Article Text

Abstract
Lung cancer remains the leading cause of cancer death worldwide despite the significant progress made by immune checkpoint inhibitors, including programmed death receptor-1 (PD1)/PD ligand 1 (PDL1)-blockade therapy. PD1/PDL1−blockade has achieved unprecedented tumor regression in some patients with advanced lung cancer. However, the majority of patients fail to respond to PD1/PDL1 inhibitors. The high rate of therapy non-response results from insufficient PDL1 expression on most patients’ tumors and the presence of further immunosuppressive mechanisms in the tumor microenvironment. Here, we sensitize non-responding tumors from patients with lung cancer to PD1-blockade therapy using highly cytotoxic expanded natural killer (NK) cells. We uncover that NK cells expanded from patients with lung cancer dismantle the immunosuppressive tumor microenvironment by maintaining strong antitumor activity against both PDL1+ and PDL1− patient tumors. In the process, through a contact-independent mechanism involving interferon γ, expanded NK cells rescued tumor killing by exhausted endogenous TILs and upregulated the tumor proportion score of PDL1 across patient tumors. In contrast, unexpanded NK cells, which are susceptible to tumor-induced immunosuppression, had no effect on tumor PDL1. As a result, combined treatment of expanded NK cells and PD1-blockade resulted in robust synergistic tumor destruction of initially non-responding patient tumors. Thus, expanded NK cells may overcome the critical roadblocks to extending the prodigious benefits of PD1-blockade therapy to more patients with lung cancer and other tumor types.
- killer cells
- natural
- lung neoplasms
- immunotherapy
- programmed cell death 1 receptor
- lymphocytes
- tumor-infiltrating
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- killer cells
- natural
- lung neoplasms
- immunotherapy
- programmed cell death 1 receptor
- lymphocytes
- tumor-infiltrating
Background
Lung cancer is the leading cause of cancer death worldwide. In 2018 alone, there were over 1.7 million lung cancer-related deaths, reflecting a dismal 5-year survival rate of less than 18%. At the time of diagnosis, the majority (80%) of lung cancer patients (LCP) already have locally advanced or metastatic disease, which continues to progress despite chemotherapy.1 As a result, the progress made by immune checkpoint inhibitors over the past decade has been revolutionary, with antibody blockade of programmed death receptor-1 (PD1) achieving unprecedented durable tumor regression in some patients with advanced lung tumors, melanoma and a growing list of other cancers. However, only ~10% of patients benefit from the therapy.2 Non-response to PD1-blockade therapy is associated with insufficient PD ligand 1 (PDL1) expression (Tumor Proportion Score, TPS) on patient tumors and additional mechanisms of immunosuppression in the tumor microenvironment.3–5 Indeed, a number of landmark trials have shown that an important component for sustained immunotherapeutic efficacy is the ability to shift the immunosuppressive tumor microenvironment to a proinflammatory milieu and restore the antitumor functions of exhausted endogenous immune cells.2 3 6–9 Thus, new immunotherapies that can both sustain strong inflammatory antitumor activity in the tumor microenvironment and increase PDL1 TPS would hold promise to extend the remarkable therapeutic benefits of PD1-blockade therapy to more patients.
The antitumor cytokine interferon-gamma (IFNγ), released by cytotoxic natural killer (NK) cells and T cells, is a critical driver of PDL1 expression on tumors and a positive predictor of clinical response to immunotherapies.8 10–12 However, the antitumor functions of NK cells and T cells are significantly hindered by the tumor microenvironment.13 14 Our previous work uncovered that NK cells from the peripheral blood and tumors of breast and patients with ovarian cancer could be expanded ex vivo for cell therapy. On adoptive transfer to mice, these expanded NK cells (exNK) were capable of sustaining antitumor activity against tumors and eliminated macroscopic ovarian tumors.15–17 Another study has also demonstrated that NK cells expanded using membrane particles were capable of upregulating PDL1 on tumor cell lines.18 In the current study, we sought to assess (1) the therapeutic potential of exNK cells against tumors from advanced-stage LCP and (2) whether exNK cells can additionally sensitize patients’ non-responding tumors to PD1-blockade therapy.
Methods
Peripheral blood, pleural effusions, and tumor pieces were obtained with written informed consent from LCP at St. Joseph’s Healthcare in Hamilton, Ontario. Pleural effusions were collected from patients via thoracentesis and tumor pieces were collected via surgical resection, both of which were conducted as part of the patients’ standard care. Peripheral blood was collected from healthy donors (HD) with written informed consent at McMaster University. NOD-Rag1null IL2rgnull (NRG) mice were originally obtained from Jackson Laboratory and bred and housed in specific pathogen-free conditions at McMaster’s Central Animal Facility.
