Article Text

Abstract
Background Hypoxia is a striking feature of most solid tumors and could be used to discriminate tumors from normoxic tissues. Therefore, the design of hypoxia-conditioned Chimeric Antigen Receptor (CAR) T cells is a promising strategy to reduce on-target off-tumor toxicity in adoptive cell therapy. However, existing hypoxia-conditioned CAR-T designs have been only partially successful in enhancing safety profile but accompanied with reduced cytotoxic efficacy. Our goal is to further improve safety profile with retained excellent antitumor efficacy.
Methods In this study, we designed and constructed a hypoxia-inducible transcription amplification system (HiTA-system) to control the expression of CAR in T (HiTA-CAR-T) cells. CAR expression was determined by Flow cytometry, and the activation and cytotoxicity of HiTA-CAR-T cells in vitro were evaluated in response to antigenic stimulations under hypoxic or normoxic conditions. The safety of HiTA-CAR-T cells was profiled in a mouse model for its on-target toxicity to normal liver and other tissues, and antitumor efficacy in vivo was monitored in murine xenograft models.
Results Our results showed that HiTA-CAR-T cells are highly restricted to hypoxia for their CAR expression, activation and cytotoxicity to tumor cells in vitro. In a mouse model in vivo, HiTA-CAR-T cells targeting Her2 antigen showed undetectable CAR expression in all different normoxic tissues including human Her2-expresing liver, accordingly, no liver and systemic toxicity were observed; In contrast, regular CAR-T cells targeting Her2 displayed significant toxicity on human Her2-expression liver. Importantly, HiTA-CAR-T cells were able to achieve significant tumor suppression in murine xenograft models.
Conclusion Our HiTA system showed a remarkable improvement in hypoxia-restricted transgene expression in comparison with currently available systems. HiTA-CAR-T cells presented significant antitumor activities in absence of any significant liver or systemic toxicity in vivo. This approach could be also applied to design CAR-T cell targeting other tumor antigens.
- receptors
- chimeric antigen
- tumor microenvironment
- immunotherapy
- cell engineering
Data availability statement
All supporting data are included in the manuscript and the online supplemental materials. Additional data are available on reasonable request to corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Hypoxia is a hallmark feature of solid tumors because of insufficient blood supply, the oxygen concentration may vary from 0.02% to 2% O2, as compared with 2% to 9% O2 in normal tissues.1 Hypoxia appears to be strongly associated with the resistance of cancer cells to radiotherapy and chemotherapy, as well as impaired functions of immune cells.2 3 Exploiting this unique environmental signal for targeted therapy is a promising strategy in cancer treatment. With an increased molecular understanding of tumor hypoxia, researchers have developed hypoxia-targeting gene therapy. Directional edited suicide genes or therapeutic genes could be activated at hypoxic tumor sites rather than normal tissues, which greatly enhances therapeutic safety.4–6 In addition, hypoxia-targeted delivery and activation of prodrug provide additional levels of selectivity to the treatment.7 To enable cells to sense and respond to hypoxia, many reporters utilized hypoxia-inducible transcription factor-1α (HIF-1α) and hypoxia response elements (HRE) to perform transcriptional and post-translational control of genes of interest (GOI).8 9 HIF-1α is degraded through hydroxylation of two prolines in an oxygen-dependent degradation domain (ODD) within its structure under normoxia.10 11 Under hypoxia, however, HIF-1α accumulates and then dimerizes with constitutively expressed HIF-1β (HIF-1β) to form HIF-1, which specifically binds to HRE sequence in the promoters of hypoxia inducible genes.2 12 Ruan et al placed the suicide gene BAX under the control of HRE and found that cells transfected with the BAX plasmid were preferentially killed through apoptosis under hypoxia.13 Collet et al used the HRE-driven soluble form of vascular endothelial growth factor receptor 2 (VEGFR2) to control angiogenesis and reduce hypoxic tumor cells growth in vitro and in vivo.14 HRE-IL-2-engineered cytotoxic T lymphocytes (CTLs) were found to produce faster and more complete tumor regression than the parental CTLs and thus increase overall survival of tumor-bearing mice.15 Other potent hypoxia-inducible transgenes also have been reported using HIF/HRE system.16–18 Besides transcriptional regulation, ODD can even directly fuse to GOI to regulate transgenes expression at the translational level. Delivery of a cytotoxic TAT-ODD-Caspase 3 fusion protein reduced tumor masses without any obvious side effects in both a mouse model and a rat ascites model.19 20 Transfection of the plasmid expressing attenuated DTAW153F/ODD or DTAH21A/ODD resulted in hypoxia-dependent apoptosis of transfected cells.21
Currently, the overlapped expression of tumor-associated antigens in cancer cells with normal tissues poses a great safety challenge in clinical applications of engineered T cells, for example, chimeric antigen receptor T cells (CAR-T) and TCR-T cells.22 23 Hypoxia-regulated CAR-T cells are proposed to increase tumor selectivity and reduce on-target off-tumor toxicity to normal tissues,22 23 and would be applicable to a wide range of tumors. HIF-CAR24 and HiCAR25 have been constructed by incorporating HRE and/or ODD into conventional CAR structure. Although these CAR-T cells exhibited well oxygen sensitivity, we observed residual CAR expression and cytolytic activity under normoxia but less-than-desirable CAR expression under hypoxia.24 25 Besides, compared with many hypoxia-activated prodrugs entering clinical trials,26 up to now, no hypoxia-regulated CAR-T cells have been progressed into clinical trial, suggesting further improvement is required.
