Article Text

Abstract
Background Immunotherapy in microsatellite stable colorectal or pancreatic cancer has not shown promising results. It has been hypothesized that targeting immunosuppressive molecules like SDF1-alpha/CXCL12 could contribute to immunotherapy and animal models showed promising results on T cell activation and migration in combination with immune checkpoint inhibition.
Methods Here, we describe the successful application of anti-CXCL12 (NOX-A12) in patients with advanced stage pretreated metastatic colorectal and pancreatic cancer (OPERA trial). The treatment consisted of 2 weeks of anti-CXCL12 monotherapy with NOX-A12 followed by combination therapy with pembrolizumab (n=20 patients) until progression or intolerable toxicity had occurred.
Results The treatment was safe and well tolerated with 83.8% grade I/II, 15.5% grade III and 0.7% grade V adverse events. Of note, for a majority of patients, time on trial treatment was prolonged compared with their last standard treatment preceding trial participation. Systematic serial biopsies revealed distinct patterns of modulation. Tissue and clinical responses were associated with Th1-like tissue reactivity upon CXCL12 inhibition. A downregulation of a cytokine cassette of interleukin (IL)-2/IL-16/CXCL-10 was associated with tumor resistance and furthermore linked to a rare, CXCL12-associated CD14+CD15+promonocytic population. T cells showed aggregation and directed movement towards the tumor cells in responding tissues. Serum analyses detected homogeneous immunomodulatory patterns in all patients, regardless of tissue responses.
Conclusions We demonstrate that the combination of CXCL12 inhibition and checkpoint inhibition is safe and grants further exploration of synergistic combinatorial strategies.
- gastrointestinal neoplasms
- cytokines
- drug therapy
- combination
- immunotherapy
- translational medical research
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- gastrointestinal neoplasms
- cytokines
- drug therapy
- combination
- immunotherapy
- translational medical research
Key messages
The anti-CXCL12 NOX-A12 is safe and well tolerated in patients with advanced stage heavily pretreated metastatic colorectal and pancreatic cancer.
For a majority of patients, time on trial treatment was prolonged compared with their last standard treatment preceding trial participation.
In serial biopsies, molecular and cellular patterns of a Th1 immune response are associated with disease stabilization and clinical benefit.
The efficacy of NOX-A12 is associated to the presence of a rare, CXCL12-associated CD14+CD15+promonocytic cell population in both entities.
Introduction
In recent years, immunotherapy has changed the therapeutic landscape for solid tumors dramatically, with the approval of new drugs in many indications within a remarkably short period. Their predominant mode of action is the activation of the adaptive immune system via checkpoint inhibition.1 However, it has been recognized that the presence and reactivity of effector cells significantly affect the efficacy of immunotherapies. Combinatorial approaches target the stromal compartment of solid tumors, resulting in migration of effector cells.2–5 These approaches act on the complex interactions in the tumor microenvironment, as was shown recently in macrophage-targeted immunotherapies.6–8
For the majority of patients with solid tumors, no effective immunotherapy strategy is yet available. Especially for microsatellite stable colorectal and pancreatic cancer, immunotherapy has thus far been ineffective.5 9 For patients with stage IV disease and limited therapeutic options and a 5-year survival of 11%–14.2% in metastatic colorectal cancer (CRC)10 11 and of 2.9% in pancreatic cancer12 and particularly in case of a significant disease burden,13 there is a tremendously high medical need for novel therapeutic options. Advanced metastatic disease typically exhibits liver metastases next to other distant metastases.14 In this setting, the development of more complex interventions into the immune landscape requires detailed knowledge of the interactions of relevant players and their response to interventions. The local immune infiltration (especially in CRC) influences the clinical course of the disease.15–18 While the vast majority of research still focuses on the tumor microenvironment in primary lesions,19 the role of infiltrating immune cells in metastases of CRC is also well established.20 21 The composition of the different populations of immune cells and their corresponding cytokines and chemokines—which reflect immune cell presence and activity—shape the local microenvironment and the subsequent clinical course.22–25 Immunosuppression and inflammation are key parameters26 and modulation of these is a main aim of new approaches. Animal experiments showed that immunosuppression can manifest as exclusion of T cells from tumor regions. Here, chemokine (C-X-C motif) ligand 12 (CXCL12) plays an important role. In mouse models, inhibition of chemokine (C-X-C motif) receptor 4 (CXCR4), a CXCL12 receptor, led to the promotion of T-cell accumulation and synergistic effects with checkpoint inhibition, leading to cancer regression.27 28 More recent data from inhibition of the CXCL12 receptor CXCR4 showed alleviation of desmoplasia, increased T-lymphocyte infiltration and an improved immunotherapeutic effect in a murine model of metastatic breast cancer.29
The majority of clinical studies addressing the CXCL12-CXCR4 axis have tackled hematological malignancies, where the neutralization of the CXCL12 gradient displaces malignant cells from the protected environment of the bone marrow towards the blood stream, where they are more susceptible to chemotherapy-mediated cell death. This has been recently seen in multiple myeloma30 or in chronic lymphocytic leukemia.31 In solid tumors, data are much scarcer, with two clinical trials that came out in 2020 in colorectal and pancreatic cancer. In the COMBAT trial, a CXCR4 antagonist (BL-8040) was given for the first time to patients with metastatic disease, in a combination with immune checkpoint blockade.32 In an independent study, another inhibitor of CXCR4, AMD3100 was administered by continuous infusion in patients with gastro-intestinal tumors as well.33
In the clinical study described here, we inhibited CXCL12 with NOX-A12 (olaptesed pegol), an L-RNA aptamer.34 35 NOX-A12 in combination with standard therapies was already employed in patients with chronic lymphocytic leukemia and multiple myeloma. In these studies, the molecule showed a favorable toxicity profile and clinical effects.31 36 37 Therefore a clinical trial in patients with pretreated advanced metastatic colorectal and pancreatic cancer was initiated (OPERA trial, Keynote-559, NCT03168139, EUDRACT 2016-003657-15), combining the anti-CXCL12 spiegelmer NOX-A12 with pembrolizumab.