Processing of lung tumors, pleural effusions and peripheral blood
Lung tumors were minced into ~1 mm3 pieces in αMEM medium containing collagenase IV and DNase I. Tumor pieces were then incubated in the media on a plate shaker at 37°C for 2 x, 1-hour intervals and pipetted vigorously in between incubations to break up the pieces. Cells were filtered through a 70 µm filter, pelleted via centrifugation, then washed with phosphate-buffered saline (PBS). Cells from pleural effusions and peripheral blood were isolated via density-gradient centrifugation with Lymphoprep (Stemcell Technologies, Vancouver, British Columbia, Canada), then washed with PBS.
NK cell expansion and isolation
NK cells were cultured in RPMI medium supplemented with 10% FBS, 1% hepes, 1% penicillin-streptomycin, and 1% L-glutamine. NK cells were expanded from the peripheral blood mononuclear cells (PBMCs) of HD, or PBMCs, pleural effusion cells, or tumors of LCP. NK cells were expanded by coculture with irradiated K562-membrane-bound--interleukin (IL)-21 feeder cells and recombinant human (rh) IL-2 as described previously.16 Expansion was conducted for at least 3 weeks prior to functional assays. For experiments using freshly isolated NK cells, NK cells were isolated via positive selection from PBMCs using a CD56+ selection kit from Stemcell, according to the manufacturer’s instructions.
Flow cytometric staining
To discriminate live/dead cells, cells were first stained for 30 min with fixable viability stain (eBioscience). Cells were then washed with 0.2% bovine serum albumin in PBS and stained with extracellular antibodies for 30 min. Cells not undergoing intracellular staining were then fixed in 1% paraformaldehyde. For intracellular staining, cells were fixed with Cytofix/Cytoperm from BD Biosciences for 20 min and then stained with intracellular antibodies in a 1X Perm/Wash solution (BD Bioscience) for 30 min. Sample acquisition was carried out on a BD LSRFortessa and analyzed on FlowJo software. See (online supplemental methods) for a complete list of the antibodies used.
Supplemental material
Functional assays and PD1/PDL1 expression
Flow cytometry-based killing and degranulation assays were conducted in complete RPMI media as described previously.15 16 Specifically, for killing assays against A549 cells, NK cells were coincubated with CFSE-labeled A549s at 1:1, 5:1, and 10:1 effector-to-target ratios for 5 hours, following which cells were stained with fixable viability stain. For killing assays against PDL1+ A549 cells, A549s were pretreated with rhIFNγ (20 ng/mL) for 48 hours to induce PDL1 expression and washed three times prior to seeding for the assay.
Killing assays against patient tumors were conducted using a transwell model ex vivo. Patient tumors were seeded on both apical and basolateral surfaces of the transwell. NK cells were added to the apical chamber at a 10:1 effector-to-target ratio and coincubated for 48 hours unless indicated otherwise. Tumors from the same patients were used across NK cell groups (HD peripheral blood (pb)NK, and LCP pbNK and tumor-associated NK). Nivolumab, a PD1 blocking antibody used clinically for the treatment of PDL1+ cancer,11 was added to wells at 1 µg/mL where indicated. Low dose IL-15 (1 ng/mL) was added as an NK cell survival factor for this extended incubation. Following incubation, cells in the apical chamber were stained to assess direct NK cell killing and cells in the basolateral chamber were stained to assess killing by endogenous tumor-infiltrating lymphocytes (TILs).
For degranulation assays and IFNγ expression against patient tumors, NK cells were coincubated with target cells at a 1:1 ratio for 5 hours. Golgi Stop (BD Biosciences) was added following the first hour of incubation.
PD1 expression on NK cells and PDL1 expression on tumor cells were assessed by co-incubating NK cells with tumor cells for 48 hours using the transwell model described above. PD1 expression was assessed by staining cells in the apical chamber for NK cell markers and PD1. PDL1 expression was assessed on live tumor cells in the basolateral chamber.
Statistics
Statistical analysis was conducted using GraphPad Prism software. Graphs with two groups were analyzed using a two-tailed unpaired t-test. Graphs with three or more conditions with one independent variable were analyzed by one-way analysis of variance (ANOVA) with Tukey correction for multiple comparisons. Graphs with two independent variables were analyzed by two-way ANOVA with Tukey correction. D’Agostino and Pearson normality test was used to determine distribution of the data. Correlation data were analyzed using Pearson correlation.