In many previous reports, investigators constructed chimeric/hybrid promoters combining tissue-specific promoters with viral minimal promoters to achieve efficient gene transcription, which showed undesirable basal expression mainly due to the strong trans-activation activity of viral promoters.25 27–29 Our main goal is to engineer CAR-T cells to exhibit low basal activity under normoxia but high inducibility under hypoxia. This might be achieved by using transcriptional amplification approaches30 31 in which GOI expression was driven by a conditionally activated trans-activator under certain circumstances, otherwise it was regulated by a weak promoter to keep low basal expression. The Gal4/upstream activating sequence (UAS) system is one of the most powerful tools for GOI expression, which consists of Gal4 protein as the transcriptional activator and UAS that is targeted by Gal4 as an enhancer.32 Although this system has been widely used, we wondered if we could design a novel trans-activator specific to UAS with refined specificity and induction levels. The Cys2-His2 zinc finger is an attractive scaffold for designing DNA binding domains (DBDs) with desired specificity. Studies have demonstrated the flexibility of finger motif and the possibility of designing zinc finger proteins (ZFPs) that can bind specifically to customized sequences.33 34 Therefore, we used this powerful tool to design our novel DBDs in this study.
In this study, we first designed and generated an effective trans-activator comprising a UAS-specific zinc finger DBD (ZFDBD) and a transcriptional activation domain,35 and then optimized the corresponding response element, that is, the minimal promoter. To make this transcriptional system oxygen-sensing, we further developed a bidirectional hypoxia-inducible transcriptional amplification system (HiTA-system) where the transactivator is regulated by both HRE and ODD. The hypoxia-inducible transactivator can bind specifically to its target sequence upstream of GOI and initiate their transcription and expression under hypoxia. Our HiTA system demonstrated a marked improvement in hypoxia-induced transgene expression while retaining low background compared with currently available systems. HiTA-CAR-T cells under control of this system exhibited hypoxia-restricted CAR expression, cytokines secretion and hypoxia-dependent tumor killing in vitro. In addition, no on-target toxicity to liver expressing human target antigen was observed in naïve mice, which further confirmed their improved safety in vivo. HiTA-CAR-T cells, not unexpectedly, achieved comparable antitumor efficacy to eukaryotic elongation factor 1-α (EF1α)-CAR-T cells in tumor-bearing mice. Overall, the HiTA system could be used to balance safety and efficacy of CAR-T cells in the treatment of solid tumors, as well as therapeutic payloads, such as bispecific T-cell engager.
Methods
Cell lines and viruses
Human acute T cell leukemia cell line Jurkat (American Type Culture Collection (ATCC), TIB-152), lung cancer cell line NCI-H292 (ATCC, CRL-1848) and ovarian cancer cell line SKOV3 (ATCC, HTB-77) was cultured in RPMI 1640 (Corning) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (Pen/Strep, Corning). Embryonic kidney cell line HEK293T (ATCC, CRL-3216), lung cancer cell line A549 (ATCC, CCL-185) and epithelioid cervix carcinoma cell line Hela (ATCC, CBP-60232) were cultured in Dulbecco’ s modified Eagle’s medium (Corning) supplemented with 10% FBS and 1% Pen/Strep. Tumor cells carrying the report gene firefly luciferase (SKOV3-Luc, NCI-H292-Luc and A549-Luc cells) were engineered as previously described.36 The cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. The hypoxia treatment (1% O2, 5% CO2 and 94 % N2) was carried out in Smartor 118 mobile three-gas control system (China Innovation Instrument) or by adding different concentration of CoCl2 into the culture medium to simulated chemical hypoxia.
