Article Text

Abstract
Background Combination therapy has been explored for advanced head and neck squamous cell carcinoma (HNSCC) owing to the limited efficacy of anti-epidermal growth factor receptor (EGFR) therapy. Increased expression and glycosylation of immune checkpoint molecules in tumors are responsible for cetuximab therapy refractoriness. The role of programmed death ligand 2 (PD-L2), a ligand of PD-1, in the immune function is unclear. Here, we examined the regulatory mechanism of PD-L2 glycosylation and its role in antitumor immunity and cetuximab therapy.
Methods Single-cell RNA sequencing and immunohistochemical staining were used to investigate PD-L2 expression in cetuximab-resistant/sensitive HNSCC tissues. The mechanism of PD-L2 glycosylation regulation was explored in vitro. The effects of PD-L2 glycosylation on immune evasion and cetuximab efficacy were verified in vitro and using mice bearing orthotopic SCC7 tumors.
Results The PD-L2 levels were elevated and N-glycosylated in patients with cetuximab-resistant HNSCC. Glycosylated PD-L2 formed a complex with EGFR, which resulted in the activation of EGFR/signal transducer and activator of transcription 3 (STAT3) signaling and decreased the cetuximab binding affinity to EGFR. The N-glycosyltransferase fucosyltransferase (FUT8), a transcriptional target of STAT3, was required for PD-L2 glycosylation. Moreover, glycosylation modification stabilized PD-L2 by blocking ubiquitin-dependent lysosomal degradation, which consequently promoted its binding to PD-1 and immune evasion. Inhibition of PD-L2 glycosylation using Stattic, a specific STAT3 inhibitor, or PD-L2 mutation blocking its binding to FUT8, increased cytotoxic T lymphocyte activity and augmented response to cetuximab.
Conclusions Increased expression and glycosylation of PD-L2 in tumors are an important mechanism for cetuximab therapy refractoriness. Thus, the combination of PD-L2 glycosylation inhibition and cetuximab is a potential therapeutic strategy for cancer.
- head and neck neoplasms
- immune evation
- biomarkers
- tumor
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data are available in the main text or the supplementary materials.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The expression of epidermal growth factor receptor (EGFR) is upregulated in head and neck squamous cell carcinoma (HNSCC). Cetuximab (cetux), an EGFR-specific antibody, is the major targeted therapy for HNSCC. However, the response rate of cetux therapy is less than 20%.1 The enhanced expression of immune checkpoint molecules (ICMs) is one of the mechanisms underlying cetux refractoriness. Therefore, the combination of ICM inhibition and cetux therapy, which has been tested in clinical trials, is a promising therapeutic approach for HNSCC.2
Programmed death ligand 2 (PD-L2), a ligand of PD-1, is a critical ICM that contributes to T cell dysfunction and immune surveillance escape.3 The expression of PD-L2 is higher than that of PD-L1 in patients with HNSCC.4 However, limited studies have evaluated the role of PD-L2 in antitumor immunity and cetux therapy.
Glycosylation plays a critical role in the stability and subcellular localization of proteins.5–7 Glycosylated ICMs contribute to various biological processes, such as immune surveillance, signal transduction, and response to targeted therapy in human cancer.8–11 Approximately 20%–90% of cell surface N-glycans exhibit fucosylation, which is catalyzed by α−1,6 fucosyltransferase (FUT8).12 13 The elucidation of the biological function of glycosylated PD-L2 and the molecular mechanism underlying PD-L2 glycosylation can aid in developing novel strategies for cancer immunotherapy.
Signal transducer and activator of transcription 3 (STAT3) promotes tumor progression through the transactivation of various downstream targets, including interleukin (IL)-6, vascular endothelial growth factor, and nuclear factor kappa light chain enhancer of activated B cells (NF-κB).14 Previously, we demonstrated that STAT3 promoted tumorigenesis and modulated cetux response in HNSCC.15 Additionally, STAT3 is reported to suppress the antitumor immune response,16 but the underlying molecular mechanisms have not been elucidated.
This study aimed to examine the mechanism involved in glycosylation-dependent regulation of PD-L2 localization and function. The findings of this study indicated that glycosylated PD-L2 modulated cetux therapy response through the regulation of the EGFR/STAT3 signaling pathway. The combination of inhibition of PD-L2 glycosylation and cetux therapy exerted beneficial effects in an orthotopic murine model of HNSCC. Thus, our findings suggest that targeting PD-L2 glycosylation can improve the response to anti-EGFR therapy in cancer.
Methods
Patient samples
Samples were obtained with signed consent form from the patients with HNSCC. None of the patients included in this study receive any treatment before cetux therapy. Tumor stage was assessed based on the American Joint Committee on Cancer (AJCC) 8th Edition. The response to cetux therapy was evaluated according to Response Evaluation Criteria in Solid Tumors V.1.1. Patients with complete or partial response were defined as ‘sensitive,’ whereas those exhibiting tumor progression were defined as ‘refractory’.
Single-cell sequencing
Single-cell preparation, library synthesis, RNA sequencing and data analysis were completed by Gene Denovo (Guangzhou, China) using 10× Genomics Chromium Single Cell Controller. Briefly, we generated full-length single-cell sequencing (scRNA-seq) profiles for biopsy from two patients with HNSCC, which had identical AJCC stage, primary site, pathological type, differentiation status, and cetux treatment refractory. Also, none of the patients received any treatment before cetux therapy. The clinical characteristics of the two patients were shown in online supplemental table 1.
Supplemental material
Fresh HNSCC tissues were dissociated with Gentle MACS Dissociator (Miltenyi Biotec) according to the manufacturer’s instructions. Next, single cells were counted using an automated cell counter and adjusted to a concentration of 1000 cells/µL, and we retained single-cell transcriptomes for 35,299 cells after initial quality controls. Cellular suspensions were loaded on a 10×Genomics GemCode Single-cell instrument that generates single-cell Gel Bead-In-EMlusion. The cells were lysed, and RNA was reverse-transcribed into cDNA. Libraries were generated from the cDNA with Chromium Next GEM Single Cell 3’ Reagent Kits V.3.1. Finally, the cDNA libraries were sequenced using the Nova 6000 Sequencing System (Illumina). The raw scRNA-seq data were processed using CellRanger V.3.0.2 (10×Genomics).
Subsequent data analysis, including principal component analysis, t-distributed stochastic neighbor embedding (t-SNE analysis) were achieved using Seurat software. Seurat embed cells in a shared-nearest neighbor graph, with edges drawn between cells via similar gene expression patterns. Unsupervised density-based clustering by density-based spatial clustering of applications with noise (DBSCAN) clustering algorithm on t-SNE analysis were performed to separate tumor cells into the distinct homogenous groups.17 The raw scRNA-seq data were uploaded to the Gene Expression Omnibus (GEO) public database (GSE173647).
Isolation and analysis of tumor-infiltrating lymphocytes
Fresh tumors obtained from mice were cut into pieces (3–5 mm3) and treated with collagenase I (1 µg/mL) and collagenase IV (1 µg/mL) for 1.5 hours at 37°C. The tissue homogenates were filtered using a 70 µm cell strainer (Falcon) before centrifugation. The samples were subjected to density gradient centrifugation using Ficoll-Paque PLUS density gradient media. The supernatant was discarded and the cells were suspended in an appropriate medium. The samples were centrifuged to obtain tumor-infiltrating lymphocytes (TILs). The TILs were stained with FITC-conjugated anti-CD3 (BioLegend), APC-Cy7-conjugated anti-CD4 (BioLegend), PE-conjugated anti-CD8 (BioLegend), PerCP-conjugated anti-CD45.1 (BioLegend), and APC-conjugated anti-interferon gamma (IFNγ) (BioLegend) antibodies. The stained samples were analyzed using a BD FACS Canto II (BD Biosciences) cytometer.
Immunoblotting and immunoprecipitation assays
Cells were prepared by lysing the cells in a lysis buffer containing 40 mM Tris-HCl (pH 7.6), 120 mM NaCl, 1% Triton X-100, and protease inhibitor Cocktail (Roche 04693116001) added before lysis. To perform immunoblotting, cell lysates were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto polyvinylidene difluoride membrane, and followed by western blotting analysis using indicated antibodies. Immunoblot detections were carried out using Horseradish Peroxidase (HRP)-conjugated secondary antibodies (7074 or 7076; Cell Signaling Technology). Quantifications of immunoblotting signal intensity were performed with ImageJ. To perform immunoprecipitation, cell lysates were incubated with antibodies with gentle rocking overnight at 4°C and then incubated with protein A/G agarose beads (Thermo Fisher) for 2 hours. After washing with lysis buffer, immunoprecipitation products were subjected to immunoblotting analysis.
