filtration through a 40 µm cell strainer (Falcon). Cells were treated with red blood cell lysis buffer (Biolegend) and resuspended in PBS including 2% bovine serum albumin (BSA; Sigma). 1×106 cells were incubated with 1 µL of anti-mouse CD16/CD32 antibody (Fc Block, BD Bioscience) for at least 15 min on ice, and stained for 60 min with antibodies to the following antigens; CD45 (30-F11), CD11b (M1/70), CD11c (N418), Ly6C (HK1.4), Ly6G (1A8), CD3 (17A2), NK1.1 (PK136), CD4 (GK1.5), CD8 (53–6.7), CD19 (6D5), H2-Kb (AF6-88.5), H2-Db (KH95), LAP/TGF-β1 (TW7-16B4), CD69 (H1.2F3), CD27 (LG.3A10), CD49a (HMa1), CD49b (DX5), NKp46 (29A1.4), DNAM1 (10E5), NKG2D (C7), Ly49A (YE1/48.10.6), Ly49C (14B11), NKG2A (16A11) from Biolegend, H2-Qa1 (6A8.6F10.1A6) from Novus, and F4/80 (Cl:A3-1) from Bio-Rad. Cells were resuspended in PBS including 2% BSA and 3 µM DAPI (BioLegend) before analysis. To detect FoxP3, cells were permeabilized by Fixation Permeabilization Reagent (eBioscience) for 30 min at room temperature, blocked with 2% normal rat serum, and stained with anti-FoxP3 antibody (FJK-16s, eBioscience) for 30 min at room temperature. Flow cytometry was performed using the LSRII cytometer (BD Biosciences) and analyzed using Flowjo software (TreeStar). For some experiments MAMs, monocytic myeloid-derived suppressor cells (M-MDSCs), and resident macrophages (RMACs) were sorted from the lung of tumor-injected mice using FACS Aria II (BD Bioscences).
In vitro NK cell cytotoxicity assay
1×103 target cells (E0771-LG:Fluc_NLR cells), 4×103 effector cells (splenic NK cells), and/or 3×103 suppressor cells (macrophages or MDSCs isolated from the lung containing E0771-LG metastatic tumors) were seeded into 96-well plates (Nunc) precoated with basement membrane extract (Geltrex, Gibco). These cells were cocultured in αMEM including 10% FBS, 1% penicillin/streptomycin, 1000 U/mL IL-2 (PeproTech), 20 ng/mL M-CSF (PeproTech), and 2.5 µM fluorogenic caspase-3 substrate (NucView 488, Biotium) at 37°C, 5% CO2, and imaged by IncuCyte Zoom Live-Cell Analysis System (Sartorius) at 10× magnification for up to 48–72 hours. The numbers of apoptotic tumor cells (nuclei showing large areas of overlapping red/green fluorescence) were counted using the IncuCyte Zoom software (Sartorius) as previously described.13 In some experiments, the following antibodies (25 µg/mL) were added in culture: anti-Ly49A (YE1/48.10.6), anti-H2-Kb (AF6-88.5), anti-TGF-β (19D8) from BioLegend, H2-Db (28-14-8), and Ly49C/I (5E6) from BD Biosciences.
Histological analysis
Perfused lungs in tumor-injected mice were isolated at day 10 after tumor injection and fixed with 10% neutral buffered formalin overnight. The lungs were embedded in paraffin and cut into 4 µm sections for immunohistochemistry (IHC) or in situ hybridization (ISH). For IHC, sections were deparaffinised, pretreated with hydrogen peroxide and incubated with Enzyme 1 (Leica, UK) for 10 min at 37°C to retrieve antigen. The sections were then stained using Rat ImmPRESS kit (Vector Labs) and Bond Intense R Detection System (Leica) with F4/80 antibody (1/200, polyclonal, Abcam). For ISH, sections were deparaffinised and pretreated with hydrogen peroxide, followed by hybridization with probes (NKp46; Mm-NCR1 or Control: Mm-UBC, Advanced Cell Diagnostics). Positive signals for Ncr1 (mRNA coding NKp46) were detected using RNAscope 2.5 LS-Brown Reagent Kit (Advanced Cell Diagnostics). Nuclei were counter stained by hematoxylin. Both staining processes were carried out using the Bond Rx Automated Stainer (Leica). Stained sections were dehydrated through graded alcohols and xylene and covered by a glass cover with DPX Mounting Media (CellPath). The stained slides were scanned by Axioscan.Z1 (Carl Zeiss) using a 40×0.95 NA Plan-Apochromat objective lens and analyzed by Tissue Studio V.4.4.2 software (Definiens). Briefly, cell nuclei showing blue color over the threshold were masked, and the region with dense nuclei localization was outlined as the tumor area. The size of each tumor area and total non-tumor area in the field were determined, and the number of brown spots that suggest Ncr1 or F4/80 expressing cells was counted in each area.
Statistical analyses
All samples were collected independently and analyzed from at least two independent experiments. Data were expressed as mean±SEM Significance was tested using the Student’s t test unless it is specified. P<0.05 was considered significant. Statistical analyzes were carried out using GraphPad Prism V.6.
Results
MAMs suppress NK cell induced tumor cell apoptosis in vitro
Using mouse models of metastatic breast cancer, we demonstrated that distinct myeloid cell populations termed MAMs and M-MDSCs in the metastatic lung suppress CD8+ T cell cytotoxicity in vitro.15 We thus hypothesized that MAMs and/or M-MDSCs also play key roles in suppression of NK cells in the metastatic tumor microenvironment.
To investigate this hypothesis, we used an experimental metastasis model where intravenously injected mouse mammary tumor cells (E0771-LG) reproducibly develop metastatic tumors in the lung of syngeneic C57BL/6 mice.11 15 In this model, numbers of M-MDSCs and MAMs, but not lung RMACs, were significantly increased in the lung containing metastatic tumors established by E0771-LG:Fluc cells (online supplemental figure 1 and figure 1A).15 Although regulatory T (Treg) cells are reported to inhibit NK cell-mediated tumor rejection in a mouse model of lung cancer,16 their contribution in our model should be minor as the number of Treg cells was reduced rather than increased in the metastatic lung compared with the normal lung (figure 1A).