Results
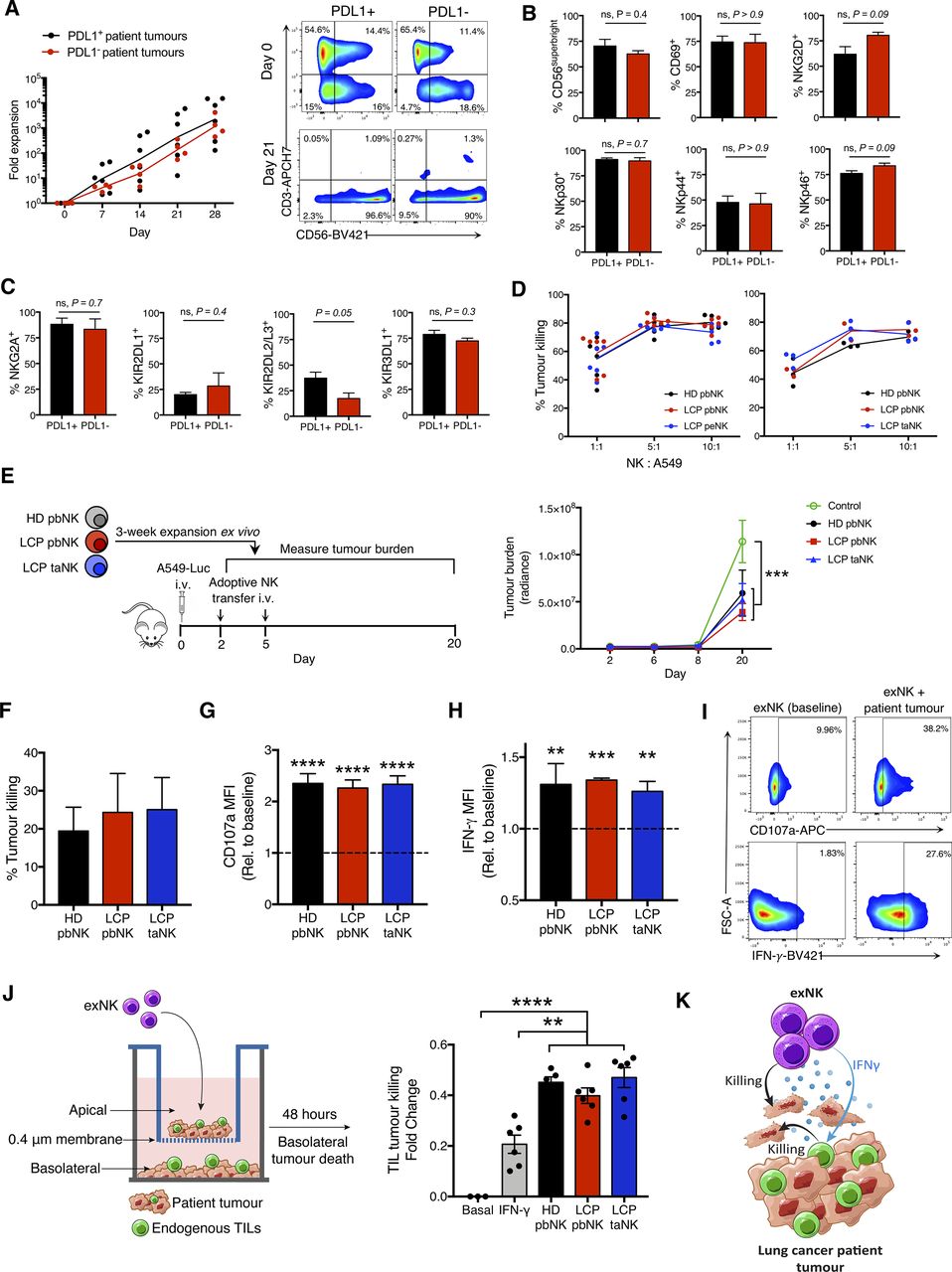
We first assessed the therapeutic potential of using NK cells expanded from the pleural effusions, surgically resected tumors, and peripheral blood of LCP to treat PDL1 +and PDL1− lung tumors. Table 1 shows the study population and tumor classification. We expanded NK cells from the above sources using irradiated K562 feeder cells genetically engineered to express membrane-bound IL-21. A recent trial demonstrated that NK cells expanded via this method had a high safety profile and remarkably reduced relapse rates in patients with high-risk myeloid malignancies.19 We found that NK cells from all sources reached clinically applicable fold expansion rates and purity (online supplemental figure S1A–C). Notably, NK cells from PDL1− lung tumors expanded comparably to those from PDL1+ tumors and had comparable and high viability (figure 1A and online supplemental figure S1D). exNK cells exhibited a CD56superbright phenotype with high activation receptor expression, which we previously found to be a phenotype associated with the greatest antitumor activity (figure 1B,C, and online supplemental figure S1E–G).16
Supplemental material

NK cells expanded from lung cancer patients (LCPs) exert strong antitumor activity against patient tumors and rescue tumor killing by endogenous TILs. NK cells were expanded from the peripheral blood (pbNK), pleural effusions (peNK) or tumors (taNK) of LCPs or peripheral blood of healthy donors (HD). (A) Fold expansion of NK cells from PDL1+ versus PDL1− tumors and representative flow plots showing NK cell (CD56+CD3-) purity pre-expansion and postexpansion. Expression of activation (B) and inhibitory (C) receptors on exNK cells from PDL1+ and PDL1− tumors at 28-day expansion. (D) Five-hour killing of human A549 lung cancer cells by exNK cells. (E) Schematic: exNK cells were adoptively transferred to NRG mice 2 and 5 days after intravenous infusion of Luciferase-expressing A549s (A549-Luc). Graph: quantification of tumor burden via bioluminescence at the