For cell surface immunoprecipitation (CS-IP) of PD-L2, 2×106 live Flag-PD-L2 overexpressing cells were incubated with Flag antibody (10 µg/mL) for 1 hour on ice. Cells were then washed with cold phosphate-buffered saline (PBS) to remove unbound antibody and resuspended in prewarmed complete medium and incubated at 37°C for 4 hours. Next cells were lysed in 1% NP-40 lysis buffer plus Roche complete protease inhibitor. The supernatants were incubated with protein A/G beads at 4°C overnight to capture antibody-bound Flag-PD-L2. After washes in 1% NP-40 lysis buffer, samples were eluted in loading buffer for 10 min at 95°C, separated by SDS-PAGE and immunoblotting.
Orthotopic mouse models and treatment
C3H mice and BALB/c nude mice (male, 4–5 weeks old) were obtained from the Animal Center at Beijing Vital River Laboratory Animal Technology (Beijing, China). SCC7 cells (3×105 cells) expressing wild type (WT) or mutant (4NQ or REY (PD-L2 REY mutant, in which the amino acid of R168, E171 and Y174 were substituted by Alanine)) PD-L2 were injected into the lateral tongue of the mice. For drug treatment, 40 mg/kg cetux or 4 mg/kg Stattic alone or in combination was injected intraperitoneally every 2 days after SCC7 cell inoculation for 3 weeks. Tumor volume was measured by luciferase activity with a Living Image system once per week.
Statistical analysis
SPSS V.24 was used for statistical analysis, and the experimental data were analyzed by one-way analysis of variance. All data are expressed as the mean±SD, representing the mean of three experiments performed in at least three replicates. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 were considered statistically significant.
Results
PD-L2 is N-glycosylated and upregulated in cetux-resistant tissues
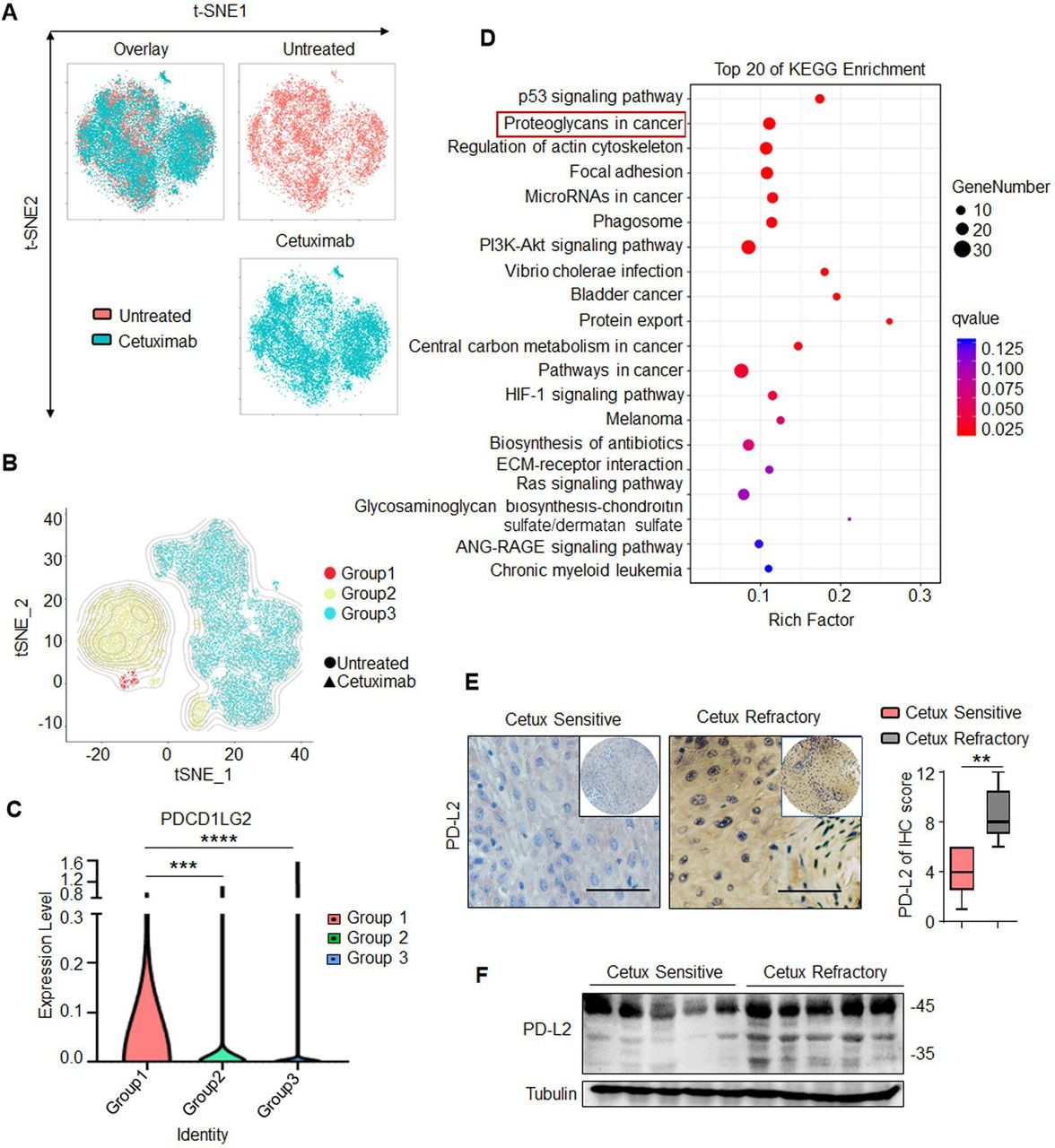
The biopsies from two patients with HNSCC were used to generating the single-cell libraries and subjected to scRNA-seq analysis before and after cetux treatment (figure 1A). The cells were separated into the following three different groups based on density-based clustering by DBSCAN clustering algorithm (figure 1B), including Group 1, Group 2 and Group 3. Group 1 represented cetux-resistant cells and was comprised of almost exclusively tumor cells from the cetux-treated group (D1). Group 2 represented cells exhibiting partial resistance to cetux and was comprised 65%–75% of tumor cells from the cetux-treated group (D1) and 25%–35% tumor cells from the untreated group (C1), represented. Group 3 represented no significant difference in the number of cells and was comprised of a mixture of tumor cells from the untreated and cetux-treated groups. The differentially expressed genes (DEGs) were selected based on the following parameters: the average gene expression by cells in Group 1 compared with other groups showed log2-fold change ≥1.0 and p value <0.05. PD-L2 was identified as one of the top five genes upregulated significantly (figure 1C and online supplemental table 2). Group 3 exhibited downregulated levels of immune markers, including SPDEF, CSF1R, TNFRSF9, TNFSF4, and CCL7, when compared with Groups 1 and 2. This indicated that the response to cetux therapy was associated with a gene transcription signature consistent with the immune response. The Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed that the DEGs of Group 1 were enriched in the ‘proteoglycans in cancer’ pathway. This suggested a close correlation between glycosylated PD-L2 and the response to cetux therapy (figure 1D).

PD-L2 is N-glycosylated and upregulated in patients with cetux-resistant HNSCC. (A) T-distributed stochastic neighbor embedding (t-SNE) plot of cancer cells in two patients with HNSCC before and after cetux treatment. (B) t-SNE plot showing three different groups identified based on density-based clustering. (C) Violin plot depicting PD-L2 expression levels in the three different groups determined using single-cell RNA sequencing. ***p<0.001; ****p<0.0001. (D) KEGG pathway enrichment analysis revealed that cetux refractoriness is correlated with the ‘proteoglycans in cancer’ pathway. (E) Immunohistochemical staining of PD-L2 in five cetux-resistant samples and nine cetux-sensitive HNSCC tissues. Scale bar, 50 µm. One-way analysis of variance (**p<0.01). (F) Protein expression of PD-L2 in clinical HNSCC samples obtained at the Tianjin Medical University Cancer Institute and Hospital. HNSCC, head and neck squamous cell carcinoma; KEGG, Kyoto Encyclopedia of Genes and Genomes; PD-L2, programmed death ligand 2; t-SNE, t-distributed stochastic neighbor embedding.
Next, the expression levels of PD-L2 in 14 clinical HNSCC samples were examined, including five cetux-resistant samples and nine cetux-sensitive HNSCC tissues. Compared with the cetux-sensitive specimen, the cetux-resistant specimen showed upregulated PD-L2 levels (figure 1E). Moreover, two PD-L2 bands were detected in most cetux-resistant tissues with a major band at approximately 48 kDa and a minor band at 35 kDa (figure 1F). The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-mediated PD-L2 knockout decreased the levels of major and minor bands (online supplemental figure 1A). Furthermore, treatment with N-glycosidase (PNGase F) (but not O-glycosidase) decreased the molecular weight of the major PD-L2 band (48 kDa) to 35 kDa (online supplemental figure 1B). Glycoprotein staining and Coomassie staining of purified PD-L2 protein revealed that the glycosylated proteins were not detected on treatment with PNGase F. This indicated that both bands represent PD-L2 and that the higher molecular weight band was the glycosylated form of PD-L2 (online supplemental figure 1C). Consistently, treatment with tunicamycin (TM), an inhibitor of N-linked glycosylation, dose-dependently inhibited PD-L2 glycosylation (online supplemental figure 1D). These results indicate that the expression of PD-L2, which exhibits N-glycosylation, is upregulated in cetux-resistant tissues.
