Article Text

Abstract
Background Targeted thorium-227 conjugates (TTCs) are an emerging class of targeted alpha therapies (TATs). Their unique mode of action (MoA) is the induction of difficult-to-repair clustered DNA double-strand breaks. However, thus far, their effects on the immune system are largely unknown. Here, we investigated the immunostimulatory effects of the mesothelin-targeted thorium-227 conjugate (MSLN-TTC) in vitro and in vivo in monotherapy and in combination with an inhibitor of the immune checkpoint programmed death receptor ligand 1 (PD-L1) in immunocompetent mice.
Methods The murine cell line MC38 was transfected with the human gene encoding for MSLN (hMSLN) to enable binding of the non-cross-reactive MSLN-TTC. The immunostimulatory effects of MSLN-TTC were studied in vitro on human cancer cell lines and MC38-hMSLN cells. The efficacy and MoA of MSLN-TTC were studied in vivo as monotherapy or in combination with anti-PD-L1 in MC38-hMSLN tumor-bearing immunocompetent C57BL/6 mice. Experiments were supported by RNA sequencing, flow cytometry, immunohistochemistry, mesoscale, and TaqMan PCR analyses to study the underlying immunostimulatory effects. In vivo depletion of CD8+ T cells and studies with Rag2/Il2Rg double knockout C57BL/6 mice were conducted to investigate the importance of immune cells to the efficacy of MSLN-TTC.
Results MSLN-TTC treatment induced upregulation of DNA sensing pathway transcripts (IL-6, CCL20, CXCL10, and stimulator of interferon genes (STING)-related genes) in vitro as determined by RNASeq analysis. The results, including phospho-STING activation, were confirmed on the protein level. Danger-associated molecular pattern molecules were upregulated in parallel, leading to dendritic cell (DC) activation in vitro. MSLN-TTC showed strong antitumor activity (T:C 0.38, p<0.05) as a single agent in human MSLN-expressing MC38 tumor-bearing immunocompetent mice. Combining MSLN-TTC with anti-PD-L1 further enhanced the efficacy (T:C 0.08, p<0.001) as evidenced by the increased number of tumor-free surviving animals. MSLN-TTC monotherapy caused migration of CD103+ cDC1 DCs and infiltration of CD8+ T cells into tumors, which was enhanced on combination with anti-PD-L1. Intriguingly, CD8+ T-cell depletion decreased antitumor efficacy.
Conclusions These in vitro and in vivo data on MSLN-TTC demonstrate that the MoA of TTCs involves activation of the immune system. The findings are of relevance for other targeted radiotherapies and may guide clinical combination strategies.
- gene expression profiling
- immunotherapy
- radiotherapy
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Targeted thorium-227 conjugates (TTCs) are emerging, innovative cancer therapies and they belong to the class of targeted alpha therapy (TAT).1 They consist of an antigen-targeting moiety, covalently attached to a 3,2-HOPO chelator, which enables stable complexation and delivery of the alpha particle emitter thorium-227, with a half-life of 18.7 days, to cancer cells on systemic administration (online supplemental figure S1). In contrast to external beam radiation therapy (EBRT), the mode of action (MoA) of TTCs relies on the targeted delivery of potent alpha particle-emitting radionuclides that emit high linear energy transfer radiation, thereby inducing difficult-to-repair DNA double-strand breaks (DSBs), forcing cells into apoptotic and necrotic cell death2 on systemic administration. To date, no cellular mechanisms of resistance have been described for TAT.3 Potent preclinical in vivo activity has been demonstrated for TTCs in monotherapy or in combination with DNA damage inhibitors in immunocompromised xenograft models.4–6 Based on these preclinical data, the safety and tolerability of several TTCs are currently being investigated in the clinic.2
Supplemental material
(Pre)clinical studies have demonstrated that EBRT causes an immunostimulatory response,7–10 resulting in increased tumor growth inhibition and increased response rates when combined with immune checkpoint inhibitors.11 However, thus far, only a few reports have described the immunostimulatory effects of TAT. In an in vitro vaccination approach where bovine serum albumin was complexed with the alpha particle emitter bismuth-213, induction of danger-associated molecular patterns (DAMPs) and immunity against a follow-up inoculation of cancer cells were observed in immunocompetent mice.12 Similarly, Malamas et al.13 demonstrated that in vitro exposure of prostate, lung, and breast cancer cells to the TAT radium-223 dichloride14 resulted in the exposure of DAMPs and MHC-1 on the cell surface, rendering cells vulnerable to T cell-mediated cell lysis. Clinical combination trials have since been pursued for radium-223 dichloride.15
Therefore, in the present study, we aimed to elucidate the immunostimulatory effects of the mesothelin-targeted thorium-227 conjugate (MSLN-TTC, thorium-227 (227Th) anetumab corixetan).4 The in vitro effects were studied on both human cancer cell lines and the murine MC38 cell line, transfected with human MSLN gene to enable binding of the non-cross-reactive MSLN-TTC. In vivo, the efficacy, MoA, and the immunostimulatory effects of MSLN-TTC were investigated both in monotherapy and in combination with an inhibitor of the immune checkpoint programmed death receptor ligand 1 (PD-L1).
Methods
Cell lines
MC38 murine colorectal cancer cells, human ovarian OVCAR-3 and OVCAR-8 cancer cells, and the lung mesothelioma cancer cell line NCI-H226 were obtained from American Type Culture Collection (ATCC) between 2012 and 2017 and authenticated using short tandem repeat DNA fingerprinting at Leibniz Institute (German Collection of Micro-organisms and Cell Cultures) before the experiments.
MC38 cells were transduced with pLenti6.3_hMSLN vector to express human MSLN or with pLenti6.3_eGFP to serve as mock-transfected control cells (NMI, Tübingen, Germany). Transfection of the MSLN gene was confirmed by flow cytometry using the MSLN-binding antibody anetumab, which detected approximately 458 000 receptors per cell. NCI-H226 cells were transfected with the luciferase gene (NMI).
Compounds
MSLN-TTC (thorium-227 (227Th) anetumab corixetan, online supplemental figure S1) and a radiolabeled isotype control were produced as described previously.4 An anti-PD-L1 antibody, based on the sequence of atezolizumab (murine IgG1), was produced in-house by Bayer AG (Wuppertal, Germany). A respective isotype control was purchased (MOPC-21, BioXCell).
Quantitative reverse-transcription (RT)-PCR, RNA sequencing, and mesoscale analysis
Altered RNA expression and secretion of cytokines were examined in cells after exposure to MSLN-TTC (5 kBq/mL), a radiolabeled isotype control, or a non-radiolabeled MSLN antibody–chelator conjugate for three (RNA sequencing (RNASeq)) or 5 days (mesoscale). Cyclic 2′−3′ GMP-AMP (cGAMP, 20 µg/mL, Sigma) was used as control. Details for the RNASeq and cytokine analyses using mesoscale are detailed in online supplemental methods.
Analysis of DAMP and immunomodulatory marker expression by flow cytometry
Cell surface expressions of DAMPs and immunostimulatory markers were determined on NCI-H226, OVCAR-3, and MC38-human gene encoding for MSLN (hMSLN) cells by flow cytometry after a 48 or 72 hours of exposure to MSLN-TTC or radiolabeled isotype control (5 kBq/mL, specific activity of 40 kBq/µg), depending on the induction of apoptosis in cells. Phosphorylation of stimulator of interferon genes (STING) was determined on MC38-hMSLN and NCI-H226 cells by flow cytometry. Induction of immunogenic cell death was performed on isolated human immature dendritic cells (DCs), cultured in media from OVCAR-3 cells exposed to MSLN-TTC (10 kBq/mL) for 5 days. The protocols are detailed in online supplemental methods.