Materials and methods
Patients’ characteristics and trial design
The OPERA phase I/II trial (olaptesed (NOX-A12) alone and in combination with pembrolizumab in colorectal and pancreatic cancer) (Keynote-559, ClinicalTrials.gov identifier NCT03168139) involves two times per week exposure to a fixed dose of olaptesed pegol (also known as NOX-A12) as a monotherapy for 2 weeks, followed by a combined therapy with 200 mg pembrolizumab one time every 3 weeks until disease progression or limiting toxicity, with a maximum of 24 months on trial treatment (figure 1). The treatment regimen with olaptesed pegol was based on safety and efficacy considerations (online supplemental methods).
Supplemental material

Schematics of the phase I/II OPERA trial. A window of opportunity, during which patients reviewed NOX-A12 (olaptesed pegol) as a monotherapy, was followed by combined NOX-A12 and pembrolizumab therapy until progression or unacceptable toxicity. SCR, screening.
Pharmacodynamics evaluating tissue alterations and immune infiltrate changes within the tumor microenvironment induced by CXCL12 inhibition with olaptesed pegol as well as safety and tolerability are the primary endpoints of this trial, being conducted according to the Declaration of Helsinki and relevant International Conference on Harmonization Good Clinical Practice guidelines. All patients had received various current standard of care treatment options and were refractory to standard chemotherapy. All patients provided written informed consent before participating in this study.
Another objective in this study was the determination of response to the combination therapy of olaptesed pegol and pembrolizumab. In the heavily pretreated populations included into this phase I/II study, the expected response rate to available standard therapies is near zero. For a sample size of 10 for each stratum, an exact 90% CI for the true response rate would be as indicated in online supplemental table 1 based on the exact binomial Clopper-Pearson method. This based the decision for the number of patients.
Biopsy collection and processing
Paired needle biopsies from liver metastases were collected during the trial from every patient: one before and a second one at the end of monotherapy with NOX-A12, in order to determine the changes in the tumor microenvironment (figure 1). Immediately after collection, biopsies were frozen in Cryo Embedding Medium (Medite) until further use. In 6 out of the 20 remaining patients, only the baseline biopsy but not the biopsy at day 14 was feasible (online supplemental figure 1).
Immunostainings
Eight micron-thick cryosections from the biopsies were fixed using either acetone/methanol ½ v/v or paraformaldehyde 4% and permeabilized with 0.1% Triton-X100. Immunohistochemistry was performed on the BOND-MAX automated stainer (Leica Biosystems, Nussloch, Germany) using the DAB-based Polymer Refine Detection Kit (Leica Biosystems) and the following antibodies: anti-CD3 (PS1) (Novocastra), anti-CD8 (4B11) (Novocastra), anti-PD-1 (NAT105) (Abcam), anti-CD11b (EP1345Y), anti-CXCL12 (EPR 1216), anti-arginase (ERP 6672(B)) and anti-iNOS (rabbit polyclonal, ab3523) from Abcam.
For immunofluorescent staining, the following primary antibodies were applied: anti-CD15 (28) and anti-CD14 (SP192) from Abcam. This was followed by secondary antibodies application (alexa488-anti-mouse and alexa594-anti-rabbit from Invitrogen), DAPI counterstain and mounting with Prolong gold antifade reagent (Invitrogen).
Histological image acquisition and analysis
Fluorescent sections were scanned using the NDP NanoZoomer scanner (Hamamatsu Photonics, Japan) at the 20× magnification. Brightfield virtual whole slide images were acquired at the 20× objective (Aperio AT2, Leica Biosystems) and semi-quantitative measurements were performed for CD3, CD11b, iNOS and arginase using the Visiomorph software platform (Visiopharm, Denmark) as previously reported 38. Briefly, the workflow allowed the quantification of cell densities across a given surface area (either whole tissue section or manually delineated liver metastasis, invasive margin and adjacent liver regions). Each image was visually inspected for exactitude and results are expressed as number of positive cells per square millimeter in the region of interest.
Multiplex cytokine profiling
Cryosections from the biopsies were collected and lysed with the Bio-Plex Cell lysis kit (Bio-Rad) according to the manufacturer’s instructions. Protein lysate concentration was measured (Pierce BCA Protein Assay, Thermo Fisher) and adjusted to 300 µg/mL. The protein concentration of 50 soluble factors was determined using the Bio-Plex ProTM human cytokine assays, as previously described 6.