PD-L2 glycosylation determines antitumor immunity and is correlated with the response to cetux
To examine the evolutionary conservation of the glycosylation sites, the NXT (-Asn-X-Ser/Thr-) motifs were examined in PD-L2 sequences of various species. Four NXT motifs were identified in this study (online supplemental figure 2A). The glycan structure of the purified PD-L2 protein was examined using liquid chromatography tandem mass spectrometry (LC-MS/MS). Glycopeptides (including the complex type) with N-glycosylation were identified for each N-glycosylation site (figure 2A and online supplemental figure 2B–D). Mutants in which each asparagine (N) was substituted with glutamine (Q) (N64Q, N157Q, N163Q, or N189Q) exhibited decreased glycosylation levels when compared with PD-L2 WT (figure 2B). PD-L2 4NQ mutant exhibited decreased intensity of the major band (approximately 48 kDa) that corresponded to glycosylated PD-L2, which was similar to the band intensity after treatment of PD-L2 WT with TM. This indicated that the glycosylation of PD-L2 was completely inhibited. These results suggest that PD-L2 is N-glycosylated at N64, N157, N163, and N189.

PD-L2 glycosylation modulates antitumor immunity and the cetux response in HNSCC. (A) Identification of N-glycopeptides by LC-MS/MS corresponds to one of four N-glycosylation sites, namely, (N64). (B) Schematic representation of several PD-L2 NQ mutants used in this study. The four hypothetical NXT patterns are marked in red. The numbers indicate the position of aspartic acid. Protein levels of wild type (WT) or NQ mutants of PD-L2. (C) Co-immunoprecipitation (Co-IP) analysis of the interaction of PD-L2 WT or 4NQ mutants of PD-L2 with PD-1 in SCC15 and SCC25 cells. (D) Representative bioluminescence images of SCC7 cells expressing PD-L2 WT or 4NQ mutant in C3H mice treated with cetux every week (n=7 mice per group). Values represent mean±SD of the number of designated mice; *p<0.05; **p<0.01; #, not significant. (E) Immunofluorescence staining of PD-L2 in the tumor mass of mice with SCC7 tumors. Scale bar, 20 µm. Bottom panel depicts the quantification, *p<0.05; **p<0.01; ***p<0.001. (F) Quantification of IFNɣ in CD8+ and CD3+ T cell populations in C3H mice bearing SCC7 tumors using intracellular cytokine staining at day 21. **p<0.01; ***p<0.001; #, not significant. Error bars denote mean±SD of three independent experiments. (G) The intracellular cetux content in the tumor mass of mice with SCC7 tumors was detected using surface plasmon resonance. **p<0.01. HNSCC, head and neck squamous cell carcinoma; IFNγ, interferon gamma; LC-MS/MS, liquid chromatography tandem mass spectrometry; PD-L2, programmed death ligand 2.
Various studies have demonstrated that glycosylation interferes with tumor immune surveillance.10 18 19 To determine the role of glycosylated PD-L2 in immunotherapy, the T cell immune response was examined by analyzing the activated T cell-mediated cytotoxic effect against tumor cells. Glycosylated PD-L2 significantly suppressed the activated Jurkat cell-mediated apoptosis by 5.1-fold and 4.4-fold compared with the PD-L2 4NQ expressing SCC15 and SCC25 cells, respectively (online supplemental figure 3A). The interaction between PD-L2 and PD-1 is critical for the cancer cells to escape T cell-mediated immune surveillance.3 20 Co-immunoprecipitation (Co-IP) analysis revealed that PD-L2 4NQ mutant significantly impaired the binding affinity to PD-1 (figure 2C). These data suggest that an intact glycan is required for the binding of PD-L2 to PD-1.
We next examined the role of PD-L2 glycosylation and evaluated tumorigenesis in response to cetux treatment of murine HNSCC SCC7 cells in C3H immunocompetent mouse models (online supplemental figure 3B). Bioluminescence imaging indicated that PD-L2 WT-transfection in tumor cells remarkably promoted tumor growth, whereas PD-L2 4NQ transfection reduced tumor growth on treatment with cetux (figure 2D). Moreover, removing the glycosylation modification of PD-L2 decreased the membrane PD-L2 levels (figure 2E) and potentiated cytotoxic T cell activity against the tumor mass (online supplemental figure 3C). The number of activated cytotoxic T cells (CD8+/IFNɣ+) among TILs in mice harboring PD-L2 4NQ-transfected tumor cells was higher than that in mice harboring PD-L2 WT-transfected tumor cells (figure 2F). Additionally, surface plasmon resonance (SPR) analysis revealed that the removal of PD-L2 glycosylation increased intracellular cetux accumulation by 3.7-fold compared with the PD-L2 WT transfected tumor cells (figure 2G). The tumor growth did not significantly change in severe combined immune-deficient BALB/c nude mice (online supplemental figure 3D). This indicated that differential tumorigenicity was dependent on immune surveillance.
These data suggest that glycosylated PD-L2 downregulates tumor-specific T cell responses and that the removal of glycosylation modification of PD-L2 restores the immune response and enhanced the antitumor efficacy of cetux therapy.
EGF/STAT3 signaling drives PD-L2 glycosylation through N-glycotransferase FUT8
As glycosylation of PD-L2 is critical for immunosuppression and response to cetux therapy, we sought to identify the upstream signaling that governs PD-L2 glycosylation. First, we evaluated the effect of multiple cytokines on inducing PD-L2 expression, including epidermal growth factor (EGF), IL-8, IL-10, tumor necrosis factor (TNF-α), IL-6, which were correlated with the response to cetux treatment in HNSCC.21–24 Among them, EGF significantly upregulated PD-L2 levels compared with other cytokines (online supplemental figure 4A). Furthermore, PD-L2 WT protein expression level was increased in response to EGF stimulation, whereas non-glycosylated PD-L2 4NQ mutant expression was not affected by EGF (online supplemental figure 4B). To examine the downstream signaling pathway involved in EGF-induced PD-L2 glycosylation, the SCC15 cells were treated with 7 µM SB203580 (MAPK inhibitor), 7 µM GF109203X (PKCα inhibitor), 7 µM rapamycin (mTOR inhibitor), 7 µM U0126 (MEK1/2 inhibitor), 7 µM Nutlin-3 (MDM2 inhibitor) and 7 µM Stattic (STAT3 inhibitor) for 4 hours. The cells were then incubated with EGF for 15 min (online supplemental figure 4C). These results indicated that PD-L2 glycosylation and STAT3 phosphorylation were significantly increased on EGF treatment. Treatment with the STAT3 inhibitor Stattic attenuated EGF-induced PD-L2 glycosylation by 40%–60% (figure 3A). Consistently, the short hairpin RNA-mediated knockdown of STAT3 attenuated EGF-induced PD-L2 glycosylation (figure 3B and online supplemental figure 4D). Additionally, treatment with Stattic dose-dependently downregulated the PD-L2 protein levels (figure 3B). Moreover, EGF enhanced the formation of the (p)STAT3/PD-L2 4NQ mutant complex (online supplemental figure 4E). Proximity ligation assay (PLA) analysis revealed cytoplasmic colocalization of STAT3/PD-L2 4NQ (red dots) was significantly increased in the presence of EGF, compared with non-treatment condition (online supplemental figure 4F). These results indicate that EGF initiates PD-L2 glycosylation by enhancing the interaction between STAT3 and non-glycosylated PD-L2.