Antitumor efficacy of MSLN-TTC and anti-PD-L1 in vivo
Female C57BL/6 mice (20–25 g, 9-week-old, Charles River) were inoculated subcutaneously with 1×106 MC38-hMSLN cells/mouse. The MC38-hMSLN cells demonstrated consistent tumor growth and take rates in comparison to the mock-transfected MC38 cells with tumor doubling times of approximately 3 (MC38-hMSLN) or 3.2 (mock-transfected MC38 cells) days, with a longer lag phase observed for MC38-hMSLN cells. MSLN expression in the established MC38-hMSLN tumors was confirmed by immunohistochemistry (IHC). PD-L1 was detectable on tumor and tumor-infiltrating immune cells (data not shown), confirming published data.16
All MSLN-TTC dosages were administered intravenously. In the monotherapy study, at an average tumor size of 88 mm3 (study day 5), mice (n=11/group) were administered a single dose of MSLN-TTC (125, 250, or 500 kBq/kg, intravenous) or radiolabeled isotype control (500 kBq/kg), both at total antibody doses of 0.14 mg/kg. Phosphate-buffered saline (PBS, 5 mL/kg) was used as the vehicle.
In the combination treatment study, at an average tumor size of 140 mm3 (study day 8), mice (n=12/group) were administered a single dose of vehicle (PBS, 5 mL/kg), MSLN-TTC (250 kBq/kg, intravenous, 0.14 mg/kg), or radiolabeled isotype control (250 kBq/kg, intravenous, 0.14 mg/kg), or anti-PD-L1 atezolizumab, administered two times per week (1.5 mg/kg, twice-weekly (Q3/4D), intraperitoneal). The combination groups received both MSLN-TTC and anti-PD-L1, whereas the monotherapy groups of MSLN-TTC and radiolabeled isotype control were dosed two times per week with the isotype control of anti-PD-L1, MOPC-21 (BioXCell, 1.5 mg/kg, Q3/4D, intraperitoneally).
Along with body weights, tumors were measured two times per week with calipers, and their volume was calculated with the formula: (length×width2)/2. For molecular analysis, tumors and lymph nodes were isolated after sacrifice 7 days after dosing (n=3/group). Molecular analysis was conducted by IHC, mesoscale analysis, or RT-qPCR as described further and in the online supplemental methods.
In the sequencing study, mice (n=12/group) received a single intravenous dose of vehicle (PBS, 5 mL/kg) or a combination of MSLN-TTC (250 kBq/kg, intravenous; 0.14 mg/kg) and anti-PD-L1 (1.5 mg/kg, Q3/4D, intravenous) or MOPC-21 (1.5 mg/kg, Q3/4D, intraperitoneal). MSLN-TTC and anti-PD-L1 were dosed simultaneously on day 8 after tumor cell inoculation, or one of the compounds on day 8 and the other on day 15.
In the CD8 depletion study, mice (n=11/group) were treated with vehicle (PBS, 5 mL/kg, intravenous) or MSLN-TTC (1×250 kBq/kg, intravenous; 0.14 mg/kg) in combination with anti-PD-L1 (1.5 mg/kg, Q3/4D, intraperitoneal) or MOPC-21 (1.5 mg/kg; Q3/4D, intraperitoneal). In parallel, one treatment arm received a single dose of rat anti-mouse CD8 monoclonal antibody (mAb) YTS.169 (BioXCell, 8 mg/kg, intraperitoneal) at the start of therapy to deplete CD8-positive T cells (CD8 T cells).
In parallel, immunodeficient MC38-hMSLN tumor-bearing Rag2/Il2Rg double knockout C57BL/6 mice (20–25 g, 7–12 weeks old, Taconic Biosciences; 1×106 MC38-hMSLN cells/mouse) were treated (n=12/group) at an average tumor size of 85 mm3 with a single dose of vehicle (PBS, 5 mL/kg, intravenous) or MSLN-TTC (125, 250, or 500 kBq/kg, intravenous; total antibody dose of 0.14 mg/kg) or a two times per week dose of anti-PD-L1 (1.5 or 5 mg/kg, Q3/4D, intraperitoneal).
Rechallenge experiment to explore immunization
Complete responders from the in vivo efficacy studies were reinoculated subcutaneously with either 1×106 MC38-hMSLN cells or 1×106 mock-transfected MC38 cells 146 days after the original inoculation. Reinoculation was performed on either the side of the primary tumor or on the contralateral flank compared with the initial MC38-hMSLN tumor cell inoculation (study day 0).
Flow cytometry analysis on isolated tumors and tumor draining lymph nodes after treatment
Tumors and tumor-draining lymph nodes (TdLNs) were harvested from mice 10 days after treatment start and were analyzed by flow cytometry as described in the online supplemental methods.
Immunohistochemistry
MSLN expression in MC38-hMSLN was determined by IHC using SP74 antibody (Spring Bioscience).4 PD-L1 expression was determined using anti-PD-L1 antibody (clone E1L3N, Cell Signaling Technology).
Murine CD8 T cells were detected using rat anti-mouse CD8 mAb (clone YTS169.4, AbD Serotec), and DNA DSBs using antiphospho-Histone H2A.X (Ser139) antibody (Cell Signaling). Fluorescent visualization was done on incubation with the Opal Fluo kit (Akoya Bio). Tissue slides were counterstained using 4’,6-diamidino-2-phenylindole (DAPI), and CD8 signals were quantified using HS Analysis Webkit tool (HS Analysis).
Statistical analyses
The statistical analyses performed are indicated in each figure legend. The differences between studied groups were considered statistically significant if p<0.05.