indicated days. (F–I) Expanded LCP taNK and pbNK cells were coincubated with LCP tumors. Five-hour (F) killing and relative increase in (G) degranulation (CD107a) and (H) IFNγ expression against patient tumors compared with expanded HD pbNK cells against these same patient tumors. (I) Representative flow plots of NK cell CD107a and IFNγ expression. (J) Schematic: patient tumors were seeded in transwell on apical and basolateral surfaces. exNK cells or rhIFNγ (20 ng/mL) or neither (basal) were added to the apical chamber. Graph shows killing of tumors in the basolateral chamber by endogenous TILs after 48 hours. (K) Schematic summarizing results. Data show means±SEM of three to eight replicates per condition. Results analyzed by two-way ANOVA (A, D, E, G, H), unpaired t-test (B, C) or one-way ANOVA (D, F, J). **P<0.01, ***p<0.001, ****p<0.000. ANOVA, analysis of variance; exNK, expanded NK; IFNγ, interferon-gamma; NK, natural killer; NRG, NOD-Rag1null IL2rgnull; ns, not significant; PDL1, programmed death receptor ligand-1; rhIFNγ, recombinant human IFNγ; TILs, tumor-infiltrating lymphocytes.
Study population and tumor characteristics
Consistent with their highly activated phenotype, LCP exNK cells showed strong killing against human A549 lung cancer cells and significantly reduced tumor burden and controlled tumor growth on adoptive transfer to NRG mice engrafted with a measurable burden of A549 tumors (figures 1D,E and online supplemental figure S2A,B). We next assessed the antitumor functions of exNK cells against patients’ own advanced tumors ex vivo, which also contain further immunosuppressive effects from regulatory immune cells. Against patient tumors, exNK cells showed strong killing and activation-induced degranulation and IFNγ production (figure 1F,G,H,I and online supplemental figure S2C). Furthermore, NK cells expanded from LCP peripheral blood and tumors showed comparable antitumor activity as HD exNK cells. These findings uncover promising therapeutic potential for LCP exNK cells to treat both PDL1+ and PDL1− tumors.