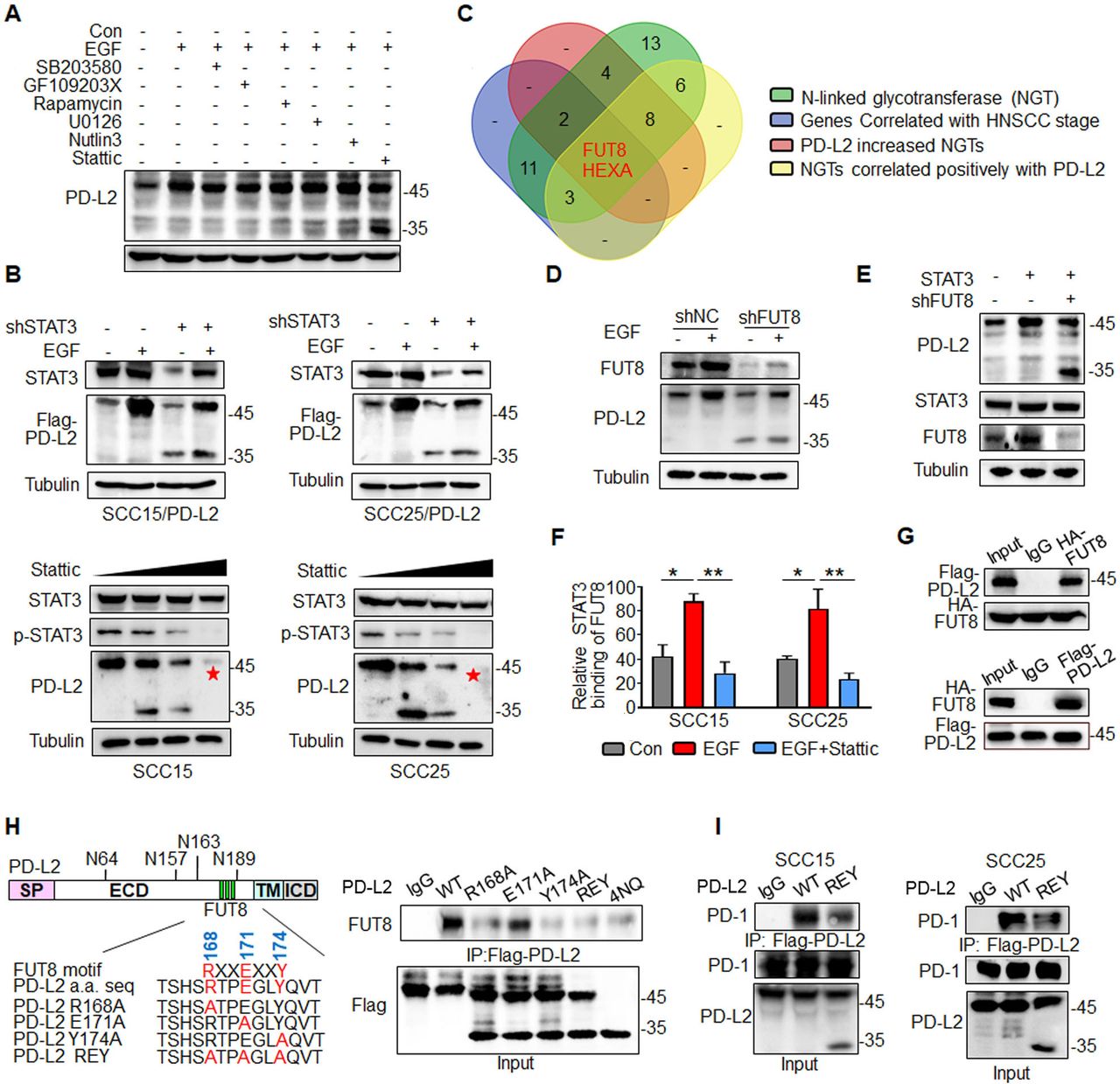
EGF/STAT3 signaling promotes PD-L2 glycosylation through the N-glycosyltransferase FUT8. (A) PD-L2 protein levels in SCC15 cells pretreated with 7μM SB203580 (MAPK inhibitor), 7 µM GF109203X (PKCα inhibitor), 7 µM rapamycin (mTOR inhibitor), 7 µM U0126 (MEK1/2 inhibitor), 7 µM Nutlin-3 (MDM2 inhibitor) or 7 µM Stattic (STAT3 inhibitor) for 4 hours and subsequently incubated with EGF for 15 min.(B) PD-L2 protein levels in shSTAT3 knockdown clones were detected using an anti-Flag antibody (upper panels). SCC15 and SCC25 cells were treated with incremental concentrations of Stattic (7–14 µM) for 4 hours and the PD-L2 levels were analyzed (lower panels). (C) Identification of NGT that potentially regulates PD-L2 glycosylation. Venn diagram demonstrating the expression pattern of distinct NGTs in HNSCC. Two potential NGTs (FUT8 and HEXA) were identified. (D) PD-L2 protein levels in EGF-treated in SCC15 cells transfected with negative control or short hairpin RNA against FUT8. (E) PD-L2 expression levels in STAT3-overexpressing and FUT8 knockdown cells. (F) Chromatin immunoprecipitation assay results revealed that Stattic-mediated inhibition of STAT3 impaired EGF-induced binding of STAT3 to the promoter of FUT8. *p<0.05; **p<0.01. (G) Co-immunoprecipitation (Co-IP) was performed to measure the interaction of FUT8 with PD-L2 in SCC15 cells. (H) Schematic diagram of FUT8-binding motifs and several mutants of the PD-L2 expression structure. ECD, extracellular domain; ICD, intracellular domain; SP, signal peptide; TM, transmembrane domain. The numbers reveal amino acid positions. The binding affinity between various PD-L2 mutants and FUT8 was analyzed using Co-IP. (I) Co-IP analysis of the interaction of PD-1 with PD-L2 in SCC15 and SCC25 cells expressing wild type (WT) or REY PD-L2. EGF, epidermal growth factor; FUT8, fucosyltransferase; HNSCC, head and neck squamous cell carcinoma; PD-L2, programmed death ligand 2; REY. PD-L2 REY mutant, in which the amino acid of R168, E171 and Y174 were substituted by Alanine; STAT3, signal transducer and activator of transcription 3.
To identify the putative N-linked glycotransferase (NGT) that catalyzes PD-L2 glycosylation, the expression of 49 NGTs was analyzed. In total, 18 genes were positively correlated with the tumor stage in HNSCC based on The Cancer Genome Atlas (TCGA) database (online supplemental figure 5A, online supplemental table 3). Of the 49 NGTs, 19 were strongly correlated with PD-L2 expression. RNA sequencing revealed that the expression of 16 NGTs was significantly upregulated in the PD-L2-overexpressing SCC15 cells (online supplemental figure 5B, online supplemental table 4). FUT8 and HEXA were recognized as overlapping genes (figure 3C and online supplemental figure 5 C,D). However, only FUT8 was involved in EGF-induced PD-L2 glycosylation (figure 3D and online supplemental figure 5E–G). Positive staining of Aleuria aurantia lectin (AAL)25 in PD-L2-expressing cells was completely abolished in FUT8-deficient cells. This indicated that PD-L2 was a substrate of FUT8 (online supplemental figure 5H).
Consistently, the loss of FUT8 reduced the glycosylated PD-L2 levels on the STAT3 induction (figure 3E). The expression of FUT8 was significantly downregulated in the stable STAT3 knockdown clones (online supplemental figure 5I). The transcription inhibitor actinomycin D significantly mitigated EGF-induced upregulation of PD-L2 (online supplemental figure 5J), which indicated that EGF-mediated glycosylation of PD-L2 is mediated by STAT3-dependent transcriptional activation. Next, the promoter sequence was analyzed using several transcription factor prediction software programs. Several candidate STAT3-binding sites were identified in the FUT8 promoter region. However, only one binding site was responsible for the transcriptional activation (online supplemental figure 5K). Moreover, the results of the ChIP assay revealed that EGF promoted the direct binding of STAT3 to the FUT8 promoter region and that treatment with Stattic markedly inhibited EGF-mediated FUT8 transactivation (figure 3F and online supplemental figure 5L). These results suggest that STAT3 upregulates FUT8 transcription and subsequently promotes PD-L2 glycosylation.
The results of the Co-IP assay revealed that FUT8 was associated with PD-L2 in HNSCC cells (figure 3G and online supplemental figure 6A). Moreover, immunofluorescence staining of HNSCC tumor tissue suggested that FUT8 colocalized with PD-L2 and that FUT8 expression was positively correlated with PD-L2 (online supplemental figure 6B). Additionally, FUT8 selectively bound to the glycosylated region of PD-L2 (online supplemental figure 6C). Moreover, the SH3 domain of FUT8 was necessary for its interaction with PD-L2 (online supplemental figure 6D). PD-L2 contains a conserved SH3 binding site (RXXEXXY, in which R, X, E, and Y denote arginine, any amino acid, glutamic acid, and tyrosine, respectively) in the glycosylation sites (figure 3H). Although similar-sized bands of PD-L2 were detected in the three mutants, the R168A and Y174A mutants (but not E171A) exhibited a significantly decreased binding affinity for FUT8, which was similar to the binding affinity of PD-L2 4NQ (figure 3H). Furthermore, the binding affinity of PD-L2 REY mutant to PD-1 markedly decreased in both SCC15 and SCC25 cells (figure 3I and online supplemental figure 6E). The PD-L2 REY mutant exhibited impaired ability to activate T cell-mediated apoptosis when compared with PD-L2 WT expressing cells. Treatment with Stattic enhanced T cell-mediated cytotoxicity against cells expressing PD-L2 WT. However, treatment with Stattic did not markedly affect the survival of cells expressing PD-L2 REY mutant (online supplemental figure 6F). These data strongly support that the NGT FUT8, as a target of STAT3, is required for EGF/STAT3 signaling induced PD-L2 glycosylation.