Statistical analysis
Statistical analysis was performed on GraphPad Prism 8 (RRID: SCR_002798). Sample groups were compared using the non-parametric Mann-Whitney U test or Wilcoxon signed-rank test when appropriate. Fisher’s exact test was used for categorial comparisons. When comparing two groups with a normal distribution (Kolmogorov-Smirnov test of normality) and similar variances, parametric tests (paired t-tests) were used. Differences were considered statistically significant provided that the (two-tailed) p value was less than 0.05. In histograms, the mean (±SEM) is represented. *, p value<0.05; **, p value<0.01; ***, p value<0.001 and ****, p value<0.0001. Recursive partitioning analysis was performed on R,39 with the rpart package.40
Results
Trial layout and safety
Twenty patients with histologically confirmed metastatic CRC (11 patients) or pancreatic cancer (9 patients) enrolled in the OPERA trial (online supplemental figure 1). All patients had microsatellite stable disease and were thus considered non-responsive to anti-PD-1 (Programmed cell death protein 1) therapy.41 42 Patient details are summarized in table 1. Patients with pancreatic cancer were required to have received at least one, those with CRC at least two previous lines of therapy. As previous therapy, all standard of care treatments were allowed (including oxaliplatinum, irinotecan, anti-EGFR (Epidermal Growth Factor Receptor) antibodies, anti-VEGF (Vascular Epithelial Growth Factor) antibodies, 5-fluorouracil/capecitabine and trifluridine/tipiracil for CRC and oxaliplatinum, irinotecan, 5-fluorouracil, gemcitabine, nab-paclitaxel or erlotinib containing treatment for pancreatic cancer). After an initial phase of monotherapy with NOX-A12, patients received additionally pembrolizumab (figure 1).
Patients’ characteristics
Treatment was generally well tolerated with 162 adverse events (AEs) in total, thereof 45.7% grade I; 37.1% grade II; 16.4% grade III; no grade IV and 0.7% grade V. Most common AEs were abdominal pain, fatigue and peripheral edema (figure 2). Immune-related AEs did not exceed the known toxicity profile of pembrolizumab.

Safety profile in the OPERA trial.AEs, adverse events; SAEs, serious AEs, CRP, C-reactive protein; INR, international normalized ratio.
Clinical outcome parameters
No objective response (ie, partial remission (PR), complete remission (CR) according to RECIST V.1.1 criteria) was observed in these heavily pretreated, end-stage patients. Colorectal patients with cancer had had a median of five and patients with pancreatic cancer had a median of three previous lines of therapy. Overall survival at 6 months was 42%, at 12 months 22%; median overall survival was 3.97 months; median progression-free survival was 1.87 months. However, 7 out of 20 patients remained on trial treatment for more than 6 months (figure 3A). Disease stabilization following NOX-A12 and pembrolizumab combination therapy was observed (27% for colorectal and 22% for pancreatic cancer), predominantly in highly pretreated and previously rapidly progressing patients (figure 3B), resulting in a disease control rate of 25% (figure 3C and online supplemental figure 2B). Clinical benefit was also seen in some patients who did not achieve stable disease with time on treatment being increased up to 10-fold in relation to their latest preceding treatment (figure 3B and online supplemental figure 2A). In summary, a significant proportion of patients remained on study treatment longer than on some of their previous standard chemotherapies (figure 3B, box).
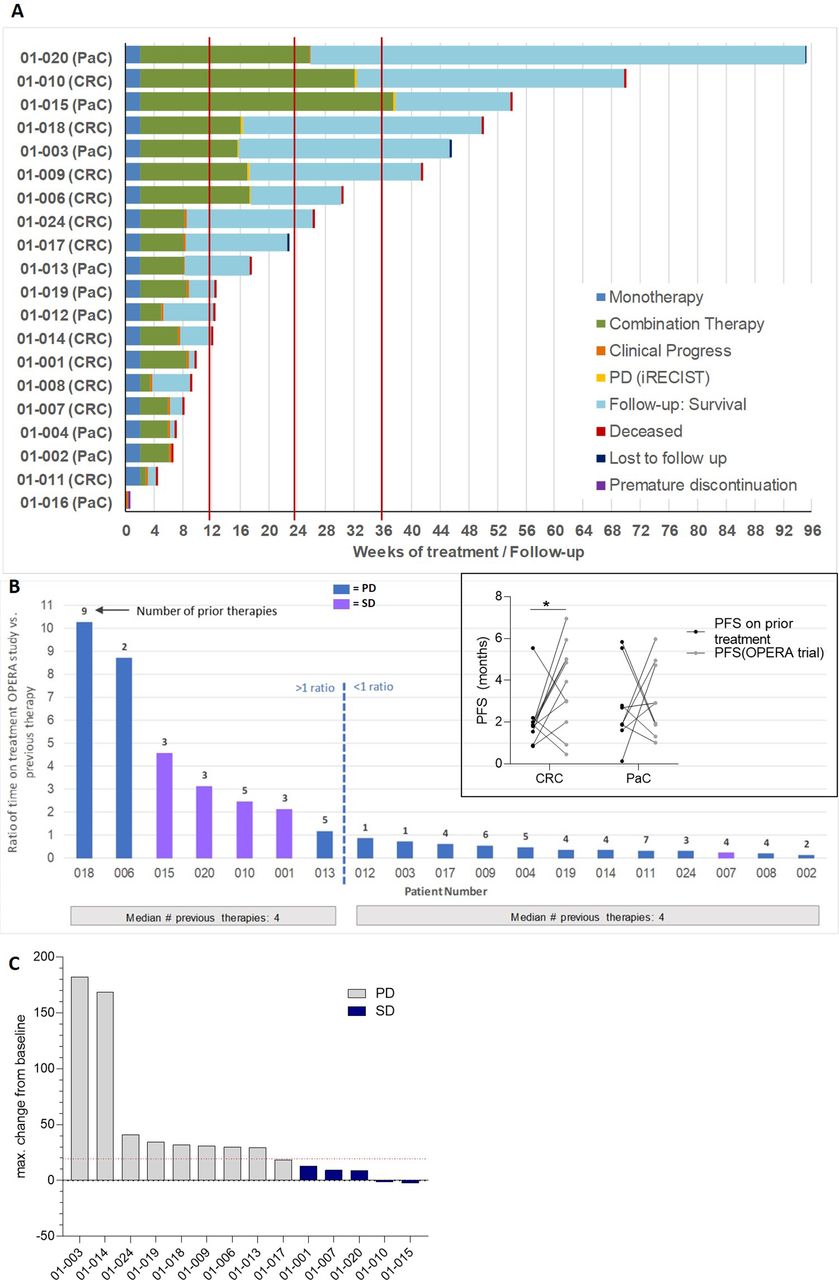
Depiction of survival and time on treatment (TOT) in the OPERA trial. The swimmer plot in (A) illustrates the follow-up of each patient in the trial, from monotherapy until discontinuation. Bar plots in (B) illustrate the TOT in all patients, as expressed in ratio of TOT in the OPERA trial versus the latest prior therapy (main graph). The insert in B illustrates the PFS in months in all patients. Finally, the Waterfall plot in (C) illustrated the objective responses in all patients in the OPERA trial at the time of best response. The maximum change in sum of diameters of target lesions from baseline at the time of best response is illustrated.CRC, colorectal cancer; PaC, pancreatic cancer; PD, progressive disease; SD, stable disease; CXCL12, chemokine (C-X-C motif) ligand 12.