Results
MSLN-TTC activates immunostimulatory pathways resulting in the secretion of (pro)inflammatory cytokines, upregulation of DAMPs, and DC activation in vitro
The molecular MoA of TTCs was investigated by non-biased gene expression profiling of human ovarian cancer OVCAR-3 and murine MC38-hMSLN cells, transfected to express human MSLN to enable binding of the non-cross-reactive MSLN-TTC as previously performed by others.17 After exposure to MSLN-TTC (0.5, 5.0, or 50.0 kBq/mL), principal component analysis demonstrated a dose-dependent deregulation of genes (data not shown). The deregulated genes mapped to the DNA sensing pathway as defined by the Kyoto Encyclopedia of Genes and Genomes database.18 Several proimmunostimulatory genes (caspase-1, IL-18, IL-6, TNF, CCL5, CCL20, NFkB, NFKBIA, NFKBIB, and IKK-γ) showed significant, dose-dependent deregulation in OVCAR-3 and/or MC38-hMSLN cells on MSLN-TTC treatment when compared with medium-radiolabeled or non-radiolabeled MSLN antibody–chelator conjugate-treated samples (figure 1A,B). Slight upregulation of genes involved in the cGAS/STING (stimulator of interferon (IFN) genes) pathway (STING, IRF3, IFN-β, TBK1, ZBP1, CXCL10, and/or TREX1) was observed in MC38-hMSLN cells. Additionally, genes described to be involved in the antiviral response (MX1, MX2 and/or OAS3) and the cytosolic pattern recognition receptor DDX58 were upregulated. In parallel, deregulation of hallmark apoptosis genes (caspase-3 and 6, FAS, BID, DNAJC3, GADD45A, GADD45B, BRCA1, the V(D)J recombination gene HMGB2, MCL-1, BIRC3, IER3, CCND2, and CDKN1A) was observed on both cell lines on MSLN-TTC treatment (online supplemental figures S2 and S3), matching our findings previously reported in in vitro assays.4 6
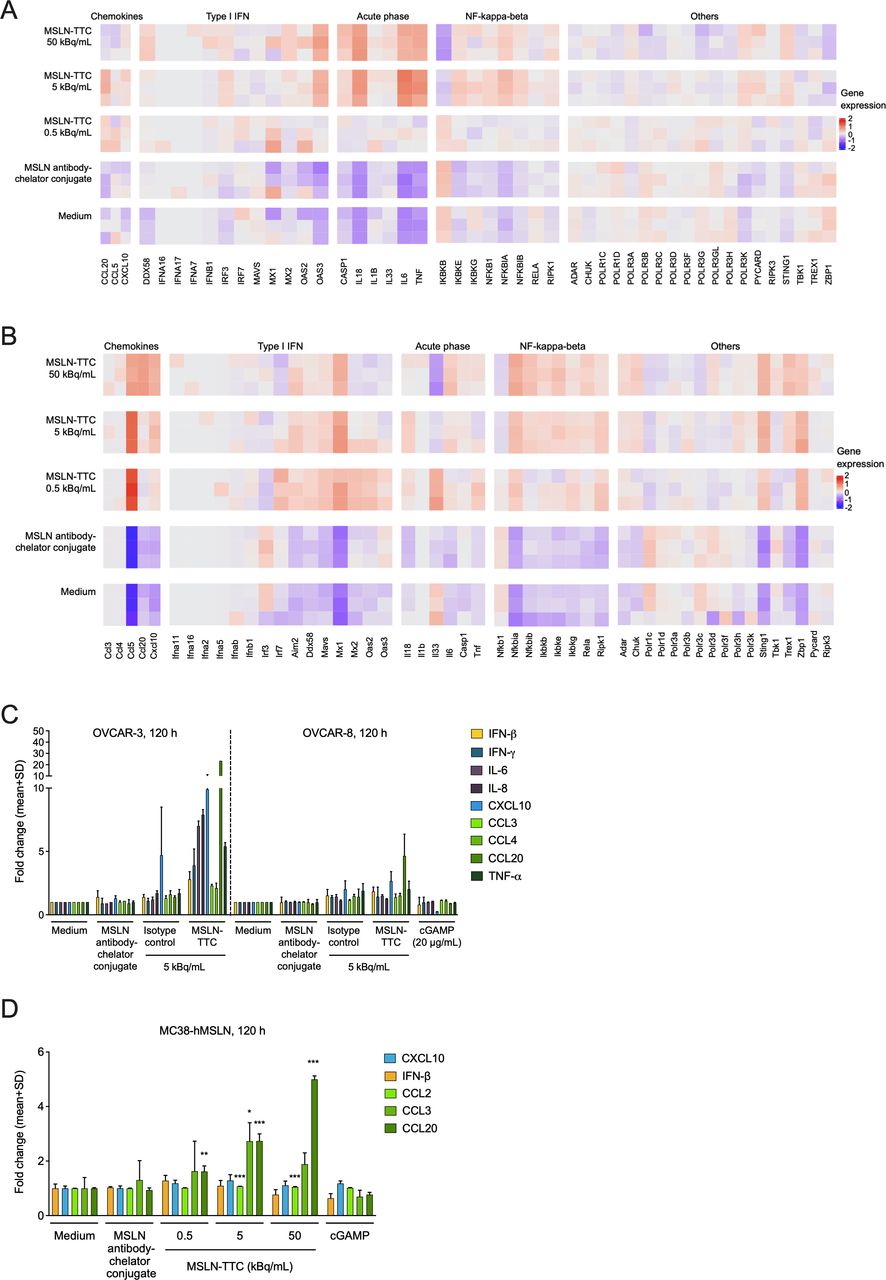
Effect of MSLN-TTC treatment on the deregulation of (pro)inflammatory cytokines in vitro on MSLN-expressing human and murine cancer cells. Cells were exposed to the radiolabeled conjugates continuously over the course of the indicated time frames at the indicated radioactivity concentrations. (A) Heatmap of deregulated genes in OVCAR-3 cells after a 72-hour exposure as determined by RNAseq analysis. (B) Heatmap of deregulated genes in MC38-hMSLN cells after a 72-hour exposure as determined by RNAseq analysis. (C) Secretion of (pro)inflammatory cytokines in OVCAR-3 and OVCAR-8 cells after a 120-hour exposure as determined by mesoscale analysis. (D) Secretion of cytokines in MC38-hMSLN cells after a 120-hour exposure as determined by mesoscale analysis. Statistical analysis was performed using one-way ANOVA. *P<0.05, **P<0.01, ***P<0.001 compared with medium. ANOVA, analysis of variance; MSLN, mesothelin; MSLN-TTC, mesothelin-targeted thorium-227 conjugate.
To confirm the deregulation of immunostimulatory genes on the protein level, two human ovarian cancer cell lines, OVCAR-3 and OVCAR-8, both expressing high levels of cell surface accessible MSLN, as well as the MC38-hMSLN cell line were exposed to MSLN-TTC. Cell supernatants were subsequently analyzed for secreted immunostimulatory chemokines. Exposure of OVCAR-3 cells to MSLN-TTC resulted in increased levels of several chemokines including TNF-α, CCL20 (MIP-3α), CXCL10, IL-6, and IL-8 (CXCL8), whereas on OVCAR-8 cells, the highest increase was observed for CCL20 (figure 1C). Similarly, in MC38-hMSLN cells, the highest secreted protein levels were detected for CCL20 and CCL3 (MIP-1α) (figure 1D). Notably, non-radiolabeled MSLN antibody–chelator conjugate did not induce a detectable increase in the chemokine levels compared with vehicle on any of the tested cell lines. In alignment with the literature, treatment with cGAMP did not result in detectable levels of IFN-β in OVCAR-3 and OVCAR-8 cells (figure 1C), both described to be defective in cGAS/STING signaling.19
Next, the status of phosphorylated stimulator of interferon genes (pSTING) in cells was analyzed by flow cytometry. Continuous exposure of cells for 48 hours to MSLN-TTC resulted in a specific 9-fold increase on human MSLN-expressing NCI-H226 mesothelioma lung cancer cells (figure 2A), and in a 4-fold increase on MC38-hMSLN cells (figure 2B).