Glycosylation blocks ubiquitination-mediated PD-L2 lysosome degradation
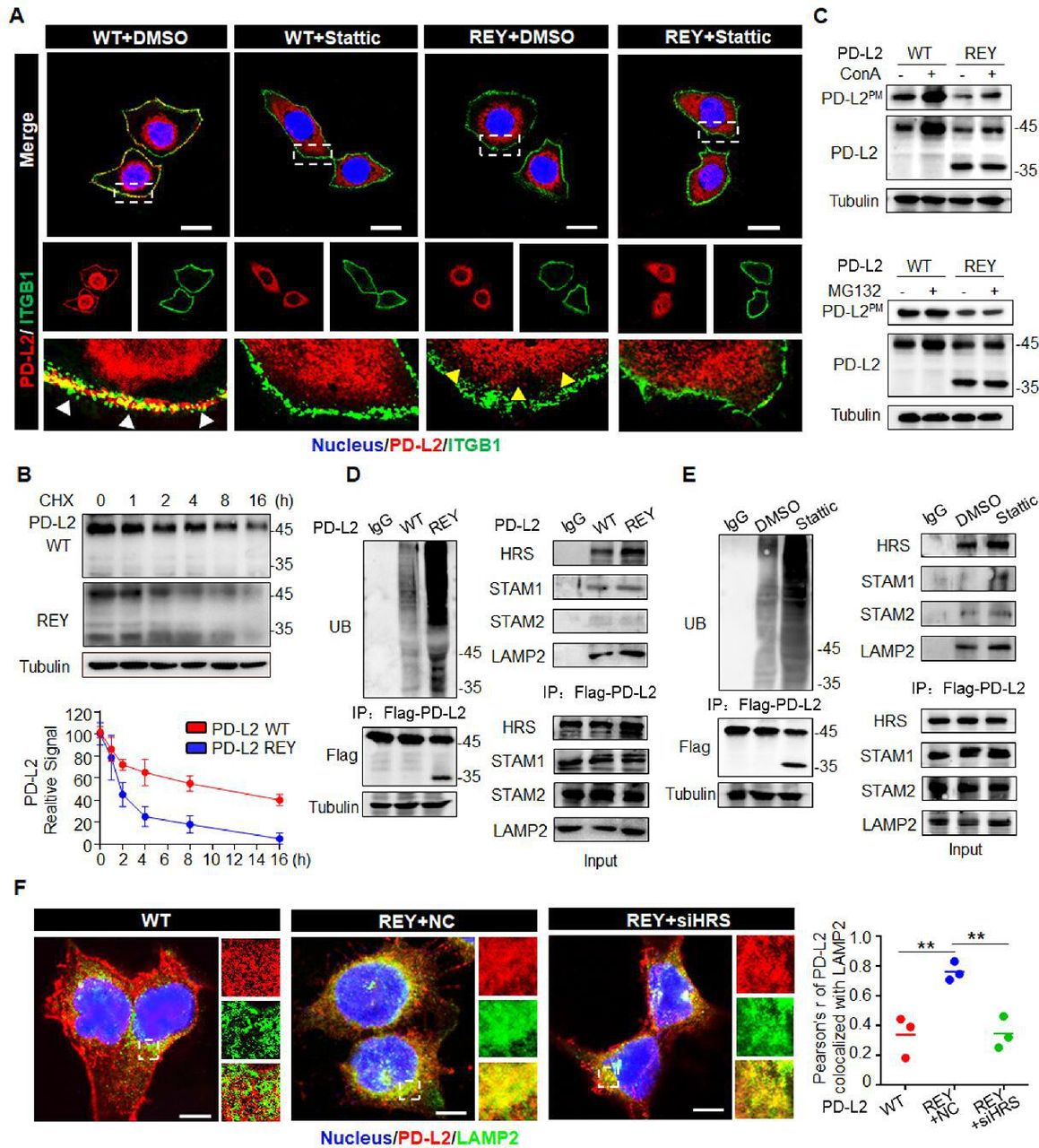
Previous studies have reported the role of glycosylation in the intracellular transport of membrane glycoproteins.26 Hence, we next examined the role of FUT8-mediated PD-L2 glycosylation in regulating PD-L2 subcellular localization after EGF stimulation. Flow cytometric analysis revealed that FUT8 deficiency impaired EGF-mediated cell membrane recruitment of PD-L2 (online supplemental figure 7A). CS-IP analysis revealed that EGF enhanced the membrane PD-L2 (PD-L2PM) levels through a FUT8-dependent mechanism (online supplemental figure 7B). Consistently, the membrane PD-L2 levels were upregulated in FUT8-overexpressing cells (online supplemental figure 7C). A remarkable reduction in cell membrane PD-L2 levels was observed in PD-L2 REY cells compared with PD-L2 WT cells (online supplemental figure 7D). Immunofluorescence staining applied ITGB1 as a plasma membrane marker.27 28 The colocalization of PD-L2 and ITGB1 was seen by yellow staining in the overlay in PD-L2 WT cells, revealing that most PD-L2 protein were localized at the cell periphery. In contrast, PD-L2 REY was localized to the cytosol with diffused signals observed on the cell surface (figure 4A). Stattic treatment promoted PD-L2 WT localization in the cytosol, which was similar to the PD-L2 REY localization pattern. However, Stattic did not affect the PD-L2 REY localization pattern. Thus, STAT3/FUT8-mediated glycosylation was crucial for PD-L2 membrane transport.

Glycosylation inhibits ubiquitination-mediated lysosomal degradation of PD-L2 and facilitates PD-L2 membrane abundance. (A) Immunofluorescence staining was performed to examine the expression pattern of PD-L2 in SCC15 cells expressing WT or REY mutant of PD-L2 with or without Stattic (7 µM) treatment for 4 hours. PD-L2 WT was mainly localized to the cell membrane (white arrow), colocalized with ITGB1. Stattic treatment resulted PD-L2 WT localized in the cytosol. PD-L2 REY mutant was primarily localized to the cytoplasm with or without Stattic treatment (yellow arrow). Scale bar, 20 µm. (B) SCC15 cells expressing PD-L2 WT or REY were treated with 20 µM CHX at the indicated time points. Bottom panel shows the quantification of the data. (C) Membrane PD-L2 (PD-L2PM) levels in cells expressing PD-L2 WT or REY treated with the lysosome inhibitor concanamycin-A or proteasome inhibitor MG132. (D) The REY mutant markedly increased PD-L2 ubiquitination when compared with WT PD-L2. Co-immunoprecipitation (Co-IP) analysis of the interaction between HRS, LAMP2, STAM1, STAM2, and PD-L2 in SCC15 cells expressing PD-L2 WT or REY. (E) Effect of the STAT3 inhibitor Stattic on PD-L2 ubiquitination. Co-IP was performed to analyze the interaction between HRS, LAMP2, STAM1, STAM2, and PD-L2 in the Stattic-treated cells. (F) Colocalization of PD-L2 with the lysosome marker LAMP2 in HRS knockdown SCC15 cells expressing PD-L2 WT or REY. Scale bar, 5 µm. Right panels show the quantification of the colocalization between PD-L2 and LAMP2. Values represent means±SD from three independent experiments. Significance was detected using one-way analysis of variance. **p<0.01. PD-L2, programmed death ligand 2; REY, PD-L2 REY mutant, in which the amino acid of R168, E171 and Y174 were substituted by Alanine; STAT3, signal transducer and activator of transcription 3; WT, wild type.
Previous study revealed that NGT played a fundamental role in determining the stability of proteins.29 We next examined the role of FUT8 in regulating PD-L2 degradation and membrane distribution. PD-L2 with a mutation at the binding motif of FUT8 (PD-L2 REY) exhibited decreased protein stability and reduced protein half-life time (figure 4B). Next, the cells were treated with concanamycin-A (ConA), chloroquine (CQ) (an inhibitor of lysosomal acidification), or MG132 (an inhibitor of the proteasome). Both ConA and CQ mitigated the enhanced degradation of PD-L2 REY expressing cells, whereas MG132 exhibited limited effects (online supplemental figure 7E). CS-IP analysis revealed that ConA increased PD-L2PM levels in the PD-L2-overexpressing SCC15 cells, whereas MG132 did not affect the PD-L2PM levels (figure 4C). Additionally, the degradation of PD-L2 at the cell membrane markedly increased in FUT8 knockdown cells. Furthermore, ConA stabilized PD-L2 in FUT8 deficient cells (online supplemental figure 7F). Thus, FUT8-mediated glycosylation suppresses PD-L2 degradation through the lysosomal pathway.