Supplemental material

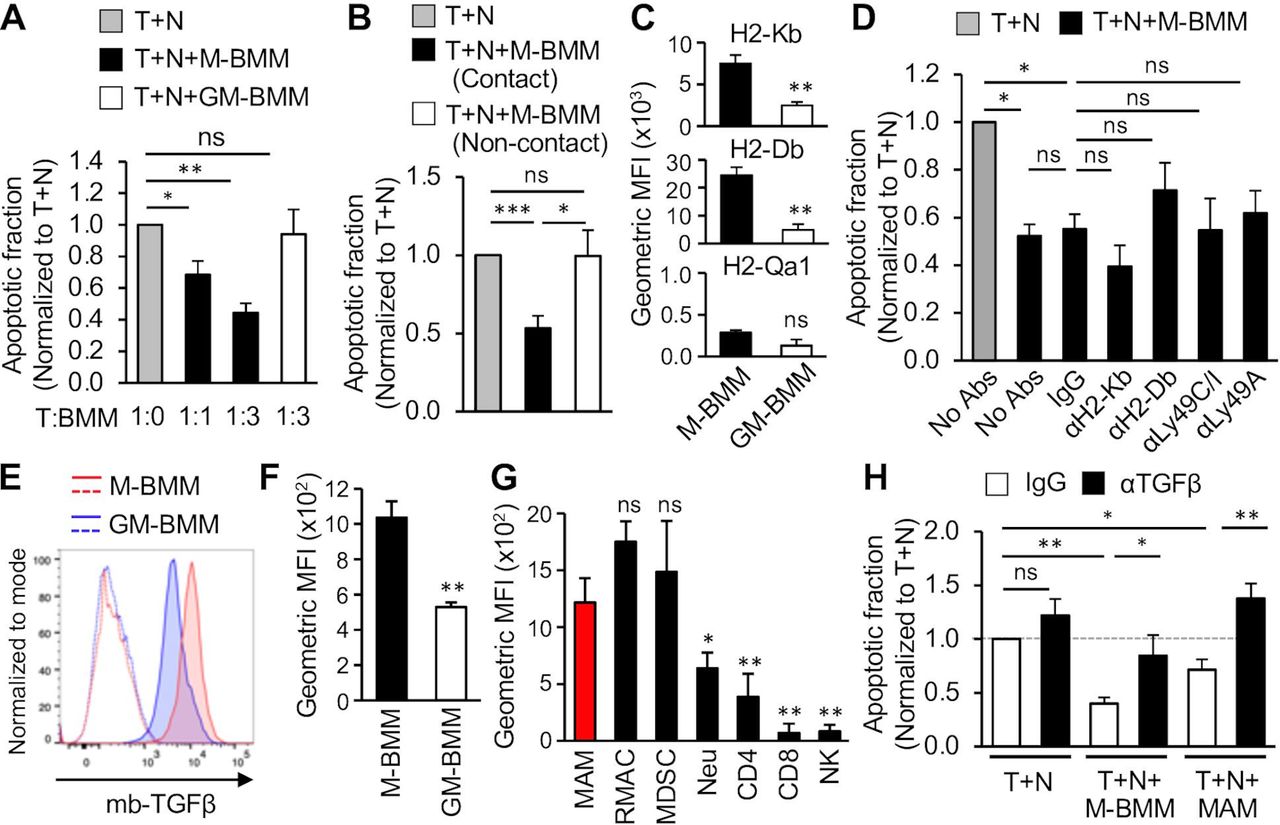
MAMs suppress cytotoxicity of natural killer (NK) cells in vitro. (A) Number of cells per milligram of lung tissue in untreated (normal; N) or tumor-injected (T) C57BL/6 mice at day 14 after tumor injection (n=3 in each group from two experiments). (B) Representative images from seven experiments showing cells in the red channel, green channel, and phase contrast merged with red and green channels. E0771-LG:NLR tumor cells were cultured with splenic NK cells at 1:4 of tumor:NK ratio in the presence of caspase-3 substrate for 24 hours. The right panel shows the same field under the red/green overlap mask where apoptotic tumor cells (large green fluorescent nuclei colocalized with red fluorescence, orange arrow heads) were distinguished from apoptotic NK cells (white arrow heads) and non-apoptotic tumor cells (blue arrowheads). Scale bar shows 100 µm. (C) Apoptotic fraction (ratio of apoptotic cells in total cells) of tumor cells (T) cultured with NK cells (N) at different T:N ratios for 24 hours (n=5 in each condition from five experiments). Data are normalized to tumor cell monoculture (T:N=1:0). (D) Apoptotic fraction of tumor cells cultured with NK cells at 1:4 of T:N ratio (T+N) or on their own (T only) for up to 54 hours (n=10 in each condition from ten experiments). Asterisk shows statistical significance compared with T only. (E) Apoptotic fraction of tumor cells cultured with NK cells (T+N) or with NK cells and myeloid cells (T+N+MDSC; T+N+MAM; T+N+RMAC) for 24 hours (n=8 in each condition from seven experiments). Tumor, NK, and myeloid cells were cocultured at 1:4:3 ratio. Data are normalized to T+N. All data show mean±SEM for all analyses: *p<0.05; **p<0.01; ***p<0.001; ns, not significant. MAM, metastasis-associated macrophage; M-MDSC, monocytic-myeloid derived suppressor cells; RMAC, resident macrophage; Treg, regulatory T cell.
We then investigated effects of MAMs and M-MDSCs on NK cell cytotoxicity against tumor cells. To this end, we established an in vitro assay where tumor cells expressing a red fluorescent protein (E0771-LG:Fluc_NLR) were cultured with normal splenic NK cells and the number of apoptotic tumor cells indicated by green fluorescence from a fluorogenic caspase-3 substrate was determined (figure 1B). In this assay, the apoptotic fraction (ie, the ratio of apoptotic (red/green double positive) tumor cells to total (red) tumor cells) was significantly elevated in accordance with increased tumor to NK cell (T:N) ratio (figure 1C) and reached the maximum at a 1:4 ratio by 36 hours of coculture (figure 1D). We thus cultured E0771-LG:Fluc_NLR and NK cells with MAMs or M-MDSCs at a 1:4:3 ratio (tumor:NK:myeloid cell) for 24 hours, and found that NK cell-induced tumor cell apoptosis (T+N) was significantly reduced in the presence of MAMs, but not M-MDSCs (figure 1E). RMACs isolated from the normal lung also did not affect cytotoxicity of NK cells. These results demonstrate that MAMs rather than M-MDSCs or RMACs play pivotal roles in NK cell suppression in the metastatic site.
Macrophages suppress NK cell cytotoxicity in vitro by TGF-β rather than MHC-I molecules
To further show that a distinct macrophage population is responsible for NK cell suppression, we investigated bone marrow-derived macrophages (BMMs) cultured with M-CSF (also known as CSF-1) or GM-CSF, called M-BMMs and GM-BMMs, respectively (online supplemental figure 2A). It is reported that the gene expression profile of GM-BMMs is closer to that of macrophages than to dendritic cells and is clearly distinct from that of M-BMMs.17 18 The phenotype of GM-BMMs (F4/80+CD11bLowCD11cHigh) is also distinguishable from that of M-BMMs (F4/80+CD11bHigh CD11cLow) (online supplemental figure 2), which represent RMACs and MAMs in the metastatic lung, respectively.19 NK cell-mediated tumor cell apoptosis was significantly reduced when M-BMMs were added into the coculture at either 1:4:1 or 1:4:3 ratios (tumor cell:NK cell:myeloid cell). In contrast, GM-BMMs did not change the NK cell-induced tumor cell apoptosis (figure 2A), suggesting that NK cell suppression is a specific feature of certain types of macrophages such as MAMs and their mimetic M-BMMs, rather than a common characteristic of all macrophage populations.
Supplemental material

Macrophages suppress NK cell cytotoxicity via membrane bound TGF-β rather than major histocompatibility complex class-I (MHC-I) molecules. (A) Apoptotic fraction of tumor cells cultured with NK cells at 1:4 of T:N ratio for 24 hours in the absence (T+N) or presence of bone marrow-derived macrophages (BMMs) cultured with M-CSF (T+N+M-BMM) or GM-CSF (T+N+GM-BMM) (n=8 in each condition from five experiments). Tumor to BMM ratio (T:BMM) is shown at the bottom. Data are normalized to T+N. (B) Apoptotic fraction of tumor cells cultured with NK cells in the absence (T+N) or presence (T+N+M-BMM) of M-BMMs under contact or non-contact conditions at 1:4:3 of T:N:BMM ratio for 24 hours (n=7 in each condition from four experiments). Data are normalized to T+N. (C) Geometric mean fluorescence intensity of H2-Kb, H2-Db, and H2-Qa1 on M-BMMs and GM-BMMs (n=5 from three experiments). (D) Apoptotic fraction of tumor cells cultured with NK cells (T+N) or with NK cells and M-BMMs (T+N+M-BMM). Cells were cultured with antibodies (Abs) against MHC-I molecules or their receptors denoted underneath (n=5 in each condition from five experiments). Data are normalized to T+N. P values were calculated by Kruskal-Wallis test. (E) Representative histogram showing membrane-bound TGF-β (mb-TGF-β) on BMMs. Dotted lines show control isotype matched IgG for each cell type (n=4 from four experiments). (F and G) Geometric mean fluorescence intensity of mb-TGF-β on BMMs (F; n=4 from four experiments) and major immune cells within the tumor-bearing lung at day 14 (G; n=6 from two experiments). Asterisk in G shows statistical significance compared with MAM. (H) Apoptotic fraction of tumor cells cultured with NK cells (T+N) or with NK cells and macrophages (T+N+M-BMM; T+N+MAM) in the presence of anti-TGF-β antibody or isotype matched IgG (n=5 in each condition from four independent experiments). Data are normalized to T+N (dotted gray line). P values were calculated by Kruskal-Wallis test. All data show mean±SEM for all analyzes: *p<0.05; **p<0.01; ***p<0.001; ns, not significant. MAM, metastasis-associated macrophage; MFI, mean fluorescence intensity; M-MDSC, monocytic-myeloid derived suppressor cells; NK, natural killer; RMAC, resident macrophage; TGF-β, transforming growth factor β.