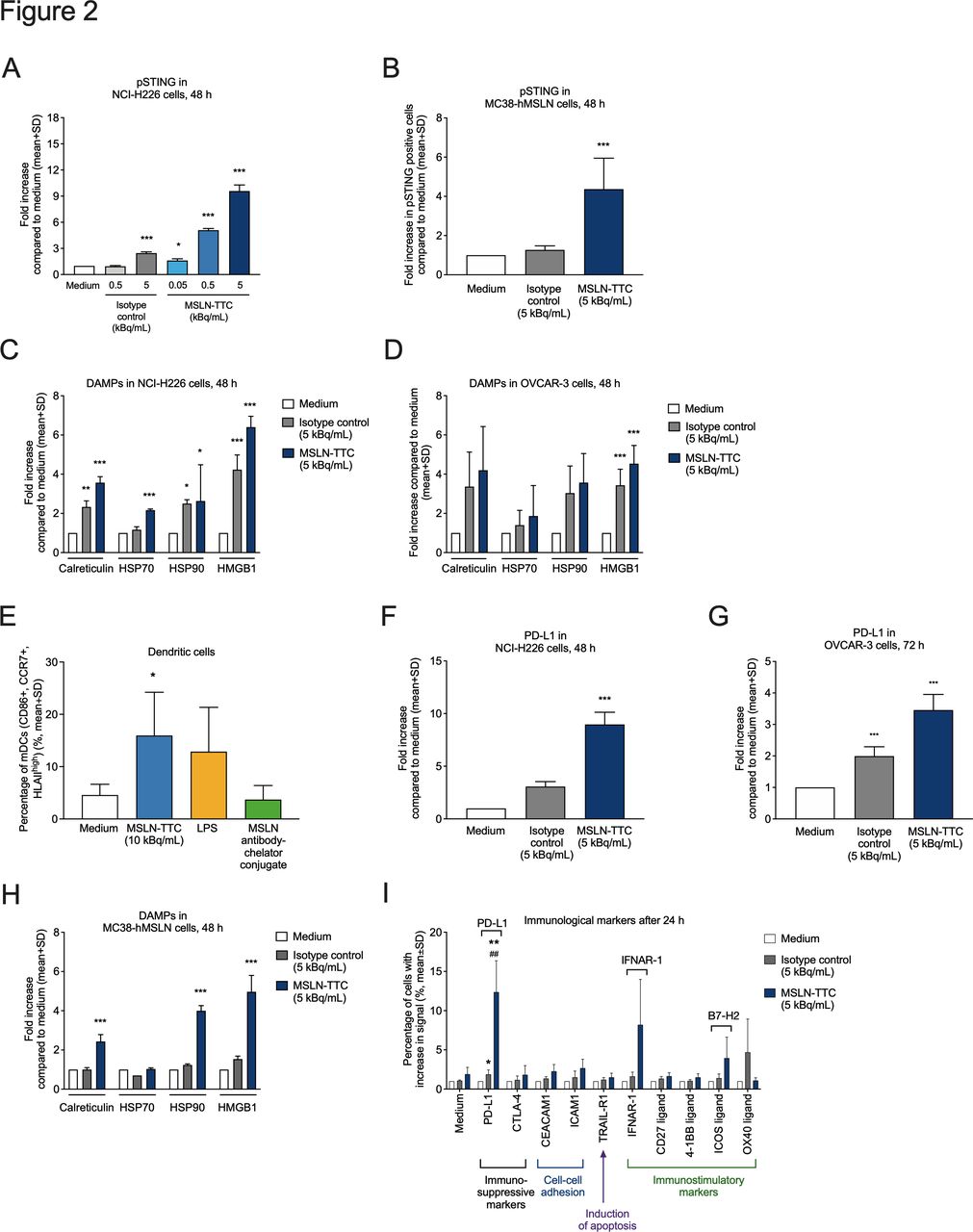
Effects of MSLN-TTC or a radiolabeled isotype control on the upregulation of phosphorylated STING, DAMPs, immune checkpoint markers, and immunostimulatory cell surface markers. Cells were exposed to the radiolabeled conjugates continuously over the course of the indicated time frames at the indicated radioactivity concentrations. (A,B) Upregulation of phosphorylated STING in (A) NCI-H226 and (B) MC38-hMSLN cells as determined by flow cytometry. (C,D) Upregulation of DAMPs in (C) NCI-H226 and (D) OVCAR-3 cells. (E) Activation of DCs on exposure of OVCAR-3 cells to MSLN-TTC, LPS, or non-radiolabeled MSLN antibody–chelator conjugate, followed by coculturing of DCs with exposed supernatants and subsequent flow cytometry analysis. (F,G) Upregulation of the suppressive immune checkpoint marker PD-L1 on (F) NCI-H226 and (G) OVCAR-3 cells as determined by flow cytometry. (H) Upregulation of DAMPs on MC38-hMSLN cells using flow cytometry. (I) Upregulation of immunomodulatory cell surface markers in MC38-hMSLN cells after 24 hours using flow cytometry. Statistical analyses were performed using repeated measures ANOVA (A–D,F,H); a linear model (ANOVA) after transforming the response to log2-scale with p values corrected for false-positive rate using Sidak’s method (G); one-way ANOVA followed by Student’s t-test (E); and a linear model of weighted least squares (I). *P<0.05, **P<0.01, ***P<0.001 compared with medium. ##P<0.01 compared with isotype. ANOVA, analysis of variance; DAMP, danger-associated molecular pattern; DC, dendritic cell; LPS, lipopolysaccharide; MSLN, mesothelin; MSLN-TTC, mesothelin-targeted thorium-227 conjugate; PD-L1, programmed death receptor ligand 1; pSTING, phosphorylated stimulator of interferon genes.
We next evaluated whether MSLN-TTC induces upregulation of DAMPs,7 the hallmarks of immunogenic cell death. As presented in figure 2C,D 48 hours of continuous exposure of the NCI-H226 lung mesothelioma cells and OVCAR-3 cells resulted in specific upregulation of cell surface-bound calreticulin (both ~4-fold), HSP70 (both ~2-fold), HSP90 (2-fold and ~3-fold), and HMGB1 (6-fold and ~4-fold), respectively. Importantly, treatment of OVCAR-3 cells with MSLN-TTC, followed by coincubation with DCs and subsequent flow cytometry analysis, resulted in a significant upregulation of DC markers, indicating DC activation (figure 2E). Furthermore, exposure of 48 or 72 hours to MSLN-TTC resulted in an approximately 9-fold and 3-fold increase of PD-L1 on NCI-H226 and OVCAR-3 cells, respectively (figure 2F,G). Specific, dose-dependent upregulation of DAMP molecules (figure 2H), PD-L1 and ICOS ligand (B7–H2) was also observed in MC38-hMSLN cells (figure 2I).
In summary, the results demonstrate that MSLN-TTC modulates the transcription and activation of several immune-stimulating genes and proteins and is capable of activating DCs in vitro.
MSLN-TTC demonstrates in vivo efficacy in the MC38-hMSLN model in monotherapy and it is potentiated when combining with anti-PD-L1
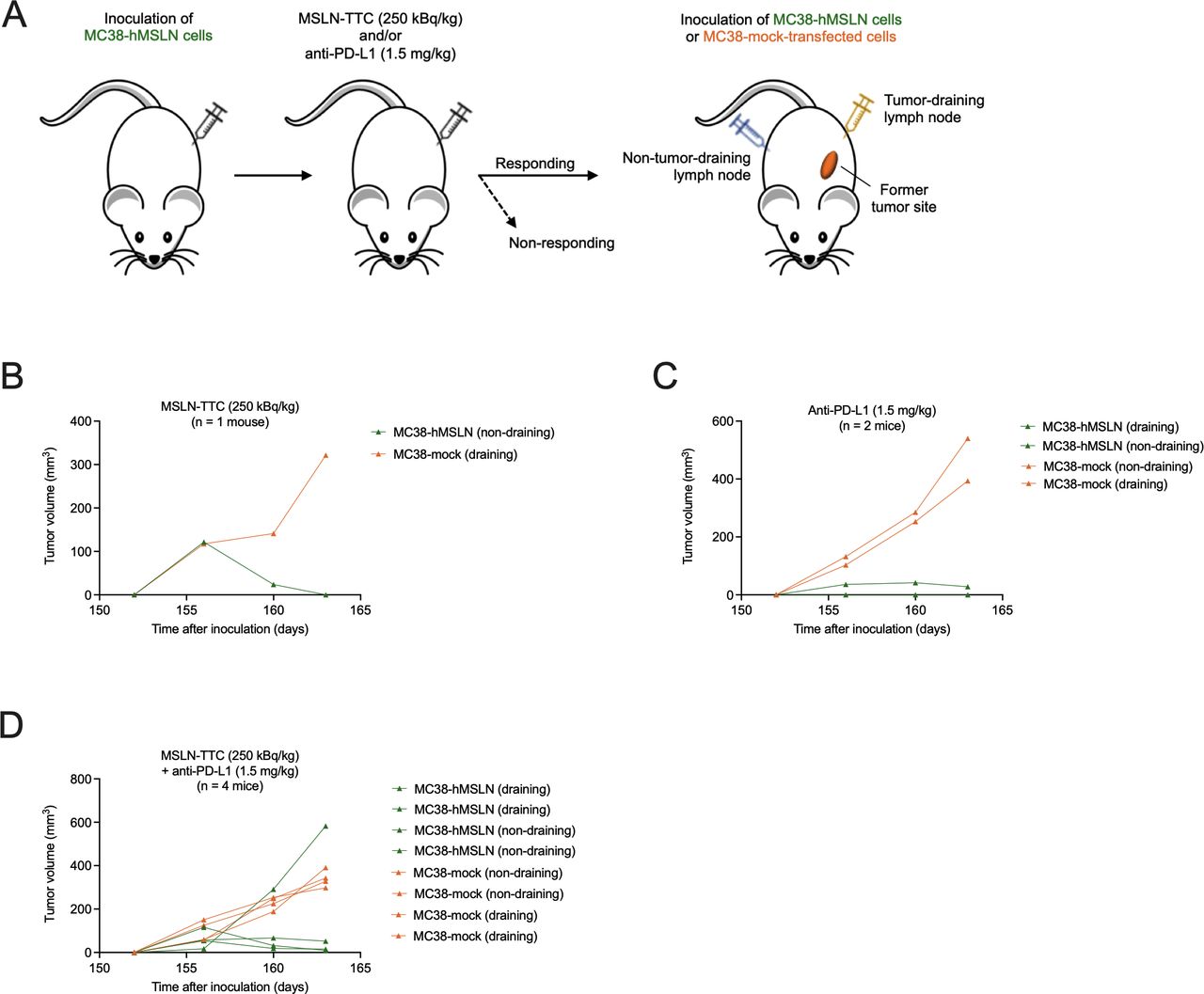
The immunostimulatory properties of MSLN-TTC were next studied in vivo in immunocompetent, MC38-hMSLN tumor-bearing C57BL/6 mice. The study design is presented in figure 3A. When administered as monotherapy at a mean tumor size of 88 mm3, a single dose of MSLN-TTC resulted in significant, dose-dependent tumor growth inhibition compared with vehicle at all the tested dose levels of 125, 250, and 500 kBq/kg (figure 3B), with a dose-dependent increase in the number of tumor-free surviving animals 121 days postadministration (online supplemental table S1). Similarly, two times per week dosing of anti-PD-L1 monotherapy (10 mg/kg) resulted in significant tumor growth inhibition (figure 3C). Both therapies were well tolerated based on body weight measurements (online supplemental file 4A,B).