Glycosylation modification maintains protein stability by attenuating ubiquitination.30 Consistently, PD-L2 REY exhibited markedly increased ubiquitination (figure 4D). Similarly, FUT8 knockdown or Stattic markedly increased PD-L2 ubiquitination (figure 4E and online supplemental figure 7G). The endosomal sorting complexes required for transport (ESCRT) complexes, including HRS, STAM1, and STAM2, are critical for recognizing and targeting ubiquitinated proteins for lysosomal degradation.31 Therefore, the role of ESCRT-0 complexes in the endolysosomal targeting of glycosylated PD-L2 was examined. Co-IP analysis revealed that the complex-forming ability of the ESCRT-0 component HRS (but not STAM1 or STAM2) with PD-L2 REY was higher than that with PD-L2 WT (figure 4D). Consistently, the interaction between HRS and PD-L2 was enhanced in the shFUT8-transfected or Stattic-treated cells (figure 4E and online supplemental figure 7G). Furthermore, HRS knockdown rescued glycosylation inhibition-induced PD-L2 degradation (online supplemental figure 7H), which further confirmed the critical role of the ESCRT-0 complex in glycosylation-induced PD-L2 stabilization. PD-L2 REY mutant exhibited enhanced colocalization with LAMP2 when compared with PD-L2 WT. HRS knockdown impaired the colocalization of PD-L2 and LAMP2 and enhanced the PD-L2 REYPM levels (figure 4F).
These findings indicated that glycosylation blocks PD-L2 polyubiquitination and consequently inhibits the sorting of ESCRT-0-mediated endolysosomal targeting of PD-L2. Consequently, the lysosomal degradation of PD-L2 is inhibited, which results in enhanced membrane accumulation and increased PD-1 binding affinity.
Glycosylated PD-L2 interacts with EGFR and upregulates EGFR/STAT3 signaling activity
NGT modulates the protein–protein interactions.32 To examine the significance of PD-L2 glycosylation in modulating the response to cetux, affinity purification and mass spectrometry were performed to identify proteins associated with PD-L2 (figure 5A). Mass spectrometric analysis revealed that EGFR is a potential interactor of PD-L2. Co-IP and PLA analyses confirmed that PD-L2 interacted with EGFR in SCC15 and SCC25 cells (figure 5B and C and online supplemental figure 8A). EGFR physically interacted with PD-L2 and the FUT8 complex in HNSCC tissues (online supplemental figure 8B,C). The glycosylation site was the critical region of PD-L2 for the binding of EGFR (figure 5D). The loss of FUT8 decreased the interaction between PD-L2 and EGFR, which suggested that the PD-L2/EGFR interaction is mediated through glycosylation-dependent mechanisms (figure 5E).

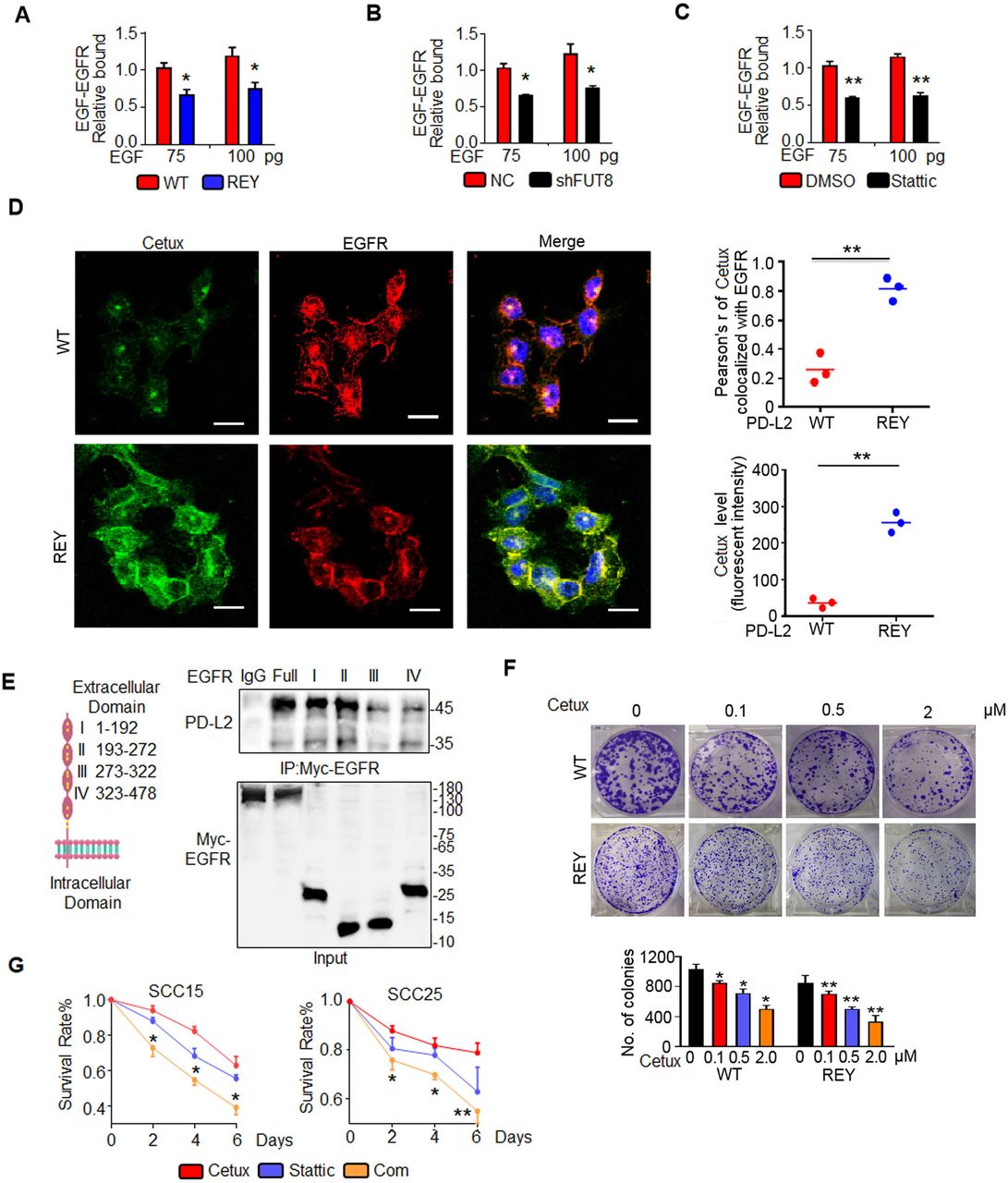
Glycosylated PD-L2 interacts with EGFR and upregulates EGFR/STAT3 signaling activity. (A) Co-immunoprecipitation (Co-IP) followed by mass spectrometry were performed to identify the proteins that bind to PD-L2. (B) Co-IP analysis of the interaction of EGFR with PD-L2 in SCC15 and SCC25 cells. (C) In situ proximity ligation assay results revealed the direct interaction of PD-L2 with EGFR in SCC15 cells. Scale bar, 20 µm. (D) Identification of the PD-L2 essential domains required for interactions with EGFR. P1, Ig-like V-type domain; P2, Ig-like C2-type domain; P3, transmembrane domain and cytoplasmic domain. (E) Silencing FUT8 abrogated EGFR/PD-L2 interaction. (F) Negative control-expressing, PD-L2 WT-expressing, or REY mutant-expressing SCC15 cells were serum-starved for 4 hours and treated with EGF (20 ng/mL). Protein expression was analyzed using the indicated antibody. (G) Cells were treated with CHX (20 µM) for the indicated times and analyzed using immunoblotting with anti-EGFR and anti-STAT3 antibodies (top panel). The intensity of each band was quantified using ImageJ software (bottom panel). EGFR, epidermal growth factor receptor; FUT8, fucosyltransferase; PD-L2, programmed death ligand 2; REY, PD-L2 REY mutant, in which the amino acid of R168, E171 and Y174 were substituted by Alanine; STAT3, signal transducer and activator of transcription 3; WT, wild type.
Additionally, PD-L2-overexpressing cells exhibited hyperactivation of EGFR/STAT3 signaling. Phosphorylated levels of EGFR and STAT3 in PD-L2 REY-expressing cells peaked at 15 min and rapidly declined thereafter. In contrast, EGF promoted the phosphorylation of both EGFR and STAT3 throughout the observation period of up to 6 hours in PD-L2 WT-expressing cells (figure 5F). Moreover, the half-life of EGFR and STAT3 proteins in PD-L2 WT-expressing cells was significantly higher than that in PD-L2 REY mutant-expressing or negative control cells (figure 5G). The secretion of EGF was not significantly different between PD-L2 WT-expressing and control cells (online supplemental figure 8D).