Supplemental material
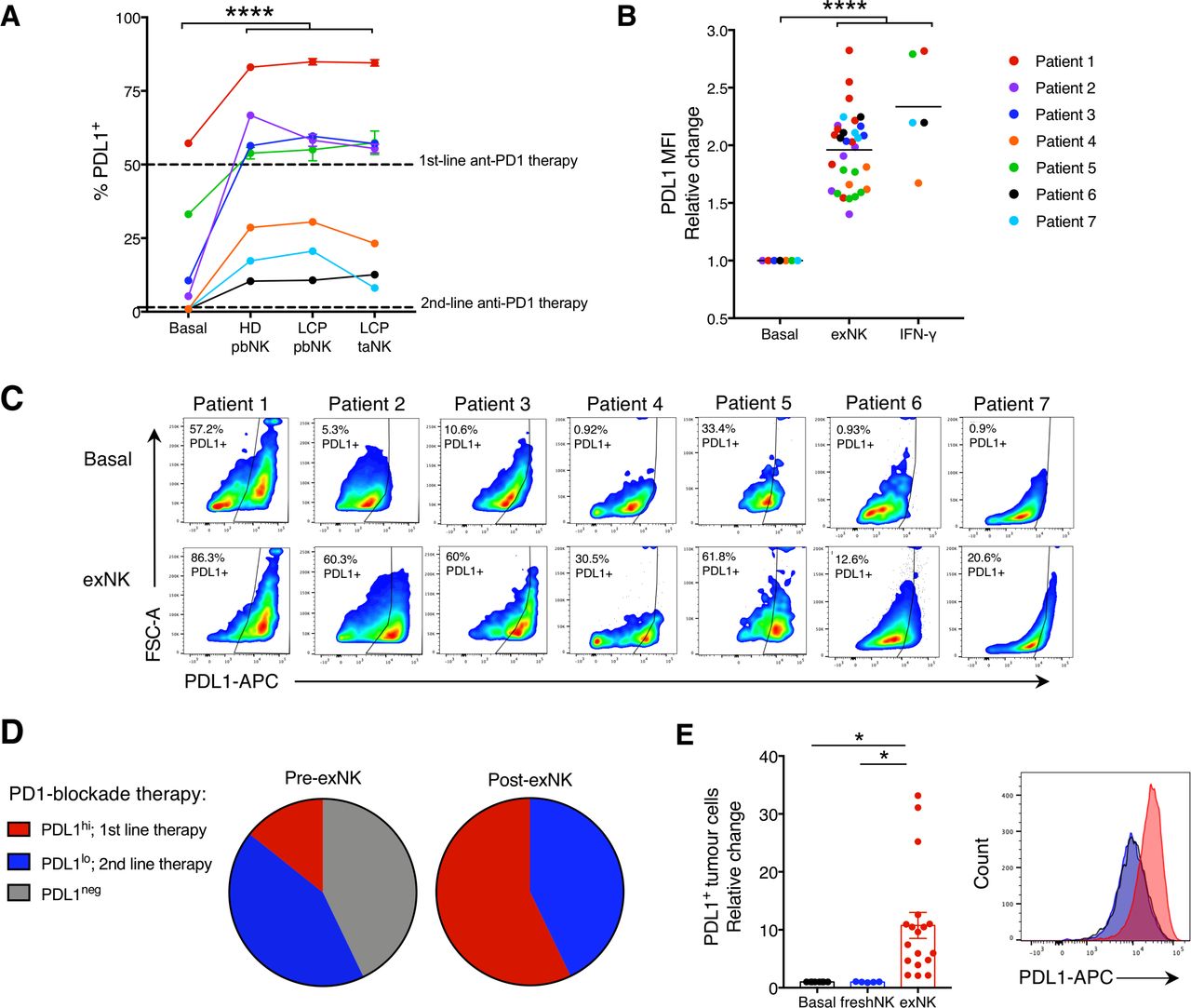
exNK cells convert lung cancer patient PDL1− tumors to PDL1+/hi. Lung cancer patient tumors were seeded in transwell on apical and basolateral surfaces. exNK cells or rhIFNγ (20 ng/mL) or neither (basal) were added to the apical chamber and incubated for 48 hours. (A) PDL1 TPS and (B) PDL1 mean fluorescence intensity (MFI) on basolateral tumor cells. (C) Representative flow plots of PDL1 on exNK-treated versus untreated patient tumors. (D) Proportion of patient tumors that were PDL1neg, PDL1lo or PDL1hi pre-exNK and post-exNK treatment. (E) Quantification and representative histogram of PDL1 expression on lung cancer patient tumors treated with expanded or unexpanded freshly isolated (fresh) pbNK cells. Data show means±SEM of 5–32 replicates per condition. A, B, E analyzed via one-way ANOVA. *P<0.05; ****p<0.0001. ANOVA, analysis of variance; exNK, expanded natural killer; pbNK, peripheral blood NK; PDL1, programmed death receptor ligand-1; rhIFNγ, recombinant human interferon-gamma; TPS, Tumor Proportion Score.
We next assessed the impact of exNK cells on endogenous immune cells in tumors. Using a transwell model in which LCP tumors were seeded on both apical and basolateral surfaces, we found that administration of exNK cells to the apical chamber activated endogenous TILs in the basolateral chamber to kill patient tumors (figure 1J). The contact-independent mechanism through which exNK cells rescued TILs from exhaustion likely involved IFNγ, as treatment of tumors with rhIFNγ alone partly rescued TIL activity. Together, these results demonstrate that expanded LCP NK cells are capable of strong anti-tumor activity on engagement with patient tumors, through both direct tumor killing and release of proinflammatory mediators that revive exhausted endogenous TILs (figure 1K).