Baseline CXCL12 and tissue parameters
Baseline levels of CXCL12 were obtained and compared with those after 14 days of treatment with NOX-A12 revealing presence and enrichment of CXCL12 in the tumor microenvironment (figure 4A–B). Analyses of baseline T cell infiltration in OPERA trial patients showed a generally lower number of T cells in the study population compared, for example, to the expected normal levels of CRC liver metastases in the literature43 (figure 4C). A multiplex detection and quantification of 50 cytokines, chemokines and growth factors was performed on the biopsies at baseline (day 0) and, interestingly, recursive partitioning analysis thereof allowed to identify a group of biomarkers with prognosis value. The respective concentrations of interleukin (IL)-17, IL-2 and IL-1b in the biopsies at baseline allow the construction of a decision tree that clusters patients according to their progression-free survival (online supplemental figure 3).

Baseline tissue parameters in the OPERA trial illustrating accumulation of CXCL12 in the CRC liver metastases and the immune alteration in the patient cohort. (A) Concentration of CXCL12 in tumor lysate of CRC primary tumor lysates and their corresponding liver metastases. (B) Concentration of CXCL12 in the primary tumors compared with their adjacent stroma and corresponding sera. (C) CD3+ T cell density in liver metastases in the OPERA trial and in a reference cohort.CRC, colorectal cancer; CXCL12, chemokine (C-X-C motif) ligand 12.
Tissue responses to CXCL12 inhibition
For n=14 patients, biopsy pairs were obtained, one at baseline (day 0) and one at the end of monotherapy with NOX-A12 (day 14). To obtain an overview of the changes in the molecular immune landscape that could be induced by NOX-A12, all biopsy pairs were used for quantification of 50 immune-related soluble factors and the results were submitted to unsupervised hierarchical clustering. This analysis, refined with the clinical annotation (ie, patients with disease stabilization), revealed the existence of patient subgroups (figure 5A). From a functional perspective, as detailed in online supplemental figure 4, three groups (‘cassettes’) of tissue responses could be classified. Cassette 1 responders were characterized by a signature of IL-2, interferon-gamma and IL-16 (patients 15, 14 and 1). Cassette 3 included patients exhibiting a decrease in Th1 cytokines (online supplemental figure 4) and a decrease of IL-16 and CXCL10 (patients 3, 2, 10, 20, 9 and 18). Lastly, cassette 2 encompassed a group of patients that did not show a specific increase or decrease in either cassette 1 or 3 cytokines/chemokines (patients 4, 6, 13, 17 and 24). Figure 5B illustrates the concentration changes of selected cytokines/chemokines in the aforementioned cassettes in tissue responders (cassettes 1 and 2) versus non-responders (cassette 3).

Local molecular changes in liver metastases at the end of monotherapy with NOX-A12 in the OPERA trial allows clustering of patients. (A) Unsupervised clustering of patients based on relative changes in the molecular immune landscape at the end of NOX-A12 monotherapy. In the heatmap, relative changes are expressed as Z-scores. (B) Concentrations of the most affected cytokines before and at the end of the monotherapy in patients clustered in tissue responders and tissue non-responders.CXCL10, chemokine (C-X-C motif) ligand 10; IL, interleukin; IFN, interferon; TNR, tissue non-responders; TR, tissue responders.
These observations were in line with the observation of increased T cell infiltration inside the liver metastasis at the end of the monotherapy, observed specifically in patients clustered as tissue responders (figure 6A–B). The density of CD8+ (figure 6C) and PD1+ T cells (figure 6D) was little affected (figure 6B). A clear change in the distribution patterns of CD3+ T cells in the tissue was observed in response to CXCL12 inhibition. Indeed, our analyses showed a trend of T cells to move towards tumor cells following CXCL12 inhibition in all patients that were classified as tissue responders (figure 6E).

Comparative immune landscape in tissues at the onset of treatment with NOX-A12 and impact of NOX-A12 in the immune landscape in responders and non-responders. (A) Density of effector T cells in liver metastases split in three regions (LM=liver metastasis, IM=invasive margin and AL=adjacent liver) in tissue responders and non-responders. (B–D) Relative changes in T cell density in the regions depicted in (A) in the corresponding patients, expressed as (density(d14) – density(d0)*100/density(d0)), for CD3 (B), CD8 (C) and PD1 stains (D). (E) Distribution/proximity analysis of T cells in the tissue biopsy collected at the onset (left) and at the end (right) of the NOX-A12 monotherapy, in tissue responders and non-responders. (F) Modification in the T cell skewness in all biopsies. CXCL12, chemokine (C-X-C motif) ligand 12.