The recombinant type 5 adenovirus expressing tumor antigen Her2 with reporter firefly luciferase (Ad5-Her2) was contracted for production to Shanghai Genechem Co.,Ltd.
DNA constructs
Transactivators plasmids
The DNA fragment Gal4VP64 consisting of the DBD of yeast transcription factor Gal4 in Gal4-UAS system fused with the activating domain VP64,35 was PCR amplified from pHR_PGK_antiCD19_SynNotch_Gal4VP64 (Addgene #79125),37 and then inserted into the vector pLVX-EF1α-IRES-ZsGreen1 (Clontech, #632187) downstream of EF1α with restriction enzyme XhoI and BamHI. The generated transactivator plasmid was abbreviated as pLV- Gal4-VP64 in the following study.
To obtain a more effective trans-activator, we designed a ZFP DBD specific to UAS according to the Zinc Finger Tools (http://www.zincfingertools.org).38 The resulted DNA cassette coding ZFP was fused with VP64 and then inserted into the vector pLVX-EF1α-IRES-ZsGreen1 as mentioned above. The newly generated transactivator plasmid was termed as pLV-ZFP-VP64.
To make this transcriptional system oxygen-sensing, the C-terminal of ZFP-VP64 was fused with ODD25 to generate a hypoxia-regulated activator ZPO. The ZPO was further put in trans upstream to five copies of HRE39 followed by cytomegalovirus minimal promoter (CMV mp) to generate ZPO-HRE. ZPO and ZPO-HRE sequences were synthesized and cloned into pLVX-EF1α-IRES-ZsGreen1with restriction enzyme 5’ XhoI and 3’ BamHI to generate pLV-ZPO and pLV-ZPO-HRE, respectively.
Responsive element plasmids
The vector pHR_Gal4UAS_IRES_mC_pGK_tBFP (Addgene #79123),40 carrying reporter gene mCherry driven by CMV mp located downstream of UAS sequence, was named as pHR_UASCMV in this study. To reduce the undesirable basal expression due to the strong transactivation activity of CMV, HIV-1 long terminal repeated (LTR)41 minimal promoter (LTR mp) was PCR-amplified from pNL4-3-luc-△Env (NIH AIDS Reagent Program #3418), and used to replace CMV mp by using ClonExpress II One Step Cloning Kit (Vazyme). The generated construct was termed as pHR_UASLTR. The primers used for LTR mp amplification included 5’- GCACTGTCCTCCGAACGACTCTGCATATAAGCAGCTGCTT-3’, and 5’- ATCTGACGGTTCACTAAACGCCAAGCTTTATT-GAGGCTTA −3’.
One-step and HiTA bidirectional plasmids
The pHR_HRELTR one-step vector consisted of a mCherry reporter gene driven by 5×HRE and LTR minimal promoter. To generate the HiTA bidirectional vector, the ZPO-HRE sequences were inserted upstream of UAS sequences in pHR_UASLTR to form pHR_ZPO-HRE-LTR plasmid. All the sequences were synthesized and confirmed by sequencing (Generay Biotech, Shanghai).
Plasmid transient transfection and western blot assay
The bidirectional plasmid pHR_ZPO-HRE-LTR were transiently transfected into 293 T cells (5×105/well) in 6-well plate. Twelve hours post transfection, the cells were divided into two groups and cultured in either normoxic (21% O2) or hypoxic (1% O2) chambers for another 24 hours and 48 hours. At the indicated time point, the cells were harvested and subjected to the detection by western blot, in which a rabbit monoclonal antibody against HIF-1a (CST #36169) and an HRP-conjugated goat anti-rabbit antibody (ZSGB-BIO # ZB-2308) were used.