Degree of tumor PDL1 expression (TPS) is associated with PD1-blockade efficacy and used to determine treatment.3 7 11 20 PD1-blockade is routinely used as first-line therapy for LCP with PDL1hi tumors (TPS ≥50%) and as second-line therapy for patients with PDL1lo tumors (TPS ≥1% and ≤49%).21 22 However, only 22% of LCP have PDL1hi tumors and only ~50% of patients have a TPS ≥1%.23 Given that IFNγ signaling upregulates PDL1 on tumor cells and that exNK cells upregulated IFNγ on engagement with tumors, we assessed whether exNK cells could induce PDL1 expression on lung tumors. Using the transwell model as in figure 1H, we seeded A549s on apical and basolateral surfaces, incubated exNK cells in the apical chamber, and assessed PDL1 expression on basolateral A549s after 48 hours (online supplemental figure S3A). Compared with untreated A549s which were PDL1−, exNK-treated A549s highly expressed PDL1, with a mean TPS of 90.1% (online supplemental figure S3B). Similar to previous reports,18 the increase in PDL1 TPS by exNK cells was consistent across other poor prognosis cancer types and independent of baseline PDL1 TPS, as exNK cells also strikingly increased PDL1 TPS and mean fluorescence intensity in OVCAR8 ovarian cancer and MDA-MB-231 triple-negative breast cancer cells which, respectively, had low and high PDL1 TPS at baseline (online supplemental figure S3C,D).
Supplemental material
We next assessed the ability of exNK cells to increase PDL1 expression on tumors from LCP via flow cytometry (see online supplemmental table S1 for baseline TPS assessment via flow cytometry and diagnostic immunohistochemistry). Treatment with exNK cells from HD or LCP peripheral blood or tumors remarkably increased PDL1 expression on tumors from all patients and comparably to treatment with rhIFNγ alone (figure 2A–C, online supplemental figure S3E). As a result, while 43% of patient tumors were PDL1− at baseline, following exNK treatment, all patient tumors were PDL1+ (figure 2D). Further, the majority of tumors became PDL1hi, surpassing the threshold for first-line PD1-blockade therapy. Notably, upregulation of PDL1 on tumor cells required NK cells capable of potent activation in response to tumors, as treatment of tumors with freshly isolated pbNK cells, known to be suppressed by tumors, had no effect on tumor PDL1 (figure 2E).14 These findings identify that exNK cells could be harnessed to increase the proportion of patients who are likely to benefit from PD1-blockade therapy.
We thus evaluated the potential for therapeutic synergy between exNK cells and PD1-blockade. It was previously reported that in addition to T cells, endogenous NK cells also respond to anti-PD1 and contribute to its immunotherapeutic efficacy.24 Although exNK cells expressed minimal PD1 at baseline, they significantly upregulated PD1 expression after exposure to patient tumors (figure 3A). Thus, although exNK cells maintain strong anti-tumor functions against established tumors, PD1-blockade may even further enhance their maximal anti-tumor capacity. To test this, we pretreated A549s with rhIFNγ for 48 hours to induce PDL1 expression, then assessed exNK cell killing of PDL1+ A549s over 5 hours with or without the clinical anti-PD1 antibody nivolumab. Treatment with nivolumab significantly increased killing by PD1+ exNK cells against PDL1+ A459 cells (figure 3B). To verify that the effect was specific to PD1-PDL1 blockade, we confirmed that nivolumab did not increase exNK cell killing against non-pretreated (PDL1−) A549s over 5 hours, a time point prior to which PDL1 expression is altered by exNK cells (figure 3C).