Finally, a distinct shift in the T cell distribution patterns towards increased ‘skewness’—defined as a heterogeneous (‘clustered’) T cell distribution in the tissue was observed. This indicates an aggregation of T cells,44 which is typically associated with enhanced antigen presentation and T cell activation (figure 6F). Interestingly, this was observed in all patients, independently of their molecular-based clustering.
Identification of a rare CD14+CD15+ promonocytic population co-localizing with CXCL12 in the OPERA trial
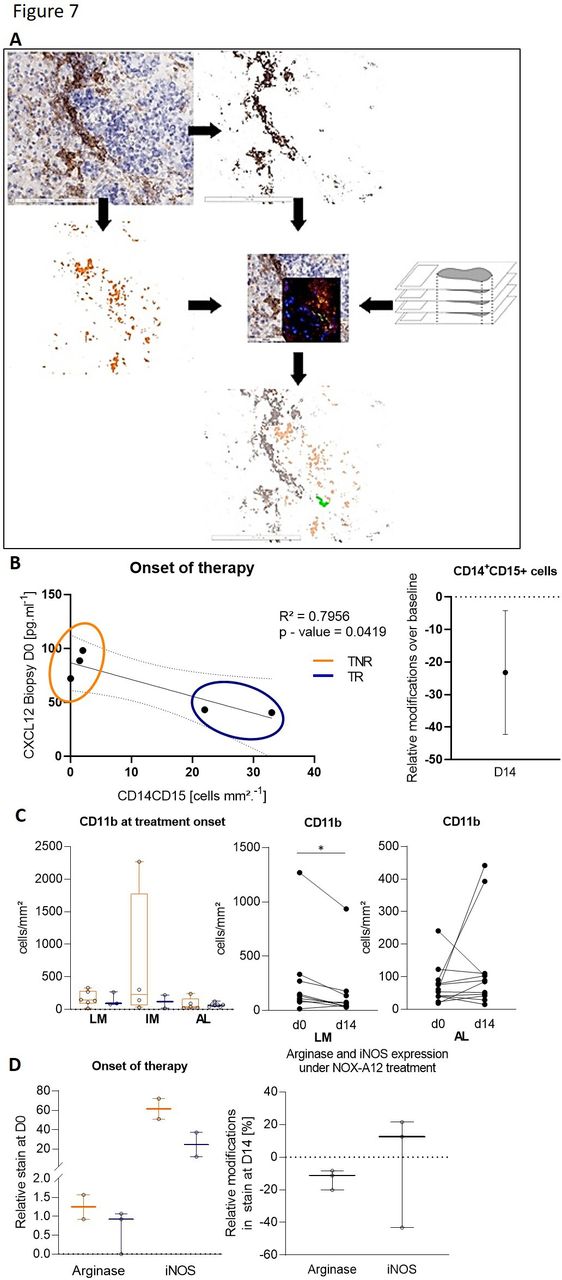
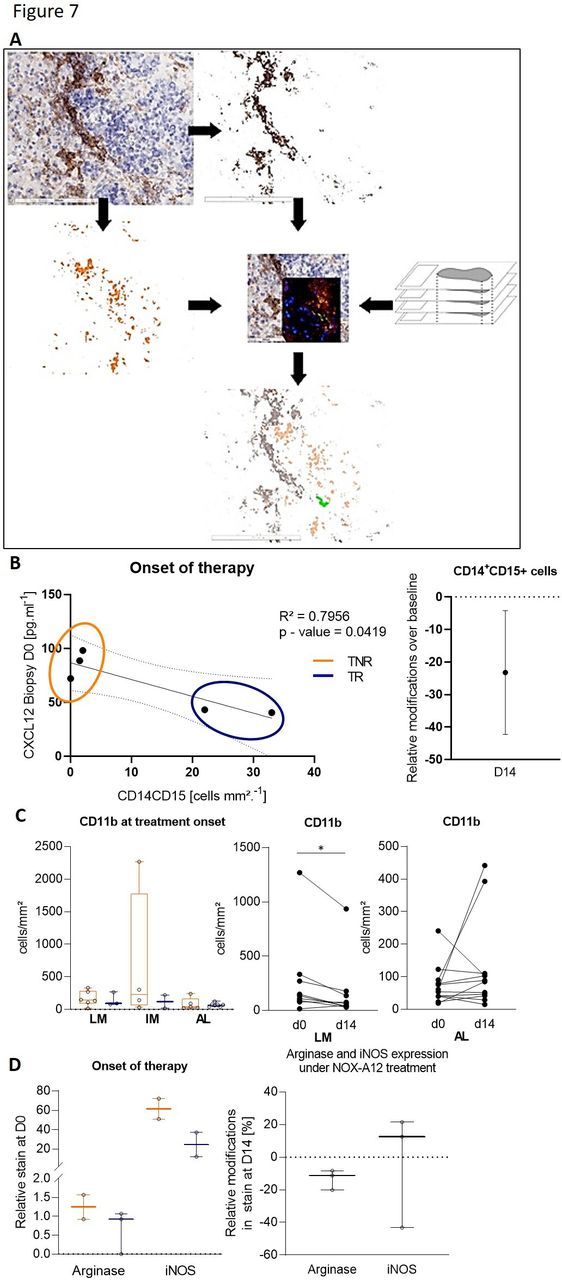
As CXCL12 has been known to influence monocyte-to-macrophage differentiation as well as macrophage polarization in several independent preclinical studies, we studied the monocyte lineage using CD11b, CD14 and CD15 markers on biopsies, at baseline and at the end of monotherapy. Monocyte analyses showed a relationship between tissue response (Th1-like patterns) and the CXCL12 levels and the presence of a specific CD14+CD15+ promonocytic precursor cell population in the tissue. This population co-localizes with CXCL12, as identified through thin section overlay image analyses (figure 7A). Intriguingly, responders displayed both higher initial numbers of CD14+CD15+ cells and lower CXCL12 concentration in the baseline biopsy, which appeared negatively correlated to each other (figure 7B). During anti-CXCL12 therapy, the numbers of these triple positive cells were slightly reduced (figure 7B). In addition, a reduced number of CD11b+ cells was also observed in the biopsies under treatment (figure 7C), where a differential effect on CD11b+ cells in the liver was seen (figure 7C) and CD11b+ cells in the liver metastasis itself and in the invasive margin being generally stable over time. No general depletion of mature macrophage populations could be identified following administration of NOX-A12. Concomitantly, both arginase and iNOS expression levels in the tissue at baseline were slightly lower in tissue responders compared with tissue non-responders (figure 7D) and while arginase expression, though low at baseline, was slightly decreased under treatment, iNOS expression showed a very light trend to an increase (figure 7D). Though these results suggest the existence of a relationship between a tissue response and a higher density of innate immune cells of the monocytic lineage, the low numbers of biopsies used impose caution at this stage and further investigations are necessary.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Identification of a CXCL12-associated CD14+ CD15 monocytic precursor cell type and myeloid markers associated with tissue response to NOX-A12 treatment. (A) Schematics of the virtual thin section overlay workflow applied to identify the CD14+CD15+CXCL12+ cells in tissue biopsies. Comparative immune landscape in tissues at the onset of treatment with NOX-A12 and impact of NOX-A12 in the immune landscape in responders and non-responders (left) and the relative changes in CD14+CD15+ cell density at day 14 (right). (B) Dot plot illustrating the concentration of CXCL12 in the liver metastasis at the onset of treatment, as well as its correlation with the density of CX14+CD15+ cells in the corresponding tissues at the same time point. (C) Density of CD11b+ cells at treatment onset and under treatment. (D) Density of cells positive for arginase and iNOS immunostains before the onset of therapy, in responders and non-responder (left) and relative changes under treatment (right).
Immunological changes in serum