We further performed the NK cell cytotoxicity assay in “non-contact” conditions where M-BMMs were placed in a Transwell chamber to physically separate them from tumor and NK cells, and found that M-BMMs under this condition did not reduce the apoptotic fraction of tumor cells cultured with NK cells (figure 2B). Therefore, macrophages require cell-to-cell contact to inhibit NK cell cytotoxicity. Since MHC-I molecules are cell surface ligands that can inhibit NK cell functions through their cognate receptors on NK cells,20 we investigated expression of MHC-I molecules detectable by flow cytometry in C57BL/6 mice (ie, H2-Kb, H2-Db, and H2-Qa1) in M-BMMs and GM-BMMs. The surface expression of H2-Kb and H2-Db was significantly higher in M-BMMs than GM-BMMs whereas H2-Qa1 expression was not significantly different between the BMM subtypes (figure 2C). We thus hypothesized that M-BMMs might suppress NK cell cytotoxicity via H2-Kb and/or H2-Db. However, blocking antibodies against H2-Kb, H-2Db, or their receptors Ly49C/I did not rescue the suppression of NK cell cytotoxicity caused by M-BMMs (figure 2D). Blockade of another inhibitory receptor, Ly49A, also did not reverse the BMM-mediated NK cell suppression (figure 2D). These results suggest that MHC-I molecules play minor roles in macrophage-mediated NK cell suppression in the in vitro models.
We then investigated the involvement of membrane-bound TGF-β (mb-TGF-β) in the in vitro macrophage models, since Treg cells and MDSCs are reported to suppress NK cell functions in a cell contact-dependent manner via mb-TGF-β.21 22 We found that mb-TGF-β expression was significantly higher in NK suppressive M-BMMs compared with non-suppressive GM-BMMs (figure 2E,F). Moreover, in metastatic tumor bearing lungs, MAMs expressed significantly higher mb-TGF-β than neutrophils, T cells (CD4 and CD8) and NK cells, whereas MDSCs and RMACs also expressed high levels of mb-TGF-β (figure 2 and online supplemental figure 3). We thus investigated the effects of an anti-TGF-β blocking antibody on macrophage-mediated NK cell suppression. In the presence of control IgG, M-BMMs and MAMs reduced NK cell-induced tumor cell apoptosis (figure 2H). However, this macrophage-mediated NK suppression was significantly inhibited by the anti-TGF-β antibody treatment (figure 2H), which suggests that macrophages suppress NK cell cytotoxicity through expression of mb-TGF-β rather than MHC-I molecules in our model.
Supplemental material
MAMs regulate activation, maturation, and accumulation of NK cells in the metastatic niche
To investigate the effects of MAMs on NK cell phenotype and functions in the metastatic tumor microenvironment, we used CSF-1 receptor conditional knockout (Csf1r-cKO) mice in which floxed CSF-1 receptor genes (Csf1rF/F) are deleted and thereby certain macrophage populations are ablated on doxycycline treatment.11 We injected E0771-LG tumor cells into the tail vein of Csf1r-cKO mice and treated with either doxycycline or vehicle from day 4 after tumor cell injection, a time point when tumor cells develop micro-metastases within the lung (figure 3A).15 As we have reported previously,15 doxycycline treatment significantly reduced the accumulation of MAMs, but not M-MDSCs or RMACs in the metastatic lungs of Csf1r-cKO mice (figure 3B).
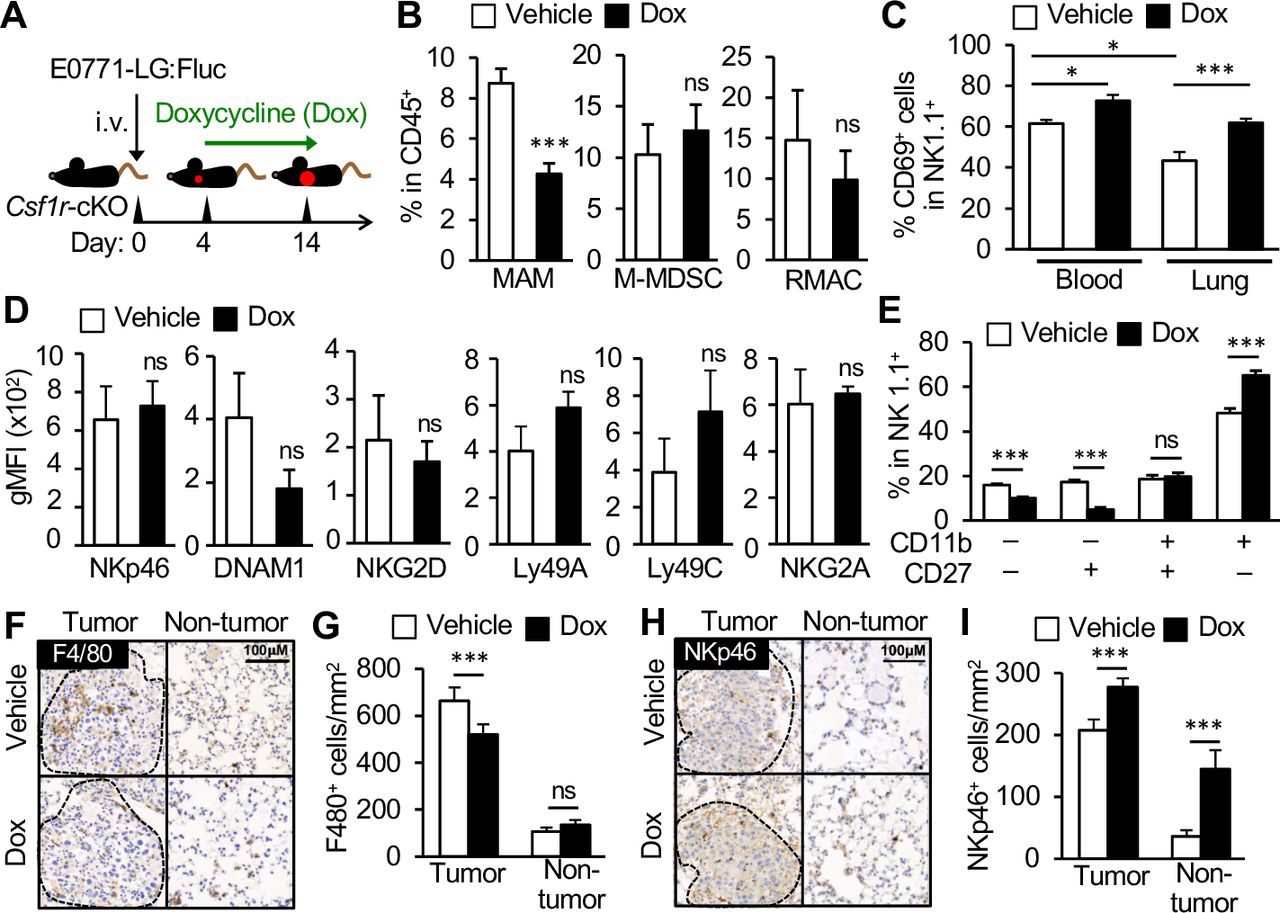
MAM depletion increases NK cell activation, maturation, and accumulation of NK cells within tumor bearing lungs. (A) A scheme of MAM depletion. Csf1r-cKO mice were intravenously injected with E0771-LG:Fluc cells and treated with doxycycline (Dox) from day 4 after tumor injection. The tumor-bearing lungs were isolated on day 14. (B) Percentage of MAMs, M-MDSCs, and RMACs in CD45+ cells in the metastatic lung of Csf1r-cKO mice treated with Dox or vehicle (n=6 in each group from two experiments). (C) Percentage of CD69+ cells in NK cells in the blood and lung with metastatic tumors in Csf1r-cKO mice treated with Dox or vehicle (n=9 in each group from two experiments. (D) Geometric mean fluorescence intensity (gMFI) of NK cell activating receptors (NKp46, DNAM1, NKG2D) and inhibitory receptors (Ly49A, Ly49C, NKG2A) on NK cells in the lung with metastatic tumors in Csf1r-cKO mice treated with Dox or vehicle (n=6 in each group from three experiments). (E) Frequency of NK cell subsets defined by CD11b and CD27 expression (denoted below) in the lung with metastatic tumors in Csf1r-cKO mice treated with Dox or vehicle (n=6 in each group from two experiments). All data show mean±SEM for all analyzes: *p<0.05; **p<0.01; ***p<0.001; ns, not significant. (F–I) Expression of F4/80 and NKp46 (Ncr1) in the lung with metastatic tumors in Csf1r-cKO mice treated with vehicle or Dox. (F and H) Representative stained images of tumor area (surrounded by dotted line) and non-tumor area. Scale bar shows 100 µm. (G and I) Number of F4/80+ or NKp46+ cells per mm2 of tumor area and non-tumor area (n=6 mice in each group. Total 111 and 152 tumors in vehicle and Dox were analyzed, respectively. All data show mean±SEM for all analyzes: *p<0.05; ***p<0.001; ns, not significant. MAM, metastasis-associated macrophage; M-MDSC, monocytic-myeloid derived suppressor cells; NK, natural killer; RMAC, resident macrophage.