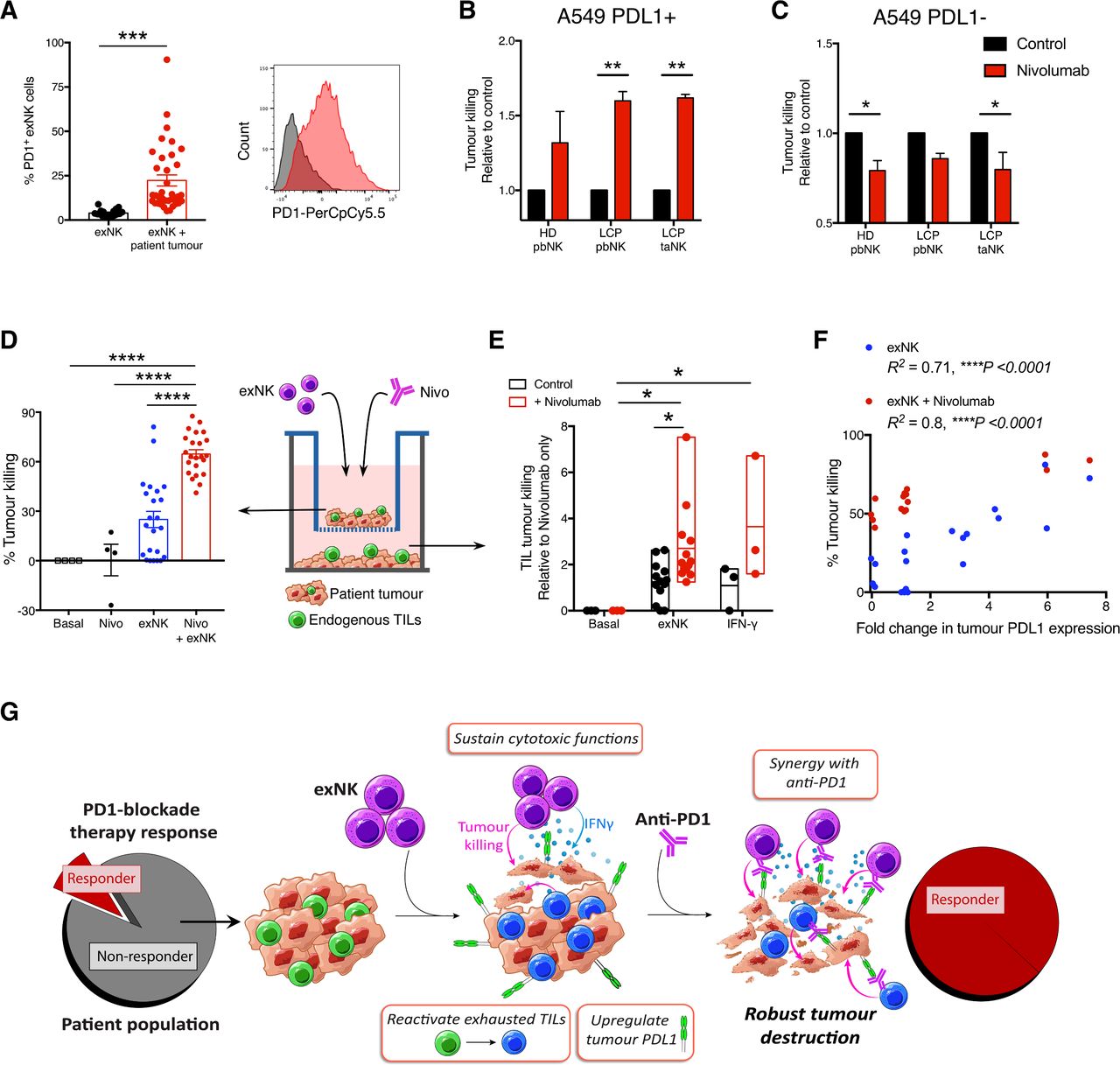
{kind=link}
{kind=link}
{kind=link}
exNK cells sensitize patients’ non-responding tumors to PD1-blockade therapy. (A) exNK cells were incubated with or without lung cancer patient tumors. Per cent PD1+ exNK cells and representative histogram of PD1 expression after 48-hour incubation. (B, C) A549s were left untreated or treated with rhIFNγ (20 ng/mL) for 48 hours to induce PDL1 expression, then washed three times and incubated with exNK cells with or without nivolumab (1 µg/mL) for 5 hours. Relative exNK cell killing of (B) IFNγ-treated (PDL1+) A549s or (C) untreated (PDL1−) A549s. (D–F) Schematic shows experimental design: patient tumors were seeded on apical and basolateral transwell surfaces. exNK cells and/or nivolumab (Nivo; 1 µg/mL) were added to the apical chamber for 48 hours. (D) Tumor killing in the apical chamber. (E) Tumor killing by endogenous TILs in the basolateral chamber. (F) Correlation between change in tumor PDL1 expression induced by exNK cells and apical tumor killing. (G) Graphical summary of the study’s main findings. Data show means±SEM of 4–36 replicates per condition. Results analyzed via unpaired t-test (A) two-way ANOVA (B, C, E), one-way ANOVA (D) or pearson correlation (F). *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; exNK, expanded natural killer; pbNK, peripheral blood NK; PD1, programmed death receptor-1; PDL1, programmed death receptor ligand-1; rhIFNγ, recombinant human interferon-gamma; taNK, tumor-associated NK; TILs, tumor-infiltrating lymphocytes.