Therefore, these data suggest that PD-L2 glycosylation is critical for EGFR ligand binding and activation of downstream signaling.
PD-L2 glycosylation attenuates cetux-EGFR binding affinity and impairs the response to cetux
Cetux, a competitive antagonist, blocks the interaction between EGFR and its ligand and consequently attenuates the EGFR signaling activity.33 PD-L2 REY decreased the affinity of EGFR to EGF by 1.7-fold compared with the PD-L2 WT expressing cells even on stimulation with 100 pg EGF (figure 6A). Consistently, FUT8 knockdown in SCC15 cells significantly decreased EGF binding to EGFR, which was similar to the Stattic-induced decreased EGF binding affinity (figure 6B and C). Confocal fluorescence images revealed that the green fluorescence signals of cetux and its colocalization with EGFR in PD-L2 REY mutant-expressing cells were higher than those in PD-L2 WT-expressing cells (figure 6D).

Glycosylated PD-L2 enhances EGF-EGFR binding affinity. (A) EGF-EGFR binding affinity in SCC15 cells expressing PD-L2 WT or REY mutant was measured using capture ELISA. Significance was determined using one-way analysis of variance. *p<0.05. (B) Loss of FUT8 decreased the binding between EGF and EGFR in SCC15 cells. *p<0.05. (C) EGF-EGFR binding affinity in SCC15 cells treated with DMSO or Stattic was measured using capture ELISA. **p<0.01. (D) Confocal microscopy of SCC15 cells expressing PD-L2 WT or REY treated with EGF for 30 min and stained with anti-EGFR antibodies and FITC-labeled cetux. Scale bar, 20 µm. Right panel shows the quantification of the colocalization between cetux and EGFR. Quantification of the cetux fluorescence intensity in (D). Values are represented as mean±SD from three independent experiments. Significance was determined using one-way analysis of variance (ANOVA). **p<0.01. (E) Description and schematic representation of EGFR indicating specific sites mutated in individual Ig-like domains. Immunoblotting of EGFR and PD-L2 in total lysates of SCC15 cells transfected with EGFR-Myc-WT or EGFR-Myc-(IgI-IV) mutants. (F) Representative images of the colony formation capability of SCC15 cells. Significance was detected using repeated-measures ANOVA. *p<0.05; **p<0.01. (G) Quantification of viability of SCC15 and SCC25 cells treated with control, cetux, Stattic, or their combination (Com) for the indicated time. Error bars represent SD (n=3). *p<0.05; **p<0.01. EGFR, epidermal growth factor receptor; FUT8, fucosyltransferase; PD-L2, programmed death ligand 2; REY, PD-L2 REY mutant, in which the amino acid of R168, E171 and Y174 were substituted by Alanine; WT, wild type.
To determine the mechanism involved in PD-L2 glycosylation-induced increased binding affinity of EGF for EGFR, the role of EGFR extracellular domain in PD-L2 binding was examined. A series of EGFR mutants was generated and subjected to immunoprecipitation. Domains I and II are critical regions for the binding of EGFR to PD-L2 (figure 6E). Domain I of EGFR is essential for EGF binding.34 Our results shown that the direct interaction between glycosylated PD-L2 and EGFR Ig I domains enhanced EGF-EGFR binding affinity and impaired cetux sensitivity.
A Matrigel colony formation assay was performed for 10 days using various concentrations of cetux (figure 6F). Glycosylated PD-L2 WT-treated cells acquired resistance to cetux. The number and size of colonies of the PD-L2 WT-expressing cells were markedly higher than those of the PD-L2 REY-expressing cells after cetux treatment. As glycosylation of PD-L2 is regulated by STAT3, the effect of Stattic on the anti-proliferative effect of cetux was examined. The results of cell proliferation assays demonstrated that Stattic-treated cells exhibited decreased cell viability and enhanced response to Cetux when compared with control cells (figure 6G).
These in vitro tests suggest glycosylation regulates the anti-EGFR therapy response by strengthening the EGF-EGFR binding affinity.
Inhibition of PD-L2 glycosylation enhances the response to cetux in an orthotopic model of HNSCC
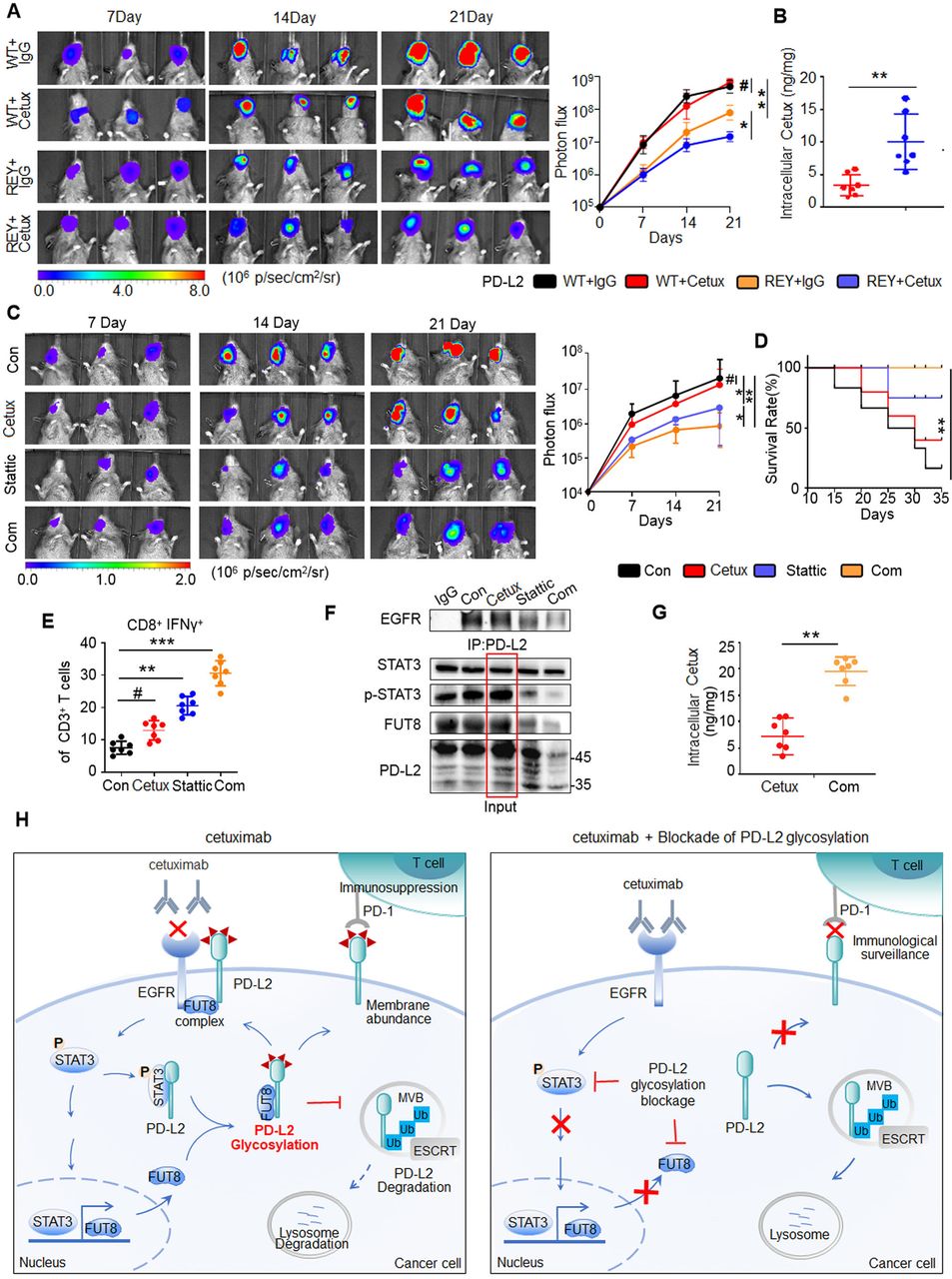
To further analyze the role of STAT3/FUT8-mediated PD-L2 glycosylation in cetux therapy response, we examined the tumorigenesis of SCC7 tumor cells expressing PD-L2 WT or PD-L2 REY mutant in C3H mice (online supplemental figure 9A). Tumor growth in mice bearing PD-L2 REY-transfected tumor cells was significantly lower than that in mice bearing PD-L2 WT-transfected tumor cells (figure 7A). Moreover, the CD8+ IFNɣ+ T cell population and granzyme B-positive staining significantly increased in cetux-treated mice harboring PD-L2 REY-transfected tumor cells (online supplemental figure 9B). SPR analysis revealed that the inhibition of PD-L2 glycosylation enhanced the intracellular cetux accumulation by 4.5-fold compared with the PD-L2 WT expressing tumors (figure 7B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Blockade of PD-L2 glycosylation enhances the response to cetux in the orthotopic model of HNSCC. (A) Representative bioluminescence images of C3H mice with tumors derived from SCC7 cells transfected with PD-L2 WT or REY mutant and treated with cetux every week (n=7 mice per group). *p<0.05; **p<0.01; #, not significant. (B) The intracellular cetux content in SCC7 tumor mass from the experiment shown in (A) was detected using surface plasmon resonance (SPR). **p<0.01. (C) Effect of cetux, Stattic, or their combination on tumor growth in C3H mice bearing SCC7 tumors. The mice were treated with STAT3 inhibitor (Stattic; 4 mg/kg bodyweight; every 2 days) with or without cetux (40 mg/kg bodyweight; once a week) for 3 weeks. The tumor burden was examined using bioluminescence imaging (n=7 mice per group). *p<0.05; **p<0.01; #, not significant. (D) Survival of C3H mice bearing SCC7 tumors from the experiment show in (C). **p<0.01. (E) Intracellular cytokine staining of interferon gamma in CD8+ and CD3+ T cell populations of C3H mice bearing SCC7 tumors from the experiment shown in (C) (n=7 mice per group). **p<0.01; ***p<0.001; #, not significant. (F) Interaction between EGFR and PD-L2 in C3H mice bearing SCC7 tumors after treatment with cetux, Stattic, or the combination of Cetux and Stattic was assessed using Co-immunoprecipitation. (G) The intracellular cetux content in SCC7 tumor mass treated with cetux alone or in combination with Stattic was detected using SPR. Significance was determined using one-way analysis of variance. **p<0.01. (H) A proposed model illustrating glycosylation of PD-L2 is an important mechanism for cetux refractoriness and immune evasion in HNSCC. Glycosylated PD-L2 forms a complex with EGFR, activating EGFR/STAT3 signaling and reducing cetuximab binding affinity to EGFR. N-glycosyltransferase FUT8, a transcriptional target of STAT3, is required for EGF induced PD-L2 glycosylation. This supports the development of a therapeutic strategy for cancer involving the combination of PD-L2 glycosylation blockade and cetux therapy. EGFR, epidermal growth factor receptor; ESCRT, endosomal sorting complexes required for transport; FUTA8, fucosyltransferase; HNSCC, head and neck squamous cell carcinoma; PD-L2, programmed death ligand 2; STAT3, signal transducer and activator of transcription 3; REY, PD-L2 REY mutant, in which the amino acid of R168, E171 and Y174 were substituted by Alanine; WT, wild type.