In vivo efficacy of MSLN-TTC (intravenous), anti-PD-L1 (intraperitoneal), and combination therapies in immunocompetent MC38-hMSLN tumor-bearing mice and CD8 T cell-depleted mice. (A) Schematic figure depicting the in vivo study design. (B) MC38-hMSLN tumor growth in mice after MSLN-TTC treatment as indicated in the figure. Statistical analysis was performed on day 19. (C) MC38-hMSLN tumor growth in mice after anti-PD-L1 treatment as indicated in the figure. (D) MC38-hMSLN tumor growth in mice after administration of MSLN-TTC, anti-PD-L1, or their combination as indicated in the figure (n=12/group). Statistical analysis was performed on values from days 22 and 19, respectively. (E) Survival of mice described in (C) during the study until study day 55. (F) MC38-hMSLN tumor growth in mice after simultaneous or sequential administration of MSLN-TTC and anti-PD-L1 as indicated in the figure. The compounds were either (1) dosed simultaneously on day 8; or (2) sequentially with MSLN-TTC on day 8, followed by anti-PD-L1 on day 15; or (3) anti-PD-L1 on day 8, followed by MSLN-TTC on day 15. Statistical analysis was performed on values from days 22 and 19, respectively. (G) MC38-hMSLN tumor growth in immunocompetent mice treated with MSLN-TTC, anti-PD-L1, or their combination as indicated in the figure (n=11/group). Dotted lines denote mice where CD8 T cells had been depleted with the rat anti-mouse CD8 mAb YTS169.4, and solid lines denote mice with intact CD8 T cells. Statistical analyses were performed using linear models estimated with generalized least squares (B,G), ANOVA (C), linear models with weighted least squares (D,F), and the Cox proportional hazards model (E). For all graphs (B–G): *P<0.05, **P<0.01, ***P<0.001 compared with vehicle; ###P<0.001 compared with isotype control; †P<0.05, ††P<0.01 compared with corresponding MSLN-TTC monotherapy; ‡‡‡P<0.001 compared with corresponding anti-PD-L1 monotherapy. ANOVA, analysis of variance; MSLN-TTC, mesothelin-targeted thorium-227 conjugate; PD-L1, programmed death receptor ligand 1.

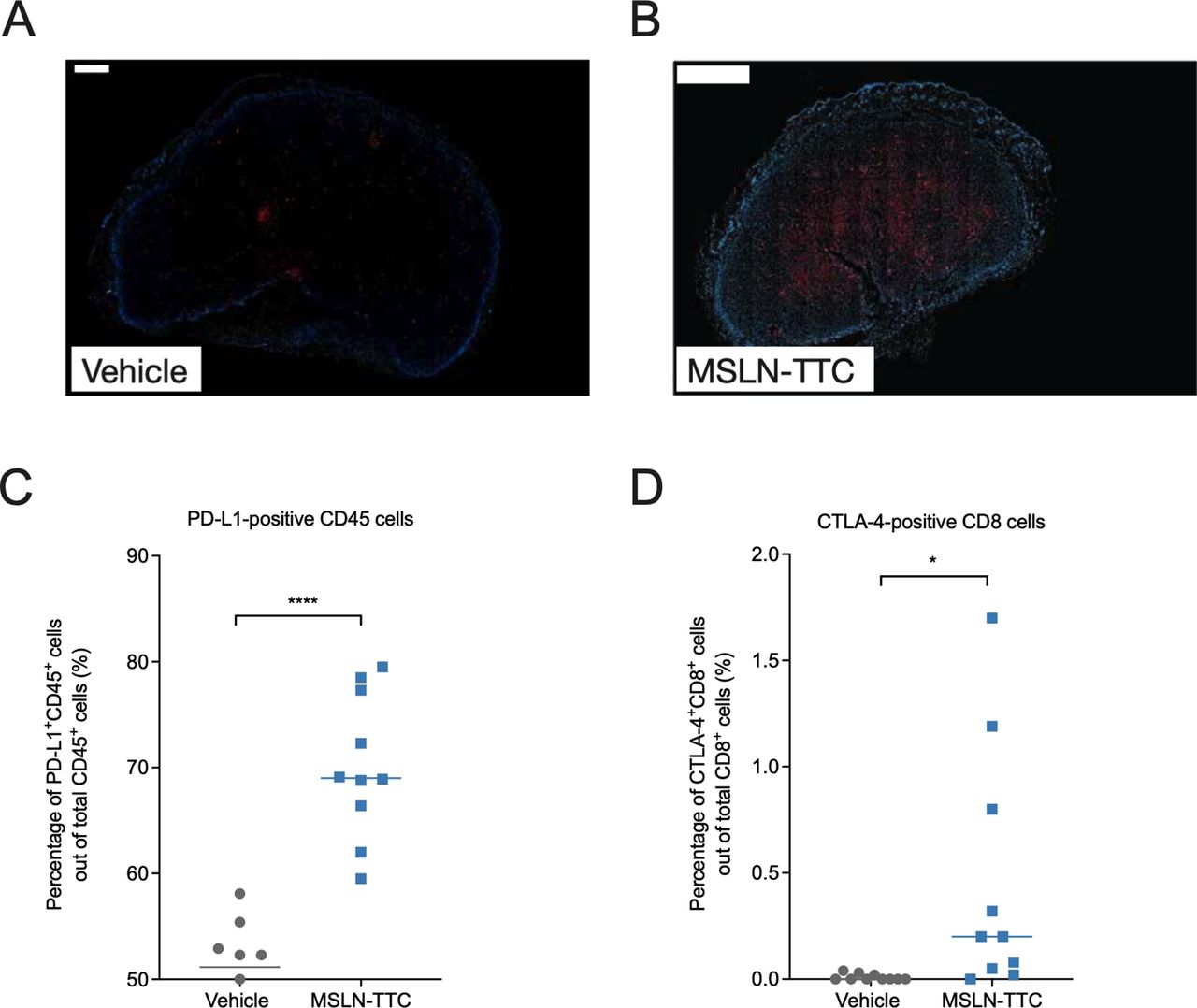
Analysis for DSBs and expression of PD-L1 and CTLA-4 in MC38-hMSLN tumors after treatment with MSLN-TTC (a single intravenous dose) by IHC and flow cytometry. (A,B) Analysis of DNA DSBs by detection of γH2AX in MC38-hMSLN tumors treated with (A) vehicle and (B) MSLN-TTC (250 kBq/kg), isolated 5 days after treatment start, using immunofluorescence staining with phosphohistone H2A.X (Ser139) rabbit mAb antibody. Blue denotes DAPI staining of the nuclei and red denotes γH2AX. Scale bars 500 and 1000 µm, respectively. (C,D) Expression of (C) PD-L1 and (D) CTLA-4 in tumors isolated 9 days after MSLN-TTC treatment (500 kBq/kg) as determined by flow cytometry. *P<0.05, ****P<0.0001 compared with vehicle. DSB, double-strand break; IHC, immunohistochemistry; MSLN-TTC, mesothelin-targeted thorium-227 conjugate; PD-L1, programmed death receptor ligand 1.