Using this model, we first investigated the effects of MAM depletion on the expression of CD69, an NK cell activation marker. In vehicle treated Csf1r-cKO mice, the percentage of CD69+ NK cells was significantly lower in tumor-bearing lungs (43%±4%) compared with blood (62%±2%), suggesting the existence of an immune suppressive environment at the metastatic site (figure 3C). In contrast, the ratio of CD69 expressing NK cells in the tumor-bearing lung was significantly increased in doxycycline treated mice compared with vehicle treated mice (figure 3C). It is reported that CD69 expression is increased on activation of NK cells and high CD69 expression correlates with high cytotoxic activity of circulating NK cells.23 24 Although tissue resident NK cells (characterized by CD49a expression) can also “constantly” express CD69,25 the number of CD49a+ NK cells was very low in the lung25 26 and was not changed by doxycycline treatment (online supplemental figure 4). Therefore, the increase in CD69+ NK cells by MAM depletion is likely to be caused by enhanced activation of tumor-infiltrating NK cells rather than accumulation of tissue resident NK cells within the metastatic lung. Together with data from our in vitro assays, these results indicate that MAMs suppress NK cell activation within the metastatic tumor niche.
Supplemental material
Using the same model, we further investigated other potential mechanisms by which MAMs might inhibit NK cell functions in vivo. We first investigated the effects of MAM depletion on the expression of NK cell activating receptors such as NKp46, NKG2D, and DNAM-1, as signaling through these receptors promotes the cytolytic ability of NK cells20 27 and other suppressor cells such as Treg cells can induce anergy of NK cells by downregulating NKG2D expression on NK cells.21 In the metastatic lung of vehicle-treated Csf1r-cKO mice, NK cells expressed detectable levels of NKp46, NKG2D, and DNAM-1. However, doxycycline treatment did not alter their expression (figure 3D). Although NK cells in the metastatic lung expressed inhibitory receptors including Ly49A, Ly49C/I, and NKG2A that negatively regulate NK cell cytotoxicity,20 27 their expression was also not changed by MAM depletion (figure 3D). It is therefore unlikely that MAMs inhibit cytotoxicity of NK cells by modulating the expression of NK cell regulatory receptors.
We next investigated the effects of MAM depletion on the maturation of NK cells defined by differential surface expression of CD11b and CD27, as mouse NK cells acquire effector functions through four consecutive maturation stages; CD11b–CD27–→CD11b–CD27+→CD11b+CD27+→CD11b+CD27–.28 In the metastatic lung of doxycycline treated mice, the percentage of “immature” NK cells (CD11b–CD27– and CD11b–CD27+) was significantly reduced and the ratio of the most mature NK subset (CD11b+CD27–) was concomitantly increased (figure 3E). These results suggest that MAMs prevent maturation of NK cells at the metastatic site.
We further investigated the effects of MAM depletion on NK cell infiltration into metastatic tumors. To this end, we detected F4/80 positive macrophages and NKp46 (Ncr1) expressing NK cells in the metastatic lung by IHC and in situ RNA hybridization, respectively. Consistent with our previous data, the number of F4/80+ macrophages in the tumor area (considered MAMs) was reduced in Csf1r-cKO mice treated with doxycycline compared with those treated with vehicle, whereas the number of macrophages in the lung parenchyma was not changed between the groups (figure 3F,G). In contrast, the number of NK cells within the tumor area was significantly higher in doxycycline treated mice compared with vehicle treated animals (figure 3H,I). The number of NK cells in the lung parenchyma (non-tumor area) was also increased by doxycycline treatment in tumor-bearing Csf1r-cKO mice. Collectively, these results suggest that MAMs suppress activation, maturation, and accumulation of NK cells in the metastatic tumor microenvironment.