Both tissue and serum CXCL12 levels increased markedly during NOX-A12 treatment (online supplemental figure 5A). This was an expected finding and is due to the binding of NOX-A12 to CXCL12, which results in an inactive complex that is subjected to slower elimination and degradation than free CXCL12.36
We did not observe any concordance between the tissue level response and the serum response, or between the serum response and the clinical outcome (progression-free survival (PFS), overall survival (OS), disease stabilization). Reaction patterns in the serum were obvious and uniform, reflecting a distinct pattern of immunomodulation (online supplemental figure 5B).
Discussion
Immunotherapy for microsatellite stable CRC or pancreatic cancer has been so far futile.5 45 46 Previous data from animal experiments indicated a beneficial effect of inhibition of the SDF1alpha/CXCL12 receptor CXCR4.27 29 The described changes included attraction of T cells into the microenvironment and subsequent activation via anti-PD-1 to induce massive tumor regression. Similarly, in tumor-stroma spheroids, CXCL12 inhibition can break the immunosuppression by paving the way for immune effector cells into the tumor.47 In order to break the negative effects of CXCL12, NOX-A12 (olaptesed pegol) was administered intravenously to patients in the OPERA trial. NOX-A12 is an L-configured aptamer (spiegelmer)48 that binds CXCL12 with high affinity and specificity across various species, including humans.49 Of note, NOX-A12 not only directly binds and inhibits but also detaches the cell-surface bound CXCL12, which also diminishes a CXCL12 gradient throughout the tissue.50 NOX-A12 inhibits the interaction of CXCL12 with both its receptors: CXCR4 and CXCR7.51 The clinical application of NOX-A12 has shown synergistic effects for combinatorial treatment with standard therapy in relapsed or refractory multiple myeloma and chronic lymphatic leukemia.31 37 The potential of NOX-A12 to treat glioblastoma in combination with radiotherapy is currently being investigated (clinical trial identifier NCT04121455).52 The OPERA trial is the first clinical trial utilizing the spiegelmer technology to inhibit selectively the chemokine CXCL12 in patients with colorectal or pancreatic cancer.
Last year, two clinical trials investigated the effect of targeting CXCR4 (one of the two receptors of CXCL12) using two independent drugs in patients with metastatic pancreatic or CRC.32 33 In the COMBAT trial, the treatment regimen was different from the current study, but also started with a phase of monotherapy followed by combination with immune checkpoint blockade. During combination therapy, patients received the CXCR4 antagonist BL-8040 every other day. This led to disease stabilization in 9 out of 37 patients and partial response in 1 out of 37 patients. With this regimen, one patient had study discontinuation because of treatment-related adverse effects.32 Independently, the small molecule inhibitor AMD3100 (plerixafor) was administered for 1 week by continuous infusion in 26 patients with metastatic, microsatellite-stable (MSS) pancreatic or CRC.33 Of course, in the latter study, a treatment duration of 1 week is too short to lead to any clinical response and the study focused on the immunobiological changes triggered by the treatment.33 53 Still, both studies identify CXCR4 inhibition by small molecule/peptide being overall well tolerated despite a continuous, high-dose regimen and observe an increase in cytotoxic T cell density in the metastasis biopsy under treatment. Regarding the NOX-A12 spiegelmer, previous clinical experience showed safe application of high-dose NOX-A12 in intermittent intervals31 37 leading to the OPERA trial design.