The efficacy of MSLN-TTC was next evaluated in combination with anti-PD-L1. In order to detect additive or synergistic activity, the doses for both therapies were reduced in the combination treatment. As presented in figure 3D and summarized in table 1, when administered at a mean tumor volume of 140 mm3, the administered lower doses (250 kBq/kg for MSLN-TTC, and 0.35, 0.75, or 1.5 mg/kg for anti-PD-L1) were less efficacious as monotherapies with determined treatment:control (T:C) values of 0.38 for MSLN-TTC and 0.42 (1.5 mg/kg) to 1.03 (0.35 mg/kg) for anti-PD-L1 on day 15. A radiolabeled isotype control administered at 250 kBq/kg showed no significant tumor growth inhibition (T:C 0.76). However, combining a single dose of MSLN-TTC (250 kBq/kg) with anti-PD-L1 (1.5 mg/kg) strongly increased antitumor activity (T:C 0.08 on day 15, figure 3D and table 1), resulting in increased overall survival (figure 3E) and a higher number of mice with complete tumor eradication on day 110 (table 1). All treatments were well tolerated based on body weight measurements (online supplemental figure S4C).
Efficacy of treatment regimens in MC38-hMSLN tumor-bearing mice
Next, we investigated whether sequencing the MSLN-TTC (250 kBq/kg) and anti-PD-L1 (1.5 mg/kg) treatments influences the outcome of the therapy. As presented in figure 3F and table 1, simultaneous administration of the compounds resulted in the strongest tumor growth inhibition. In contrast, when either of the compounds was administered 1 week prior to the other, the combined antitumor activity decreased. As MC38-hMSLN tumors were found to coexpress both targets, the decrease in efficacy might be explained by target clearance when therapies are administered sequentially. This hypothesis was supported by the fact that even though the MSLN expression was lower in tumors simultaneously treated with MSLN-TTC and anti-PD-L1 than in vehicle-treated tumors, the expression was still higher than that seen in the sequentially treated group (online supplemental figure S5).

Immune cell populations in MC38-hMSLN tumors after treatment with MSLN-TTC, anti-PD-L1, or their combination as determined by flow cytometry. MSLN-TTC was administered at 500 kBq/kg (a single intravenous dose) and anti-PD-L1 at 10 mg/kg (Q3/4D, intraperitoneal). Tumors and tumor-draining lymph nodes were isolated 10 days after first treatment (n=5–10/group). (A,B) Percentage of (A) CD4-positive T cells and (B) CD8 T cells in tumors. (C,D) Percentage of CD103-positive cDC1 cells in (C) tumors and (D) tumor-draining lymph nodes. (E–G) Percentage of tumor-associated (E) M1 and (F) M2 macrophages and (G) the calculated M1/M2 ratio in tumors. Statistical analysis was performed using one-way ANOVA. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 compared with vehicle. ANOVA, analysis of variance; MSLN-TTC, mesothelin-targeted thorium-227 conjugate; PD-L1, programmed death receptor ligand 1.
In summary, the data demonstrate that MSLN-TTC shows in vivo efficacy as a single agent, and the efficacy is further enhanced in combination with anti-PD-L1 in immunocompetent mice.
In vivo tumor growth inhibition of MSLN-TTC is decreased in CD8 T cell-depleted mice and in immunocompromised Rag2/Il2rg knockout mice
To study the role of CD8 T cells in potentially mediating the observed efficacy, CD8 T cells were depleted by the administration of an anti-CD8 antibody to MC38-hMSLN tumor-bearing immunocompetent C57BL/6 mice. Based on T:C ratios and tumor doubling times, a decrease in efficacy for all treatments was observed in comparison to corresponding treatment groups with intact CD8 T cells indicating a role for CD8 T cells in MSLN-TTC and anti-PD-L1 monotherapy as well as combination therapy (figure 3G and online supplemental table S2).
Furthermore, MC38-hMSLN cells were inoculated into immunocompromised Rag2/Il2rg knockout mice, lacking T cells, B cells, and NK cells. Tumor growth inhibition of MSLN-TTC compared with vehicle was observed only at the highest dose level (T:C values 0.63, 0.5, and 0.26 with 125, 250, and 500 kBq/kg, respectively; online supplemental figure S6). As expected, anti-PD-L1 treatment at 1.5 or 5.0 mg/kg showed no tumor growth inhibition resulting in T:C values of 0.8 and 1.0, respectively.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rechallenge of mice with initial complete tumor eradication 146 days after start of experiment. (A) Tumor-free survivors from experiments described in figures 3 and 4) were inoculated with MC38-hMSLN and mock-transfected MC38 cells and treated with MSLN-TTC and anti-PD-L1 as indicated in the schematic figure. Inoculation was performed on the side of the primary tumor or on the contralateral flank in parallel. (B–D) Tumor growth in rechallenged mice initially treated with (B) MSLN-TTC, (C) anti-PD-L1, or (D) MSLN-TTC/anti-PD-L1. Green color denotes mice that were inoculated with MC38-hMSLN cells and orange color denotes mice with mock-transfected MC38 cells. MSLN-TTC, mesothelin-targeted thorium-227 conjugate; PD-L1, programmed death receptor ligand 1.
MSLN-TTC induces DNA DSBs, upregulation of immune cell markers, and increases the number of CD8 T cells in monotherapy and in combination with anti-PD-L1 in vivo
As the main MoA of MSLN-TTC is the induction of clustered DNA DSBs, the expression of the DNA damage marker γH2AX was examined by IHC. As presented in figure 4, the MC38-hMSLN tumors in MSLN-TTC-treated, immunocompetent mice showed increased levels of γH2AX in comparison to vehicle-treated tumors (figure 4A,B). Isolated tumor cell populations from mice treated with MSLN-TTC also demonstrated a significant upregulation of the immunosuppressive markers PD-L1 (mirroring the in vitro findings in figure 2I) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) compared with vehicle-treated tumors (figure 4C,D).