MAM targeting enhances efficacy of NK cells in suppressing metastatic tumor outgrowth
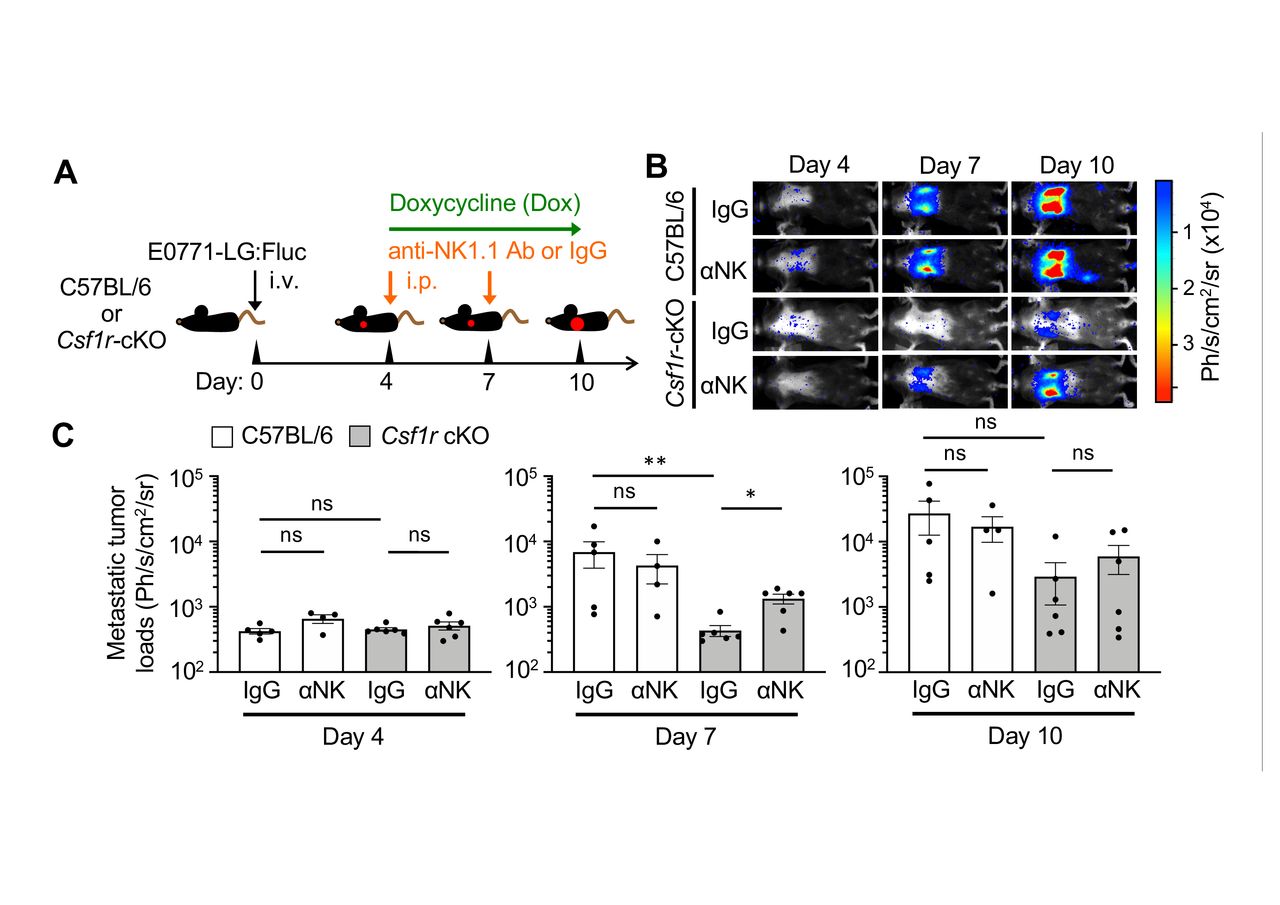
We thus hypothesized that depletion of MAMs would suppress metastatic tumor outgrowth by promoting NK cell functions. To investigate this hypothesis, tumor-injected Csf1r-cKO mice were treated with doxycycline and injected with anti-NK1.1 antibody, which depletes MAMs (figure 3B) and NK cells29 in the metastatic lung, respectively. As a MAM-intact control, syngeneic C57BL/6 mice were given the same treatments. In order to trace the metastatic tumor growth in the lung, E0771-LG cells expressing firefly luciferase (E0771-LG:Fluc) were injected into the tail vein of mice. We have reported that intravenously injected E0771-LG:Fluc cells reproducibly extravasate into the lung by day 2, develop into small metastatic foci by day 7, and grow into macro-metastatic lesions by day 14.11 15 We thus started doxycycline and/or anti-NK1.1 antibody treatment from day 4 after tumor cell injection in order to investigate effects of the treatments on the expansion of metastatic tumor cells after their seeding (figure 4A).
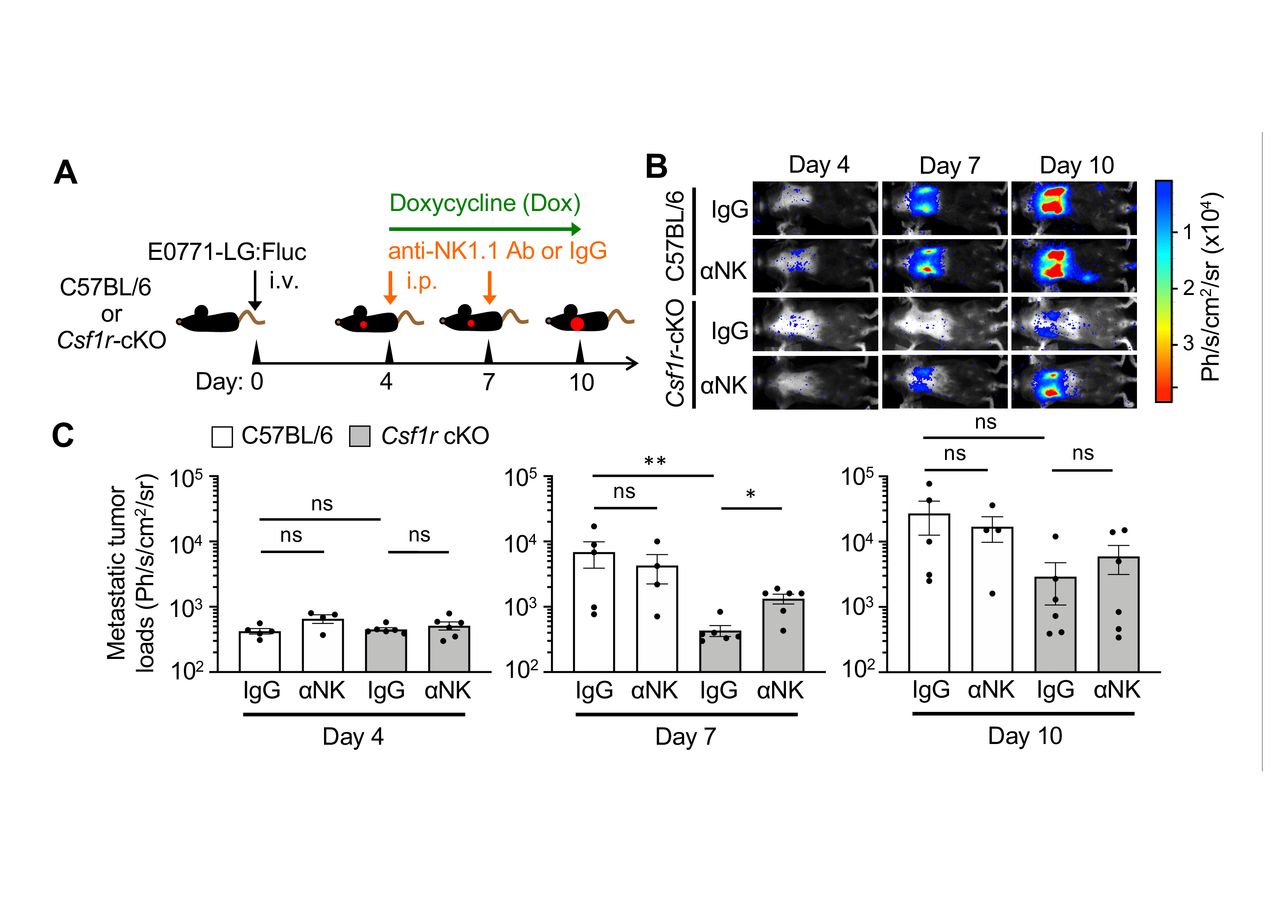
Depletion of MAMs suppresses metastatic tumor expansion via natural killer (NK) cell-mediated mechanism. (A) A scheme of metastasis-associated macrophage and NK cell depletion. Csf1r-cKO mice or control C57BL/6 mice intravenously injected with E0771-LG:Fluc cells were treated with doxycycline (Dox) from day 4 to day 10 after tumor injection. The animals were also injected with anti-NK1.1 antibody (αNK) or control IgG into the peritoneum on days 4 and 7. (B and C) Representative bioluminescence images (B) and mean metastatic tumor loads (C) on days 4, 7, and 10 after tumor injection (n=5, C57BL/6:IgG; n=4, C57BL/6:αNK; n=6, Csf1r-cKO:IgG; n=6, Csf1r-cKO:αNK from two experiments). P values were calculated by Kruskal-Wallis test. All data show mean±SEM; *p<0.05; **p<0.01; ns, not significant.