Indeed, similarly to the two aforementioned studies, the overall treatment with either NOX-A12 as monotherapy or the combination with pembrolizumab was very well tolerated, even by patients who had more than four previous lines of chemotherapy. The profile of AEs in the study was comparable with the safety profile for pembrolizumab and typical for the underlying diseases colorectal and pancreatic cancer, with abdominal symptoms being the most prevalent. Overall tolerability was excellent with 83.8% of AEs being grades 1 and 2, similarly to other trials.31 Although the response rate for partial or complete remissions was 0%, the clinical course for many patients was remarkable. Especially the prolonged duration of trial treatment and disease stabilization in comparison to patients’ previous chemotherapies was unprecedented. For instance, in four out of five patents with disease stabilization under NOX-A12-pembrolizumab combined treatment, the best response in any previous line of treatment had been progressive disease. This is especially surprizing, as the common experience in solid tumor treatment shows shortening intervals for treatments with advanced lines of therapy.54 55 For immunotherapy, the typical expectation is that there is, with every line of treatment, a shortened disease stabilization but prolonged overall survival.56 57 Here we report the opposite: immunotherapy with NOX-A12 and pembrolizumab is able to stabilize the disease in heavily pretreated microsatellite stable patients for prolonged periods, for almost a quarter of the patients overall survival close to 12 months could be reached. Of the 10 patients who were alive for more than 3 months, 80% were alive beyond 24 weeks and 60% beyond 36 weeks.
One of the questions arising is: what makes up the microenvironment in the patients that do have a clinical benefit from the dual inhibition. Increased CXCL12 levels in all post-treatment tumor biopsies as well as in serum were consistent with penetration of NOX-A12 into the tumor tissue, confirming the in vitro data.58 Indeed, CXCL12 is normally rapidly engulfed in CXCR7+ cells by endocytosis and degraded, thereby maintaining a functional gradient.58 This is blocked in the presence of NOX-A12, which detaches CXCL12 from all its receptors.36 A higher amplitude of CXCL12 level changes in tissue observed on monotherapy not only indicates a more complete CXCL12 neutralization but also correlates with a favorable cytokine profile and clinical benefit.
Systematic analyses of serial biopsies revealed the presence of three main groups of responses after 2 weeks of monotherapy with NOX-A12. Unsupervised clustering splits patients into three distinct groups, based on the modification of a few cytokines of which the changes indicate the local establishment of a Th1 immune response: IL-2, interferon (IFN)-γ, IL-16 and CXCL10. Based on these cytokines, the patients cluster into tissues responders, exhibiting an increase in IL-2, IFN-γ and IL-16 and a group of tissue non-responders, of which the tumor expression of IL-2, IL-16 and CXCL10 is decreased. In the group defined as tissue responders, one would assume an existing proliferation of T cells (based on the IL-2 increased expression) as well as their chemotaxis (in response to cancer cell-derived and monocyte-derived IL-16) and activation (in response to IFN-γ). In the group defined as non-responders, one would assume the opposite trend, predicted by the decreased IL-2, CXCL10 and IFN-γ. Accordingly, the immunohistochemistry data illustrates an increased CD3+ T cell density inside the liver metastases, restricted to the responders. Furthermore, there was an agglomeration of T cells (termed ‘skewness’ of distribution,44 indicating activation and enhanced antigen presentation.59 60 Finally, the proximity between T cells and tumor cells increased during treatment in the tissue responders. In summary, the patients that could mount a Th1-like response in the tissue with influx of T cells and interferon production—rendering the tumor ‘hotter’ for combinatorial immunotherapy—had the biggest benefit and showed tissue response in terms of long-term stabilization of the disease.
By and large, NOX-A12 triggers an immune response that is visible at the cellular and molecular level in paired biopsies sampled under the monotherapy treatment with application of NOX-A12 two times per week. The results from these tissue investigations show—as expected from previous experiences from clinical trials with NOX-A12—an aggregation of the target cytokine. This observation might account for the extended progression-free survival under the subsequent combined immune checkpoint blockade. Interestingly, similar results were observed in the study by Biasci and collaborators.33 The authors reported an increase in CD8+ cytotoxic T cell density in the liver metastasis as well as RNA-seq data pointing at T cell accumulation and activation. Also, similarly to the clustering of T cells in the OPERA trial, the authors reported RNA data indicating the formation of tertiary lymphoid structures.33 These results are partially in accordance since in the OPERA trial, we observed an increase in CD3+T cell numbers but not in CD8+T cell numbers. Of course, the study populations were somewhat different since (i) Biasci and collaborators selected patients with a leukocyte count above the lower limit of normal, while the study population in the OPERA trial exhibited low tumor-infiltrating T cell density (figure 4) and (ii) the patients in Biasci et al had a median of two previous lines of chemotherapy, whereas patients in the OPERA trial had a mean of five (CRC) or three (pancreatic cancer(PaC)) lines of treatment.