Flow cytometry analysis demonstrated a significantly lower percentage of CD4-positive T cells in MC38-hMSLN tumors treated with MSLN-TTC/anti-PD-L1 when compared with the vehicle-treated group (figure 5A). The percentage of CD8 T cells was significantly increased in tumors treated with MSLN-TTC or anti-PD-L1 monotherapies, and further elevated in MSLN-TTC/anti-PD-L1-treated tumors, compared with vehicle (figure 5B). The increase of CD8 T cells in tumors was also detectable by IHC (online supplemental figure S7A,B). In tumors isolated from vehicle-treated mice, CD8 T cells localized in the outer periphery, whereas in the MSLN-TTC, anti-PD-L1, or MSLN-TTC/anti-PD-L1-treated tumors, they localized more centrally, indicating CD8 T cell infiltration on treatment.
DCs play a central role in cross-presentation of tumor cell antigens, thereby leading to an activation of the adaptive immune system, particularly via CD8-positive and CD103-positive conventional type 1 dendritic cells (cDC1). No significant changes were detectable in the percentage of intratumoral CD8-positive cDC1 cells on any treatment (data not shown). However, migratory CD103-positive cDC1 cells were significantly decreased in MC38-hMSLN tumors in all treatment groups compared with vehicle, with the strongest effect observed for MSLN-TTC and MSLN-TTC/anti-PD-L1 treatments (figure 5C). In TdLNs, no changes in CD8-positive cDC1 cells were observed (data not shown), but instead, a trend in increasing levels of CD103-positive cDC1 cells was detectable in MC38-hMSLN tumors in mice treated with MSLN-TTC, anti-PD-L1, or MSLN-TTC/anti-PD-L1 in comparison to the vehicle group (figure 5D).
As EBRT is known to induce an immunosuppressive M2 macrophage phenotype,20 treated MC38-hMSLN tumors were analyzed for immunostimulatory M1 and immunosuppressive M2 macrophages by flow cytometry. MSLN-TTC and MSLN-TTC/anti-PD-L1 treatments caused non-significant decreases in M1 macrophages (figure 5E), whereas the percentage of CD206-positive M2 macrophages increased significantly in MSLN-TTC and MSLN-TTC/anti-PD-L1 groups compared with the vehicle-treated group (figure 5F). Overall, a trend towards an M2 macrophage phenotype induction was observed in MSLN-TTC and MSLN-TTC/anti-PD-L1-treated tumors compared with vehicle (figure 5G).
RT-qPCR was used for detecting changes in immunological markers in treated tumors (online supplemental figure S8A). The transcript levels of IL-2 and the M1 macrophage marker iNOS were increased significantly in MSLN-TTC/anti-PD-L1-treated tumors and IFN-γ in anti-PD-L1 monotherapy group compared with vehicle. Furthermore, trends in increased levels of programmed death receptor 1 (PD-1), PD-L1, and TNF-α were observed in MSLN-TTC/anti-PD-L1-treated tumors, and increases of IL-6 in monotherapy groups. A 1.5-fold upregulation of CXCL10 was detected in MSLN-TTC-treated tumors, whereas increased transcript levels for IL-10 and the immunosuppressive markers TGF-β and FOXP3 were detected in anti-PD-L1 and MSLN-TTC/anti-PD-L1, but not in MSLN-TTC-treated tumors. Transcripts for STING, IFN-β, IFNAR, and TREX1 were not found to be deregulated.
On the protein level, MSLN-TTC, anti-PD-L1, and MSLN-TTC/anti-PD-L1 treatments increased the levels of the immunostimulatory markers IFN-γ, CCL2, and CCL3 in tumors, with the strongest significant increase observed for MSLN-TTC/anti-PD-L1 treatment (online supplemental figure S8B–D and table S3). In addition, the combination treatment induced significant upregulation of IL-10 (online supplemental figure S8E) and a trend in increase of IL-2, IL-5 and CCL4 (online supplemental figure S8F–H).
In summary, these data demonstrate that MSLN-TTC as a single agent induces tumor infiltration of CD8 T cells which is enhanced in combination with anti-PD-L1. Furthermore, the data indicate that MSLN-TTC treatment results in the migration of CD103-positive cDC1 cells from the tumor to TdNLs.
Rechallenge of mice with complete tumor regression demonstrated immunity against MC38-hMSLN
To test whether treatment with MSLN-TTC, anti-PD-L1, or MSLN-TTC/anti-PD-L1 had resulted in systemic immunity against MC38-hMSLN cells in mice with complete tumor regression, the same mice were rechallenged by inoculating MC38-hMSLN cells and MC38-mock-transfected cells into the same animal simultaneously, 146 days after the first tumor inoculation (figure 6A). As presented in figure 6B–D, mice initially treated with MSLN-TTC, anti-PD-L1, or MSLN-TTC/anti-PD-L1 rejected the growth of MC38-hMSLN cells, but not the growth of MC38-mock-transfected cells. Furthermore, the tumor rejection was irrespective of the site of cell inoculation (primary tumor or the contralateral flank), indicating that the respective initial treatments had resulted in a systemic immunization against MC38-hMSLN tumors but not against MC38-mock-transfected tumors. This suggests that the acquired immunity is only developed against the cell line with the same ‘genetic background’ after systemic MSLN-TTC therapy.
In summary, these data further corroborate our findings that immune cells contribute to the efficacy of MSLN-TTC.
Discussion
The immunostimulatory effects caused by EBRT have been extensively studied in preclinical models,21 22 supporting optimized clinical protocols in combination with immune checkpoint inhibitors and resulting in improved patient outcomes.10 23 24 Several targeted radiotherapies have been approved or are in clinical development for various cancer types, in particular for metastatic castration-resistant prostate cancer (NCT03511664, NCT03658447, and NCT038055942 14 25). However, only a few preclinical studies have investigated the underlying immunostimulatory effects of targeted radiotherapy or they have lacked a detailed analysis of the underlying pathways. Still, if available, such knowledge could guide clinical development.12 26–28
Here, we demonstrate that beyond apoptotic cell death induced by difficult-to-repair clustered DNA DSBs, TTCs are capable of activating the innate and the adaptive immune systems involving several signaling pathways. MSLN-TTC treatment activated multiple immunostimulatory genes in a dose-dependent manner in vitro such as chemokines, type I interferons, molecules of the acute phase as well as genes involved in DNA sensing pathways, including antiviral response genes, pathogen-associated molecular patterns (PAMPs), and STING-related genes. Interestingly, the gene for DNA exonuclease TREX1, known to suppress immunostimulatory effects by clearing cytosolic double-stranded DNA fragments,9 was upregulated in MC38-hMSLN cells at high TTC dose levels. In parallel, we observed activation of pSTING on protein level in NCI-H226 and MC38-hMSLN cells, but not in OVCAR-3 and OVCAR-8 cells, the latter ones described to be defective in cGAS/STING signaling.19 Furthermore, secretion of several proimmunostimulatory proteins was observed on these two ovarian cancer cell lines, matching the effects of EBRT on human breast cancer cell lines.29 Importantly, most of the deregulated genes and upregulated proteins from underlying pathways were enriched in all studied cell lines, although at different intensities, which could be explained by differences in the genetic background of the cells.
Furthermore, in vitro exposure of cells to MSLN-TTC led to an increase in cell surface expression of DAMPs,2 4 resulting in DC activation, demonstrating that TTCs are capable of activating the innate immune system. This is in line with previously published data for the TAT radium-223 dichloride, which has been described to upregulate calreticulin, leading to subsequent activation of CD8 T cells in various cancers.13 In addition, exposure to MSLN-TTC increased cell surface levels of the immunosuppressive marker PD-L1 on several tested cell lines, along with upregulation of the costimulatory marker ICOS ligand and the type I interferon sensing receptor IFNAR on MC38-hMSLN cells in vitro.