CAR and HiTA-CAR design
The DNA fragment of Her2-CAR described previously25 was inserted into pHR_ZPO-HRE-UASLTR plasmid with XbaI and Pfl23II to replace the reporter gene mCherry. The Her2-CAR expressing bidirectional plasmid was termed as pHR_HiTA-CAR. The plasmid containing Her2-CAR driven by EF1α constitutively25 was named as pHR_EF1α- CAR in the following study.
T cells transduction and expansion
Customized lentiviruses LV-HiTA-CAR and LV-EF1α-CAR, were produced by pHR_HiTA-CAR and pHR_EF1α-CAR lentiviral vectors, respectively (Kanglin Biotech., Hangzhou), and were used for T cell transduction as described previously.36 Briefly, T cells derived from human peripheral blood mononuclear cells of healthy donors, which were delinked before the study, were activated by anti-hCD3 and anti-hCD28-coated immunobeads at cell/bead ratio of 1:1 and cultured in T cell growth medium (TCM), consisting of X-VIVO 15 medium (Lonza #BE02-060F), human IL-7 (R&D systems #P13232), human IL-15 (R&D systems #P40933) and human IL-21 (Novoprotein #GMP-CC45)36 at 37°C for 48 hours. The activated T cells were then transferred into a 48-well plate and transduced with the lentivirus (multiplicity of infection (MOI)=5) by centrifugation at 1000×g at 32°C for 1.5 hours. Thereafter, the transduced T cells (HiTA-CAR-T and EF1α-CAR-T) expanded at least for 7 days when they were rested and could be used in assays. During expanding period, the cell density was adjusted to 0.5–2×106/mL by supplement of fresh TCM every 3 days. The untransduced T cells (UTD), as control cells, were treated the same.
In vitro evaluation of CAR-T cells function
Quantitation of cytokine
SKOV-3 cells (1×105/well) were seeded in 96-well flat-bottom plate. After cultured 12 hours, CAR-T cells were added at an effector/target (E/T) ratio of 3:1, and then continuously cultured in hypoxic condition (1% O2) or normoxic condition (21% O2) for 24 hours. IL-2 and interferon γ (IFN-γ) levels in the culture supernatant were determined using the Human IL-2 ELISA Set (BD Biosciences #555190) and Human IFN-γ ELISA Set (BD Biosciences #555142), respectively.
Validation of CAR-T cells cytotoxicity
CAR-T cells cytotoxicity was assessed using Luciferase Assay System as described previously.36 Briefly, tumor cell lines carrying the report gene firefly luciferase were plated in a 96-well black flat-bottom plate in triplicate and grown for overnight. HiTA- CAR-T and EF1α- CAR-T cells were added into the plate at the indicated E/T ratio and cocultured for another 24 hours, respectively. The culture supernatant was removed, and the viability of the remained tumor cells was assessed by quantifying the luciferase activity unit (LU) with a GloMax 96 reader (Promega #E1501). The formula is as follows:

Animal study
The mice used in this study were 8–10 weeks old female NOD-PrkdcscidIl2rgtm1/Bcgen (B-NDG) mice, which were purchased from Biocytogen (Shanghai, China) and maintained under specific pathogen-free conditions at the animal facilities of Shanghai Public Health Clinical Center (SPHCC) (Shanghai, China). There were at least four mice analyzed at each collection time point. All animal experiments were performed in accordance with the Institutional Animal Care and Use Committee of SPHCC.
For in vivo safety evaluation, mice were injected intraperitoneally (i.p.) with 1×109 plaque forming unit of recombinant adenoviruses Ad5-Her2. After 7 days, the mice were intravenously injected with five million of the CAR-T cells. Mouse peripheral blood was collected from orbital vein bleeding at indicated time as shown in figure 5 and left to clot. After centrifuged at 8000 g for 20 min, the resulting serum was collected and stored at −80°C for aminotransferases assay and cytokines assay. To investigate the effect of injected CAR-T on Her2 expressing tissues, 125 µL of D-luciferin (3 mg/mL) were injected (i.p.) into mice anaesthetized with 1% pentobarbital sodium (i.p.). After 8 min, the mice were subjected to observe luminescence using in vivo imaging system NightOWL II LB 981 (Berthold Technologies). At the end of study, the blood samples and the single cell suspension of the digested tissues (liver, spleen, lung) collected from the mice were subjected to T cell analysis with flow cytometry. In addition, 1/4 part of the liver excision was paraformaldehyde-fixed followed by paraffin embedding and sectioning for Immunohistochemistry and immunofluorescence assay.