Upon treatment with doxycycline and control IgG, metastatic tumor loads within the lung area were significantly reduced in Csf1r-cKO mice compared with C57BL/6 mice at day 7 after tumor injection (figure 4B,C). However, the reduction in metastatic tumor load seen in Csf1r-cKO mice was significantly reversed by depletion of NK cells (figure 4B,C). The average tumor load at day 10 in Csf1r-cKO mice treated with IgG was also lower than that of control C57BL/6 mice, although this was not statistically significant. These data suggest that MAM depletion suppresses the expansion of metastatic tumor cells by activating NK cells during the early phase of metastasis, whereas activation of intrinsic NK cells is not sufficient to block later metastatic tumor expansion.
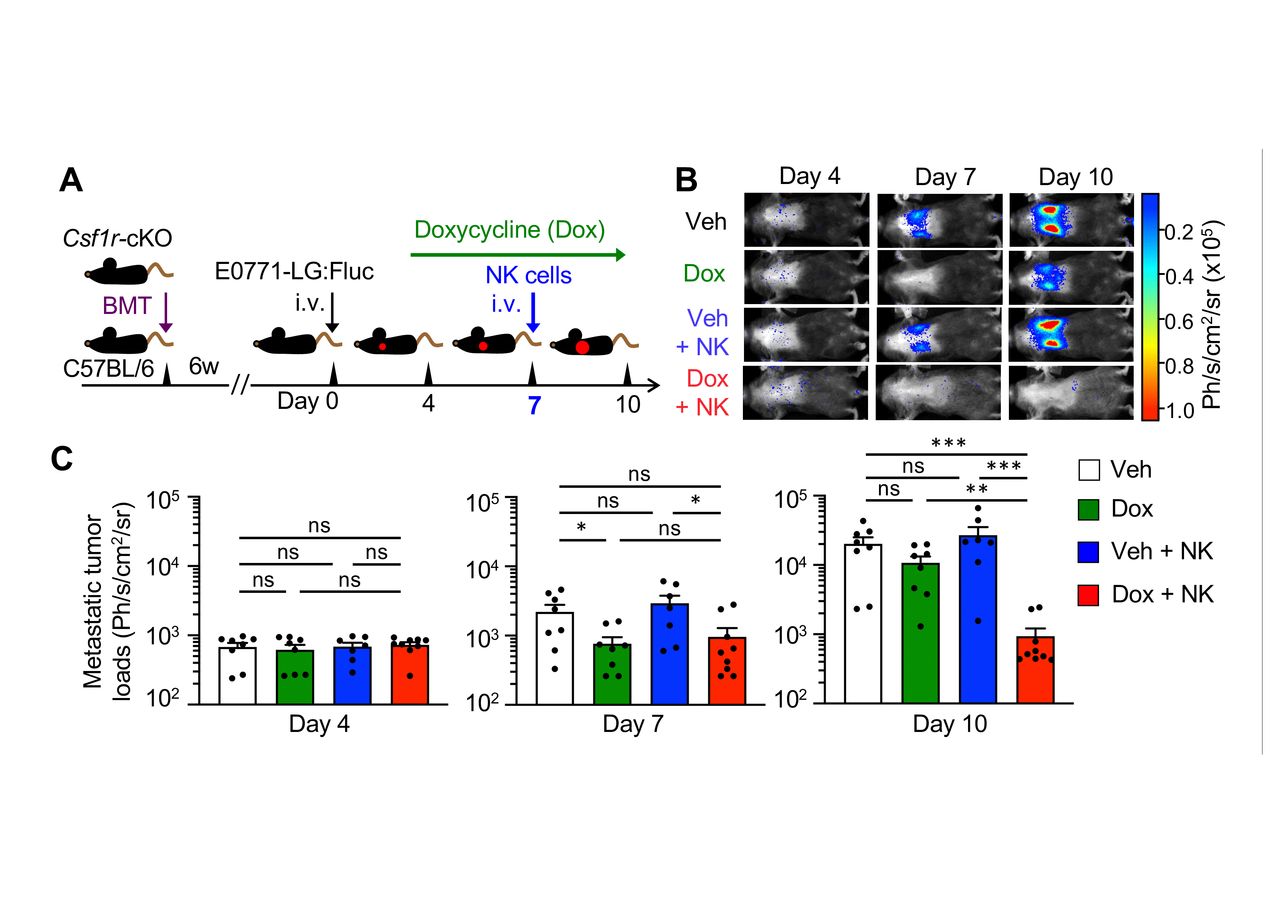
We then investigated whether adoptive NK cell transfer can inhibit metastatic tumor outgrowth in the presence or absence of MAMs. In order to restrict genetic alterations to blood cells, we transferred bone marrow cells from Csf1r-cKO mice into lethally irradiated recipient C57BL/6 mice in this experiment (figure 5A). Consistent with our previous results (figure 4), metastatic tumor growth in Csf1r-cKO:BMT mice treated with doxycycline was suppressed by day 7 after tumor injection whereas it was increased by day 10 (figure 5B,C). Importantly, adoptive transfer of NK cells significantly reduced the metastatic tumor load at day 10 in doxycycline treated mice, but not in vehicle treated mice (figure 5B,C). Therefore, the depletion of MAMs not only enhances intrinsic NK cell activity but also improves the efficacy of transferred NK cells, which can suppress metastatic tumor expansion.

NK cell infusion in combination with metastasis-associated macrophage (MAM) depletion suppress metastatic tumor outgrowth. (A) A scheme of MAM depletion and NK cell infusion. C57BL/6 mice that received bone marrow transplantation (BMT) from Csf1r-cKO were intravenously injected with E0771-LG:Fluc cells after 6 weeks from BMT and treated with doxycycline (Dox) or vehicle (Veh) from day 4 after tumor injection. Cohorts of animals were intravenously injected with ex vivo cultured NK cells on day 7. (B and C) Representative bioluminescence images (B) and mean metastatic tumor loads (C) on days 4, 7, and 10 after tumor injection (n=8, Veh; n=8, Dox; n=7, Veh+NK; n=9, Dox+NK from two experiments). P values were calculated by Kruskal-Wallis test. All data show mean±SEM; *p<0.05; **p<0.01; ***p<0.001; ns, not significant. NK, natural killer.