What could be allowing the improved T cell response observed in the OPERA trial? We observed two distinct effects of NOX-A12 on myeloid immune cells. A subgroup of immune cells co-localize with CXCL12 in the microenvironment: a rare CD14+CD15+ promonocytic cell population. Under treatment, these cells diminish in number, being suggestive of the therapeutic effect in the modulation of the microenvironment. How this decreased number is mitigated is currently unclear. On the other hand, we also do not understand how these cells are being generated a priori. A co-localization of CD14+CD15+ cells and the CXCL12 protein might indicate that either these cells are producers of CXCL12 in the tumor microenvironment (in addition to the well-described FAPα+ cancer-associated fibroblasts). Alternatively, the co-localization may suggest that CXCL12 binds to its receptor at their surface. These complex and reproducible findings show that NOX-A12 has capabilities beyond increasing quantities of T cells. The precise effects of NOX-A12 on myeloid/monocytic cells is unclear and warrants further investigations. Simultaneously, the CD11b+ monocytic population was decreased in numbers in the tumor core and increased in the adjacent liver. Notably, there were strong associations between a low number of CD11b+ monocytes, a higher number of CD14+CD15+ promonocytic precursor cells, lower arginase and iNOS activity and tissue response. Together, these results highlight the (pro-)monocytic compartment landscape as key in predicting the tissue response and suggest that an alteration of the activity of macrophages takes place in the tissue responders under NOX-A12 treatment.
In our cohort of heavily pretreated, patients with MSS, we did not observe any objective response in terms of RECIST V.1.1 criteria in spite of the consistent modifications in the tumor immune contexture. One could argue that at this late stage, the remaining local antitumor immune response is scarce. This is in line with the analyses of baseline T cell infiltration in OPERA trial patients, which showed a generally lower number of T cells in the study population (figure 6), compared, for example, to the expected normal levels of CRC liver metastases in the literature.43 Nonetheless, it was encouraging to observe that the inflammatory and immune responses are not totally absent in our cohort, as indicated by the recursive partitioning analysis (online supplemental figure 3). Indeed, on one hand, a strong inflammatory reaction (reflected by high concentrations of IL-17 at baseline) is indicative of a shorter PFS, accordingly to other studies with earlier stage pancreatic cancer61 or in CRC.62 63 On the other hand, some patients displayed moderately elevated IL-2 levels in their metastasis at baseline, indicative of some Th1 effector activity, and this was associated to a longer PFS.
Besides the limited sample size of our single-arm study, there are a few other limitations. One is certainly the fact that no biopsy was sampled after combined therapy with NOX-A12 and pembrolizumab, so one cannot predict the changes in the tumor microenvironment under such therapy. Furthermore, the dosing schedule was suboptimal in hindsight as the assumption at the time of study design was that continuous inhibition of CXCL12 by olaptesed pegol would not be required. The mode of action of olaptesed pegol was thought to facilitate the influx of immune effector cells into solid tumors to allow effective response to immune checkpoint inhibition, and once within the tumor and activated by recognition of the antigen on the target cells, it was expected that the immune effector cells would be able to expand and to perform serial killing for a prolonged period of time. Therefore, a 7-day window of immune effector cell influx into the tumor within a 21-day treatment cycle with pembrolizumab was chosen. However, in a recent clinical study, continuous infusion of the CXCR4 inhibitor plerixafor induced an integrated immune response that was even predictive of a clinical response to T cell checkpoint inhibition.33 Also, in the COMBAT study the CXCR4 inhibitor BL-8040 was given three times weekly resulting in a more continuous signaling blockade leading to a partial response in one of the patients.32 These new data and the excellent toxicity profile of the applied NOX-A12 regimen in combination with checkpoint inhibition allows for a more dose-dense regimen, like a continuous blockade of the CXCL12/CXCR4 axis. Further exploration of this potential in combination with PD-1 inhibition can be imagined. In light of these results, a number of adjustments would make sense in further studies. A small cohort of heavily pretreated, advanced stage patients is undoubtedly mostly informative for exploratory studies, but a follow-up study would maybe focus on one tumor entity and also include patients with fewer lines of treatment.
In summary, the novel approach of specifically inhibiting CXCL12 through a spiegelmer showed activity alone and in combination with checkpoint inhibition in patients who were confirmed microsatellite stable. It was well tolerated and long-term stabilization of disease could be reached—with a disease control rate of 25%. The mechanisms of NOX-A12-mediated transformation of the tumor immune microenvironment clearly included a specific cytokine signature consisting of IL-2, IL-16 and IFN-γ as indicator for activation in the tumor tissue, which is in accordance with the observed cellular activation and clustering of T cells and their migration towards the tumor core. Future studies will allow to further integrate anti-CXCL12 in coherent modulatory approaches for solid tumors.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This trial has approval from the ethics committee of the University of Heidelberg.
Acknowledgments
The authors thank Jana Wolf for technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MeggySuarez, @halama_immuno
MS-C and AW contributed equally.
Contributors DE, JUJ, DJ and NH conceived and designed the study. AW, JS, NH and JK planned and implemented the clinical trial. AW, JS, NH, JK, DJ and NH took care of patients and collected clinical data. MS-C and UP acquired experimental data. MS-C, AF, DB, DE, JUJ, AM and NH analyzed and interpreted the data. NH drafted the manuscript. MS-C, DE, DB, AF and JK provided critical revision of the manuscript. All authors read and approved of the manuscript before submission.
Funding The University Hospital Heidelberg received funding from NOXXON Pharma AG for the implementation of the clinical trial.
Competing interests The University Clinics in Heidelberg received funding from NOXXON Pharma for the implementation of the first clinical trial, of which we here present the results, using combined CXCL12 inhibition and pembrolizumab-mediated immune checkpoint blockade in patients with microsatellite stable colorectal and pancreatic cancer.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.