To further validate the in vivo function, tumor-bearing mice were treated with Stattic and/or cetux. Cetux alone did not markedly decrease the tumor burden in mice. However, the Stattic/cetux combination markedly decreased tumor burden without affecting the bodyweight of C3H mice (figure 7C and online supplemental figure 9C). Additionally, the survival of mice treated with the Stattic/cetux combination was higher than that of mice treated with cetux or Stattic (figure 7D). The Stattic/cetux combination decreased PD-L2 expression and enhanced cytotoxic T cell activity in the tumor mass (online supplemental figure 9D). Among TILs, the proportion of cytotoxic CD8+ T cells in the mice treated with the Stattic/cetux combination was approximately 1.3-fold to 2.1-fold higher than that in the mice treated with single cetux (figure 7E). Additionally, cleaved caspase 3 staining revealed that the combination therapy promoted the death of tumor cells when compared with cetux monotherapy (online supplemental figure 9E).
To confirm the function of STAT3, the tumor lysates were subjected to immunoprecipitation analysis. The association between EGFR and PD-L2 was significantly attenuated in the Stattic/cetux combination treatment group. Moreover, continuous administration of cetux increased the PD-L2 levels (figure 7F). Stattic mitigated cetux-induced PD-L2 upregulation by disrupting FUT8 transcription. This explains the decreased efficacy of cetux against the tumor and the synergistic anticancer activity of the Stattic/cetux combination. SPR analysis revealed that the cetux levels increased by 1.8-fold to 4.4-fold in the cells treated with Stattic/cetux combination compared with the single cetux treated group (figure 7G). Thus, STAT3/FUT8-mediated PD-L2 glycosylation is a master regulator of immune evasion and anti-EGFR therapy response in HNSCC (figure 7H).
Discussion
Drug resistance remains the determining factor for the failure of anti-EGFR therapy. Recently, ‘immunogram’,35 the suppressive tumor microenvironment, was suggested to be mechanistically linked to responses to cetux therapy. This study uncovered that PD-L2 glycosylation is a major regulator of immune evasion and the response to cetux in HNSCC, reinforcing the key role of post-translational modification in ICM and demonstrating its potential as a novel source of therapeutic targets. In addition, our findings underscore the intimate connections between immune response and clinical benefit prediction from anti-EGFR therapy in HNSCC patients.
To date, most studies have focused on intrinsic resistance to anti-EGFR therapy in a tumor-intrinsic strategy, and we developed an approach to overcome tumor resistance to anti-EGFR treatment through a tumor-extrinsic strategy, reactivating the immune response. We showed that PD-L2 interacts with EGFR in an FUT8-dependent manner. FUT8-mediated PD-L2 glycosylation emerges as a synchronized program that orchestrates EGFR signaling dysregulation and bolsters tumor immunosuppressive networks and the response to anti-EGFR therapy. Therefore, PD-L2 glycosylation is expected to serve as a new biomarker to predict the response to cetux treatment in patients with HNSCC. Our study extends the clinical significance of PD-L2 glycosylation in cancer therapy, suggesting that targeting this modification may be a powerful marker for personalized combination therapy to enhance the anti-EGFR therapy response in HNSCC.
In the current study, we discovered that EGFR/STAT3 signaling promotes PD-L2 glycosylation by recruiting the N-glycosyltransferase FUT8. Core fucosylation expression is comparatively higher in HNSCC cancer tissues than in normal tissues, suggesting that FUT8-mediated glycosylation of PD-L2 is a cancerous event.36 37 Our Co-IP and PLA assays demonstrated that non-glycosylated PD-L2 binds to (p)STAT3 in response to EGF stimulation. STAT3 was identified as the upstream regulator of FUT8, which is the key N-glycosyltransferase required for PD-L2 glycosylation and stabilization. Importantly, enhanced expression of STAT3 and FUT8 was linked to high PD-L2 expression in patients with HNSCC tumor tissue. Therefore, we uncovered oncogenic signaling and found that the key N-glycosyltransferase orchestrates the glycosylation process.
Glycosylation is reported to increase ICM protein stability by blocking proteasome-induced degradation.38 However, the role of glycosylation in PD-L2 stability and activity remains unknown. Our findings identified that the glycosylation sites N157, N163 and N189, but not N64, are responsible for PD-L2 protein stability. PD-L2 glycosylation and inhibited PD-L2 polyubiquitination, thereby blocking its association with the core components of the ESCRT complex HRS. Blockage of PD-L2 glycosylation by the PD-L2 REY mutant of Stattic treatment facilitated PD-L2 lysosome-mediated degradation and decreased its cell surface abundance, which resulted in loss of binding affinity for PD-1 and activation of the T cell-mediated immune response, constituting a striking antitumor effect in a murine model.
In summary, our results define the unrecognized function of PD-L2 glycosylation in predicting the response to cetux therapy. We clarified the oncogenic signaling modulating PD-L2 glycosylation initiation and uncovered the mechanism of glycosylation-induced PD-L2 stabilization. Our findings provide a rationale for combining PD-L2 glycosylation blockade and cetux therapy.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data are available in the main text or the supplementary materials.
Ethics statements
Patient consent for publication
Ethics approval
The study involving human subjects was approved by Institutional Ethical Boards of Tianjin Medical University Cancer Institute and Hospital (EK 2019179) and informed consent was obtained from all patients whose tissues were used for this study. All animal experiments were performed in accordance with the guidelines approved by the Animal Care and Use Committee of Tianjin Medical University (TMUaMEC 2017016).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
YX, ZG and RH contributed equally.
Contributors The conception and design of the article: XZhou, ML, YR, MM. Collection and aggregation of data from article: YX, ZG, RH and YQ-W. Data analysis and interpretation of the article: All authors. Manuscript writing: All authors. Final approval of the manuscript: All authors. Responsible for the overall content as the guarantor: XZhou.
Funding This work was supported by grants from the National Science Foundation of China (NSFC) through 81872495 (to YR), 82172764 (to YR), 81872206 (to XZhou), 82073010 (to XZhou) and 81673026 (to MM), the Key Laboratory of Immune Microenvironment and Disease (Tianjin Medical University), Ministry of Education.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.