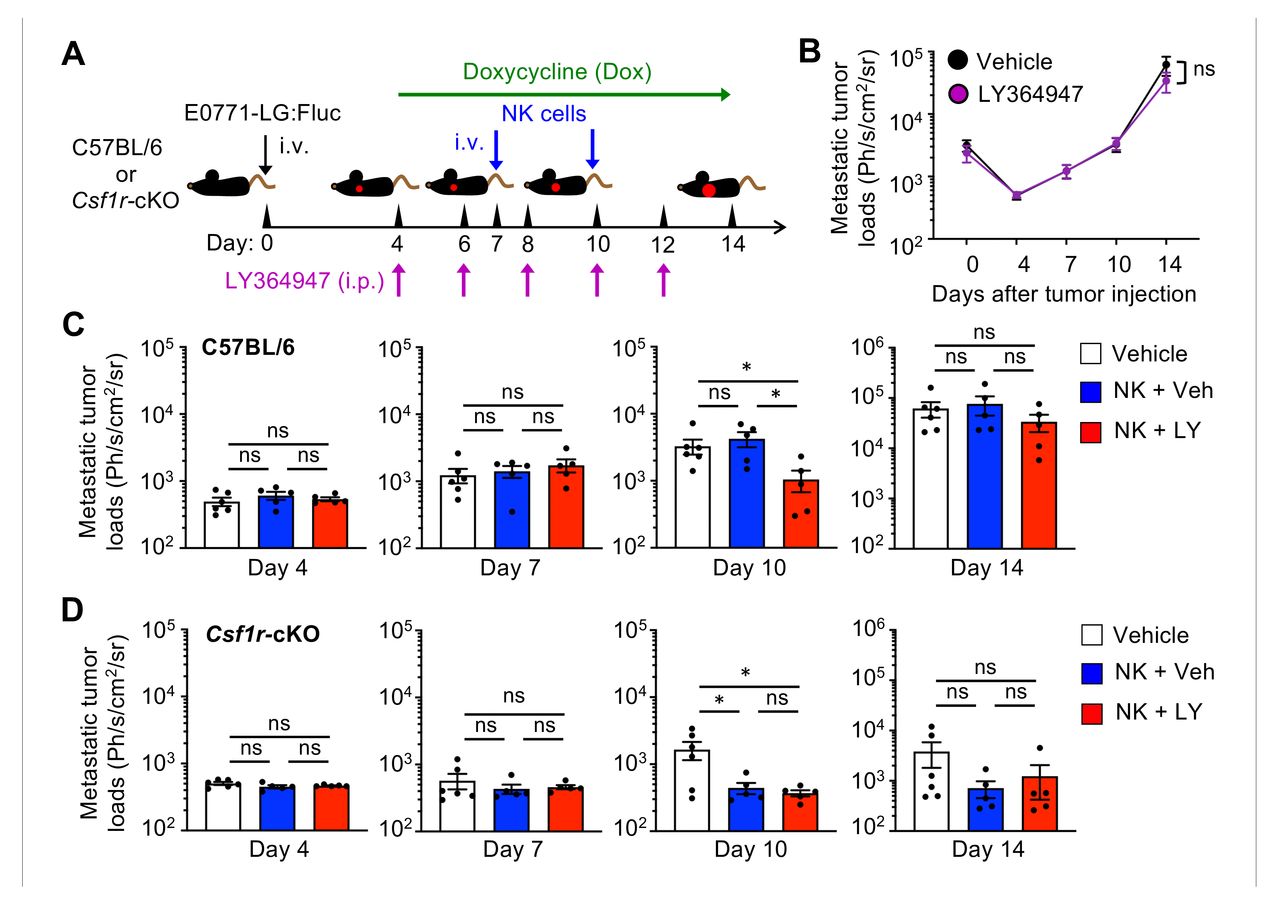
To investigate the involvement of TGF-β in the MAM-mediated constraint of infused NK cell efficacy, tumor injected C57BL/6 or Csf1r-cKO mice were treated with the potent and selective TGF-β receptor antagonist (LY364947) and doxycycline from day 4 after tumor injection (figure 6A). This treatment by itself did not change metastatic tumor growth in C57BL/6 mice (figure 6B). We then adoptively transferred NK cells into a cohort of mice on days 7 and 10 and found that NK cell infusion in combination with LY364947 treatment significantly reduced metastatic tumor load at day 10 after tumor injection (figure 6C). In contrast, LY364947 treatment did not affect the efficacy of transferred NK cells in the macrophage-depleted Csf1r-cKO mice (figure 6D), which strongly suggests that MAMs suppress efficacy of infused NK cells in part through expression of TGF-β. In this model, NK cell injection with LY364947 treatment or macrophage depletion did not significantly suppress the late metastatic tumor growth from day 10 to day 14 (figure 6C,D). Collectively, these data demonstrate that MAM targeting (eg, depletion of MAM or blockade of TGF-β signal transmission by MAMs) promotes NK cell cytotoxicity and thereby enhances efficacy of NK cell infusion in suppressing “early” metastatic tumor expansion, whereas further improvement of NK cell efficacy is required to suppress ”late” stage of metastatic tumor growth.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transforming growth factor β (TGF-β) receptor antagonist enhances efficacy of NK cell infusion in suppressing metastatic tumor expansion. (A) A scheme of treatments. Tumor injected C57BL/6 or Csf1r-cKO mice were treated with doxycycline (Dox) from day 4 to day 14 after tumor injection and injected with TGF-β receptor antagonist (LY364947) or vehicle (Veh) every 2 days from day 4. Cohorts of animals were intravenously injected with ex vivo cultured NK cells on day 7 and day 10. (B) Mean metastatic tumor loads in C57BL/6 mice on days 4, 7, 10, and 14 after tumor injection (n=5 in each group from two experiments). (C) Mean metastatic tumor loads in C57BL/6 mice on days 4, 7, 10, and 14 after tumor injection (n=6, vehicle; n=5, Veh+NK; n=5, LY+NK from two experiments). (D) Mean metastatic tumor loads in Csf1r-cKO mice on days 4, 7, 10, and 14 after tumor injection (n=6, vehicle; n=5, Veh+NK; n=5, LY+NK from two experiments). P values were calculated by Kruskal-Wallis test. All data show mean±SEM; *p<0.05; ns, not significant. NK, natural killer.
Discussion
NK cell infusion therapy has been identified as a novel immunotherapy to treat malignant tumors including metastatic breast cancer that are resistant to conventional therapies. However, NK cells do not exhibit their full cytotoxic capacity in vivo compared with in vitro and are not effective in treating most solid tumors.30 Thus, identification and thereby blockade of immune suppressive factors in the tumor microenvironment represents a key issue in order to design effective NK cell-based therapies for solid tumors. However, the major component of the NK cell suppressive tumor microenvironment seems to be different between tumor types. For example, NK cell-mediated tumor rejection in the metastatic lung is suppressed by Treg cells in a mouse model of melanoma,16 whereas NK cell activation is suppressed by MDSCs rather than Treg cells in mouse models of liver and lung cancer.22 31 Therefore, a better understanding of target cell types characteristic for metastatic breast cancer is important to improve the therapeutic effects of NK cells on this lethal disease. Here we have shown, to our knowledge for the first time, that MAMs are the main negative regulator of NK cells in the metastatic tumor niche established in a mouse model of metastatic breast cancer.