The immunostimulatory effects of MSLN-TTC were further evaluated in vivo in immunocompetent MC38-hMSLN tumor-bearing mice. MSLN-TTC monotherapy showed dose-dependent efficacy resulting in a higher number of mice achieving complete tumor eradication at increasing MSLN-TTC doses. Detailed MoA analysis demonstrated that MSLN-TTC monotherapy caused induction of DNA DSBs as evidenced by the DNA damage marker γH2AX, and further resulted in an upregulation of the immune checkpoint markers PD-L1 and CTLA-4, priming them for combination therapies such as anti-PD-L1. Indeed, the combination of MSLN-TTC with anti-PD-L1 strongly potentiated the efficacy in comparison to the respective monotherapies when administered simultaneously, but not sequentially. This might be due to potential target clearance on administration of, for example, anti-PD-L1 prior MSLN-TTC.
The increased antitumor efficacy of the combination treatment was demonstrated to rely on intratumoral changes of immune cells in vivo. A strong decrease in CD4-positive cells was observed in MSLN-TTC/anti-PD-L1-treated tumors. Furthermore, MSLN-TTC induced infiltration of CD8 T cells in tumors in monotherapy, which was further potentiated in MSLN-TTC/anti-PD-L1-treated mice. These changes in CD8 T cells were detectable by flow cytometric and IHC analyses. As such, CD8 T cells also tended to localize centrally in MSLN-TTC and MSLN-TTC/anti-PD-L1-treated tumors, whereas in vehicle-treated tumors, a more peripheral localization was observed. A similar relocalization of CD8 T cells on anti-PD-L1/PD-1 treatment has been demonstrated in MC38 tumors by positron emission tomography (PET) imaging.30 Notably, activation of CD8 T cells has also been observed for other TTCs in syngeneic models in our laboratories, including the murine breast cancer model 4T1 and the Lewis lung carcinoma model (data not shown). The detected cellular changes were accompanied by increased levels of proinflammatory cytokines and chemokines as detected by ex vivo protein and mRNA analyses of the tumors. Strongest upregulation of IFNγ, CCL3, CCL4, IL-2, IL-5, and IL-10 was observed for the MSLN-TTC/anti-PD-L1 combination treatment. Interestingly, IL-10 has been shown to enhance antitumor responses by the suppression of CD4-positive T cells31 32 which matches our flow cytometry observations on CD4-positive T cells. The combination treatment also led to upregulation of transcripts of the immunosuppressive markers TGF-β and FOXP3 in vivo; however, no changes in STING or TREX1 were detectable in MSLN-TTC-treated tumors, which contrasts our in vitro findings. We explored the potential involvement of type I IFN signaling with an intratumoral administration of an IFNAR-blocking antibody to MC38-hMSLN tumor-bearing mice (data not shown). The blockage had no impact on the efficacy of the MSLN-TTC/anti-PD-L1 combination treatment or the MSLN-TTC monotherapy. In contrast to this, a recent study demonstrated the involvement of the pSTING pathway and type I IFN signaling after systemic administration of an yttrium-90-NM600 targeted radiotherapy very elegantly in tumor-bearing immunocompetent mice.33 Thus, further studies are warranted to study the immune activation on TTC therapy in vivo.
In parallel to increased levels in CD8 T cells, we also detected a significant decrease in the level of migratory CD103-positive cDC1 cells, described to transport cellular antigens from the periphery to the lymph nodes to activate CD8 T cells,34 35 in tumors treated with MSLN-TTC or MSLN-TTC/anti-PD-L1. In parallel, a slight increase of CD103-positive cDC1 cells was observed in the respective TdLNs.
The involvement of CD8 T cells boosting the efficacy of TTCs was further highlighted in a CD8 T cell depletion experiment in MC38-hMSLN tumor-bearing immunocompetent mice, resulting in a strong decrease in the antitumor activity of MSLN-TTC and MSLN-TTC/anti-PD-L1. Similarly, only weak antitumor activity was observed when immunosuppressed Rag2/Il2Rg double knockout MC38-hMSLN tumor-bearing mice were treated with MSLN-TTC. Furthermore, rechallenge experiments which would be difficult to perform in human peripheral blood mononuclear cell-engrafted mice36 and where we therefore used MC38-hMSLN and MC38-mock-transfected cells instead, demonstrated that the mice had developed a systemic immune memory response against the transfected MC38-hMSLN, but not MC38-mock-transfected, cells. This indicates that the transfected human MSLN protein is the ‘neoepitope’, resulting in tumor immunity after MSLN-TTC therapy. Similarly, it has been recently demonstrated that tumor-bearing mice initially treated with a combination of an immune checkpoint inhibitor and the targeted radiotherapy yttrium-90-NM600 rejected only tumors of the same ‘genetic background’ in a rechallenging experiment.33 Overall, these experiments further support the hypothesis that immune cells contribute to the efficacy of TTCs in immunocompetent mice, which extends the knowledge on TAT beyond previously published data.1 12–15
EBRT is known to induce an immunosuppressive M2 macrophage phenotype.20 A non-significant increase of M2 macrophages in tumors after MSLN-TTC treatment was observed here as well, which, however, did not further increase in combination with anti-PD-L1, suggesting that the effect might be elicited by MSLN-TTC alone. Therefore, to circumvent potential TTC-mediated immunosuppressive phenotypes, additional studies should focus on optimized dose schedules,for example, by using fractionated dosing, and explore combinations of TTCs with macrophage-depleting agents.20
Strong MSLN expression is observed in biopsies of malignant pleural mesothelioma (MPM), ovarian cancer,37 and non-small cell lung cancer (NSCLC),38 and the highest co-expression of MSLN and PD-L1 can be found in NSCLC samples.39 40 Immune checkpoint inhibitors targeting the PD-1/PD-L1 signaling axes have shown clinical efficacy in MPM,40–42 and the combination of nivolumab and ipilimumab was recently approved based on findings from CheckMate 743 (NCT02899299). Thus, there would be a rationale of combining MSLN-TTC with anti-PD-L1 in the clinic.
In summary, we demonstrate that TTCs, such as the examined MSLN-TTC, activate the innate and the adaptive immune systems via multiple immunostimulatory pathways. Furthermore, these data highlight that immune cells, in particular CD8 T cells, contribute to the efficacy of TTCs in vivo. In addition, TTCs may increase the expression of immunomodulatory cell surface markers which primes tumors for combination treatment with immune checkpoint inhibitors. These data may therefore guide clinical combination strategies.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Animal experiments were performed under the national animal welfare laws in Germany and Denmark and approved by the local authorities.
Acknowledgments
We thank Manuela Brand, Kathleen Stadelmann, Jochen Hilbig, Stefanie Mai, Manuela Steinbach, and Maria Spelling for excellent technical assistance and HS Analysis GmbH (Karlsruhe, Germany) for developing the HS Analysis Webkit software used for IHC quantification. Aurexel Life Sciences Ltd. www.aurexel.com is acknowledged for editorial support funded by Bayer AG.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors UH and PL conceived the study and designed the experiments, acquired, analyzed, and interpreted data, wrote and revised the manuscript. LB, VC, AM, AS, ABL, CK, MJ, JJ, CN, and SS conceived the study and designed the experiments, acquired and analyzed data. AS, CK, CS, SH, JJ, CN, CS, SB, SZK, SH, and JK interpreted data, wrote and revised the manuscript. HH, AC, and DM wrote and revised the manuscript.
Funding The study was conducted by Bayer AG.
Competing interests PL, VC, ABL, AS, AM, LB, SB, SZK, CK, SS, SH, CS, HH, JK, AC, DM, and UH are employees of Bayer AG or Bayer AS. AS, SB, SH, CS, HH, and DM are shareholders of Bayer AG. PL, SH, JK, AC, and UH hold patents on targeted thorium-227 conjugates.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.