Given our findings that exNK cells convert PDL1− patient tumors to PDL1+/hi, we assessed whether exNK cells could induce a response to PD1-blockade therapy in even initially PDL1− patient tumors. As anticipated, treatment of PDL1− tumors with nivolumab alone for 48 hours had no significant effect on tumor killing (figure 3D). However, addition of exNK cells with nivolumab synergistically increased tumor cell death compared with treatment with either nivolumab or exNK cells alone. In addition to increasing exNK cytotoxicity, combination therapy of exNK cells with nivolumab significantly enhanced the reactivation of tumor killing by endogenous TILs compared with either treatment in isolation, and to a similar degree as rhIFNγ (figure 3E). Notably, the ability of exNK cells to kill patient tumors with and without nivolumab correlated significantly with their ability to upregulate tumor PDL1 expression (figure 3F), indicating that change in PDL1 expression may serve as a predictor for tumors that will be most responsive to exNK cell therapy.
Discussion
Patients with advanced tumors that do not respond to checkpoint blockade immunotherapy face a dearth of effective treatment options. Although studies have predominantly focused on the role of T cells to mediate the antitumor effects of immune checkpoint blockade therapy, there is a growing understanding for the important role of other immune cell types, including NK cells and B cells, in mediating this response.24 25 Notably, NK cell depletion was shown to reduce response to PD1/L1-blockade therapy in a syngeneic murine model of colon carcinoma.24 A previous study also showed that NK cells expanded from induced-pluripotent stem cells increased PDL1 TPS on tumor cell lines.18 Our study identifies highly cytotoxic exNK cells as a promising therapy for lung cancer that may also further extend the benefits of PD1-blockade therapy to patients with non-responding tumors (figure 3G). A significant barrier to developing broadly effective immunotherapies against solid tumors has been the inability to sustain cytotoxic and proinflammatory immune cell functions in the tumor microenvironment.3–5 Our findings that NK cells expanded from LCP maintain strong tumor killing and IFNγ production, both in xenograft models in vivo and over prolonged exposure to patient tumors ex vivo, suggest that exNK cells overcome the critical hurdle of tumor-induced suppression. Further, our results that exNK cells restore the antitumor activity of endogenous TILs indicates a striking ability to not only resist, but dismantle, the immunosuppressive tumor microenvironment and unleash the patient’s own antitumor immunity.
Previous work by our group showed that NK cells expanded from patients with breast and ovarian cancer have comparable antitumor activity against the patients’ own autologous tumors as NK cells expanded from HD.15–17 The present study extends these findings to NK cells from LCP, identifying exNK cells as a promising autologous cell therapy for lung cancer.
A seminal study recently found that anti-PDL1−mAbs can directly activate the cytotoxic effector functions of PDL1+ NK cells, irrespective of tumor PDL1 status.26 These findings together with the results in the present study, identify NK cells as critical effectors for inducing responses to PD1/L1-blockade therapy in initially PDL1− tumors.
A recent randomized control trial in patients with PDL1+ non-small-cell lung carcinoma found that combination treatment of NK cells with the PD1 inhibitor pembrolizumab was well-tolerated and improved overall and progression-free survival in patients compared with pembrolizumab treatment alone.27 Importantly, the trial found that there were no adverse events associated with the addition of NK cell therapy. A limitation of our current study is the relatively small study population. Nevertheless, the proven safety profile shown in this previous trial, together with the robust synergistic tumor killing we observed on combination treatment of exNK cells and nivolumab against initially PDL1− tumors, suggests that such combination treatment should be investigated in patients with advanced PDL1− tumors.
Acknowledgments
We thank Rebecca Long for administrative assistance and all lung cancer patient and healthy donors who donated samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SMP and AAA conceived the project and designed the experiments; YS provided intellectual input, contributed to experimental design and guided clinical sample acquisition; SMP, TR and IYF performed experiments. AE-S, ALP, RB-A, ER and MC contributed to performing experiments. YS and RB-A obtained the clinical samples. SMP curated and formally analyzed the data. SMP and AAA wrote the manuscript; TR and YS edited the manuscript; AAA secured funding and supervised the project.
Funding This work was supported by the Canadian Institutes for Health Research (CIHR) (20 009 360 to AAA). AAA holds a tier 1 Canada Research Chair in Natural Immunity and NK Cell Function. SMP is supported by a CIHR Vanier Canada Graduate Scholarship.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All research involving human samples was approved by the Hamilton Integrated Research Ethics Board at McMaster University. All research involving animals was approved by the Animal Research Ethics Board at McMaster university.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplemental information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.