In mouse models of breast cancer, MAMs are recruited to the tumor challenged lung and promote extravasation and survival of metastasizing tumor cells, which accelerates the establishment of lethal metastatic foci.19 32 Therefore, blockade of MAMs have been considered an important therapeutic strategy to prevent metastatic tumor outgrowth.33 34 In this study, we have shown that depletion of MAMs can inhibit metastatic expansion of breast cancer cells not only by restricting pro-metastatic tumor cell functions but also by promoting NK cell mediated antitumor immune reactions in the metastatic site. In contrast to MAMs, RMACs in the normal lung did not suppress NK cell cytotoxicity. Interestingly, we have found that BMMs cultured with GM-CSF also do not suppress NK cell-induced apoptosis of mammary tumor cells, whereas BMMs cultured with M-CSF do. Since CSF-1 signaling is essential for the differentiation and survival of MAMs but not RMACs in the metastatic lung,15 19 these data suggest that CSF-1 dependent macrophage populations including MAMs acquire NK cell suppressive functions and can be a therapeutic target to boost antitumor NK cell functions. In contrast, RMACs in the lung of normal mice are reported to promote the cytotoxicity of NK cells in vitro,35 suggesting that RMACs in the lung have potential to activate rather than suppress antitumor NK cell functions. In fact, RMACs in the tumor-challenged lung activate intrinsic NK cells and thereby restrict pulmonary metastasis formation on treatment with blocking antibodies for CD39 in a mouse model of melanoma.36
We suggest here that one of the major mechanisms behind NK cell suppression by MAMs and M-BMMs is the expression of mb-TGF-β. Consistent with our results, bone marrow derived macrophages cultured with M-CSF in combination with IL-4 and IL-13 can inhibit NK cell expression of a degranulation marker CD107a in a TGF-β and contact-dependent manner.37 Although precise mechanisms by which mb-TGF-β suppress NK cells have not been identified yet, TGF-β is reported to reduce the expression of an NK-activating receptor NKG2D in NK cells, which is associated with impaired NK cell cytotoxicity.38 Another study suggests that TGF-β promotes the conversion of NK cells (CD49a–CD49b+) into less cytotoxic type I innate lymphoid cells (CD49a+CD49b–), which can also inhibit NK cell-mediated immune surveillance.39 However, it is unlikely that MAMs use these mechanisms in our model since depletion of MAMs did not change the expression of activating and inhibitory receptors on NK cells or percentage of CD49a+CD49b– cells in the metastatic lung. Thus, further studies are needed to delineate how mb-TGF-β limits NK cell-mediated antitumor immune reactions. Interestingly, mb-TGF-β was expressed also by M-MDSCs at comparable levels to MAMs, whereas M-MDSCs did not suppress NK cell cytotoxicity toward cancer cells in vitro. Since M-MDSCs can differentiate into MAMs at the metastatic site,15 it is possible that MAMs acquire additional molecules that suppress NK cells synergistically with mb-TGF-β during their differentiation in the tumor microenvironment. The enhancement of NK cell recruitment into the parenchyma of metastatic lungs by MAM depletion also suggests the existence of additional mechanisms other than the mb-TGF-β dependent NK cell suppression. In mouse models of pulmonary metastasis, NK cells introduced into the blood migrate evenly to the non-tumor area first, and then redistribute within the organ toward the metastatic tumor area.40 Based on these results, it is possible that MAMs prevent the recruitment of circulating NK cells into the parenchyma of the metastatic lung, which subsequently reduces their accumulation within metastatic tumors. Our data suggest that MAMs also prevent differentiation of NK cells within the metastatic site. Although NK cells develop in the bone marrow and exit as mature cells, peripheral NK cell populations can also originate from site-specific immature NK cells.41 Although maturation status or origin of NK cells in human cancer is still unclear, a mouse study has shown that NK cells in primary mammary tumors express lower levels of CD27 compared with NK cells in the spleen,42 suggesting the accumulation of immature NK cells in the tumor microenvironment. Since CD27low NK cells demonstrate lower cytotoxicity against target cells compared with CD27high NK cells,43 these results suggest that prevention of NK cell maturation within the metastatic site may be another mechanism behind MAM mediated suppression of intrinsic NK cells in vivo.
This study also highlighted the importance of MAM targeting to enhance therapeutic efficacy of adoptively transferred NK cells. On the other hand, several studies have shown that MDSCs can suppress NK cell functions and are thus important targets to improve NK cell-based immunotherapies.44 MDSCs include two major subsets called monocytic (M-) and polymorphonuclear (PMN)-MDSCs that are characterized in mice as CD11b+Ly6ChighLy6G− and CD11b+Ly6ClowLy6G+, respectively. However, most of the former studies used an anti-Gr-1 antibody that recognizes both Ly6C and Ly6G to identify MDSCs. Since PMN-MDSC but not M-MDSC subset inhibits the activation of NK cells in an in vitro model of vaccinia virus infection,45 it is possible that PMN-MDSCs rather than M-MDSCs play major roles in NK cell suppression in the particular tumor microenvironment. In line with this notion, blockade of PMN-MDSC recruitment enhances therapeutic efficacy of transferred NK cells in a mouse model of oral cancer.46 However, depletion of Ly6G+ cells did not alter the number of metastatic foci developed by E0771-LG cells in the lung,11suggesting minor contribution of PMN-MDSCs in metastatic formation in our model. These results emphasize the importance of the selection of target cell types appropriate to each cancer type in order to design an NK cell infusion therapy in combination with suppressor cell targeting.
In our experimental metastasis model, intravenously transplanted E0771-LG cells extravasate and colonize in the lung by day 4 after tumor injection, form small metastatic foci by day 7, and develop into lethal macro-metastatic lesions by day 14.11 15 Here, we have shown that NK cell infusion at day 7 reduces metastatic tumor growth by day 10 after tumor injection when macrophages are depleted or TGF-β signaling is blocked from day 4. These results indicate that NK cell infusion combined with MAM targeting can suppress expansion of metastatic tumor cells that have disseminated into the metastatic site and established small metastasis foci. In this model, however, the development of macro-metastatic lesions by day 14 was not suppressed by NK cell infusion combined with the TGF-β receptor antagonist, which suggests the involvement of other suppressive mechanisms in the later stage of metastatic tumor expansion. It has been reported that several cytokines such as IL-6, G-CSF, and activin-A can attenuate NK cell functions in vitro.46–48 It is possible that these cytokines are upregulated in macro-metastatic lesions and can also contribute to the reduced efficacy of infused NK cells. On the other hand, it is worth noting that the number of NK cells infused in this study (1×106) was much lower than that used in previous studies (5–10×106).49–52 It has been reported that NK cells in C57BL/6 mice can inhibit subcutaneous tumor growth when 1×103 of cancer cells are inoculated but fail to suppress tumor development initiated by 1×105 of cancer cells,53 suggesting the requirement for a certain number of NK cells to eliminate a large amount of cancer cells. It is therefore possible that efficient delivery of increased numbers of infused NK cells in addition to MAM targeting is necessary to suppress the outgrowth of established tumor lesions especially at later stage of metastasis.
In summary, this study indicates that MAMs are critical NK cell suppressors in metastatic tumors and provides a proof of concept that MAM targeting can improve the efficacy of NK cells. Further investigation of the mechanism behind MAM-mediated NK cell suppression, along with definition of efficient NK cell delivery methods, will lead to the improvement of therapeutic efficacy of NK cell-infusion therapy for metastatic breast cancer.
Acknowledgments
The authors thank Dr Mihich for providing E0771 cells, Dr Webb for maintenance of mouse colonies, Dr Winchester for critique of the article, and Dr Pollard for scientific advice. Flow cytometry data were generated with support from the QMRI Flow Cytometry and Cell Sorting Facility, The University of Edinburgh. Histological data were generated with support from the Histology Facility, CRUK Beatson Institute.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DB and TK designed and carried out experiments and wrote the manuscript. DB, DD-S, TK, and NC established and performed the in vitro NK cell cytotoxicity assay. CN and LC generated histological data. DS carried out computational analyses of histological data.
Funding This study was funded by a Wellcome Trust Seed Award in Science (109657/ Z/15/Z) and a Medical Research Council (MRC) Career Development Award (MR/S006982/1) to TK. LMC laboratory is supported by Cancer Research UK (A17196 and A23983). NC laboratory is supported by Cancer Research UK funding (C42454/A28596 and C42454/A24892). The MRC Centre for Reproductive Health at the University of Edinburgh receives a core funding support from MRC (MR/N022556/1).
Competing interests No, there are no competing interests.
Patient consent for publication Not required.
Ethics approval All animal experiments were performed in accordance with the Animals (Scientific Procedures) Act 1986, the EU Directive 2010/63, and under licensed permission from the UK Home Office (P526C60B3) and local ethics committee (BVS, University of Edinburgh).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data sharing not applicable as no datasets generated and/or analyzed for this study. Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.