For in vivo efficacy evaluation, mice were subcutaneously (s.c.) inoculated with tumor cells NCI-H292 cells (5×106) or SKOV3 cells (2×106). To confirm the degree of hypoxia of tumors at the time of CAR-T infusion, mice were given the hypoxyprobe pimonidazole (Hypoxyprobe, #HP1) by i.p. injection at the dose of 60 mg/kg. Two hours later, tumor samples were obtained and fixed followed by paraffin embedding and sectioning for immunofluorescence assay. Then HiTA-CAR-T cells (green fluorescence protein positive (GFP)+) (5×106 cells resuspended in 125 µL phosphate buffered saline (PBS)) were injected intravenously into the mice 10- and 15-days post the tumor cells inoculation. T cells without transduction and transduced with EF1a-CAR were also transferred to mice as negative and positive control, respectively. The tumor size was monitored with calipers every 3 days after T cells transfusion, and its volume (V) was determined using the following equation: V = (length × width2)/2. At day 34, mice were sacrificed. Tumor tissues of SKOV3 were carefully separated, weighted and enzyme-digested for further T cell analysis with flow cytometry.
Flow cytometry analysis
To examine the effects of ZFP-VP64 on the specific UASLTR promoters, 250 ng of pLV-ZFP-VP64 and pHR_UASLTR were cotransfected into HEK-293T cells using Turbofect transfection reagent (Thermo Scientific #R0531) according to the manufacturer’s instructions. In control groups, pHR_UASLTR was transfected alone or cotransfected with pKL-Gal4-VP64. Forty-eight hours post-transfection, the cells were assayed for mCherry expression using fluorescence microscopy and then digested and harvested for mCherry analysis.
To detect the Her2 CAR expressed on T cells surface in vitro, T cells (5×105) were harvested and stained with 0.5 µL PE-anti-DYKDDDDK (Biolegend # 637310) at 4°C for 30 min, washed twice with fluorescence activated cell sorting (FACS) buffer (1×PBS containing 2% FBS), and then resuspended in FACS buffer for detection.
To analyze T cells from the mice tissues, ex vivo single-cell suspensions were stained with a fixable live/dead blue stain (Cat #: L23105, Invitrogen), and the antibodies included anti-CD45-PerCP, anti-CD8-APC, and anti-DYKDDDDK tag-PE (Biolegend, California, USA). Data were collected on LSRFortessa (BD Biosciences) and analyzed using FlowJo software V.10.
Serum levels of aminotransferases and cytokines
Serum levels of mouse alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured by ALT Assay Kit and AST Assay Kit (Nanjing Jiancheng Bioengineering Institute), respectively. The serum cytokines were measured using BD Human Th1/Th2/Th17 Cytometric Bead Array (BD Bioscience #560484).
Immunohistochemistry and immunofluorescence
Paraffin section slides were stained with anti-Her2/ERBB2 antibody (Sino Biological #10004-T56) to detect Her2 expression on liver cell surface or stained with H&E for the verification of inflammatory infiltration. T cells and CAR +T cells were stained with anti-CD3 antibody (Abcam #11089) and purified anti-DYKDDDDK tag antibody (Biolegend #637301) to perform immunofluorescence. Paraffin section slides of tumor tissues were stained with antipimonidazole monoclonal antibody (Hypoxyprobe, #HP1) and goat antimouse secondary antibody (Maokang Biotechnology, Shanghai), #MM51006) to detect hypoxic region in tumors by immunofluorescence. For staining quantification, slides were scanned digitally using TissueFAXS 200 (TissueGnostics).
Statistics
Statistics were analyzed using Prism V.8 (GraphPad Software). The Student’s t-test was used to compare two groups. The one-way analysis of variance (ANOVA) was used for comparison of three or more groups in a single condition. Statistical analysis for cytotoxicity and tumor volume was performed using two-way ANOVA. The statistical test used for each figure was described in the corresponding figure legend. Data were transformed when needed to normalize variance. Symbols (*) indicate statistical significance as follows: *p<0.05; **p<0.01, ***p<0.001 and ****p<0.0001.
Results
Generation of a novel ZFP-VP64 trans-activator and corresponding response element for effective gene induction
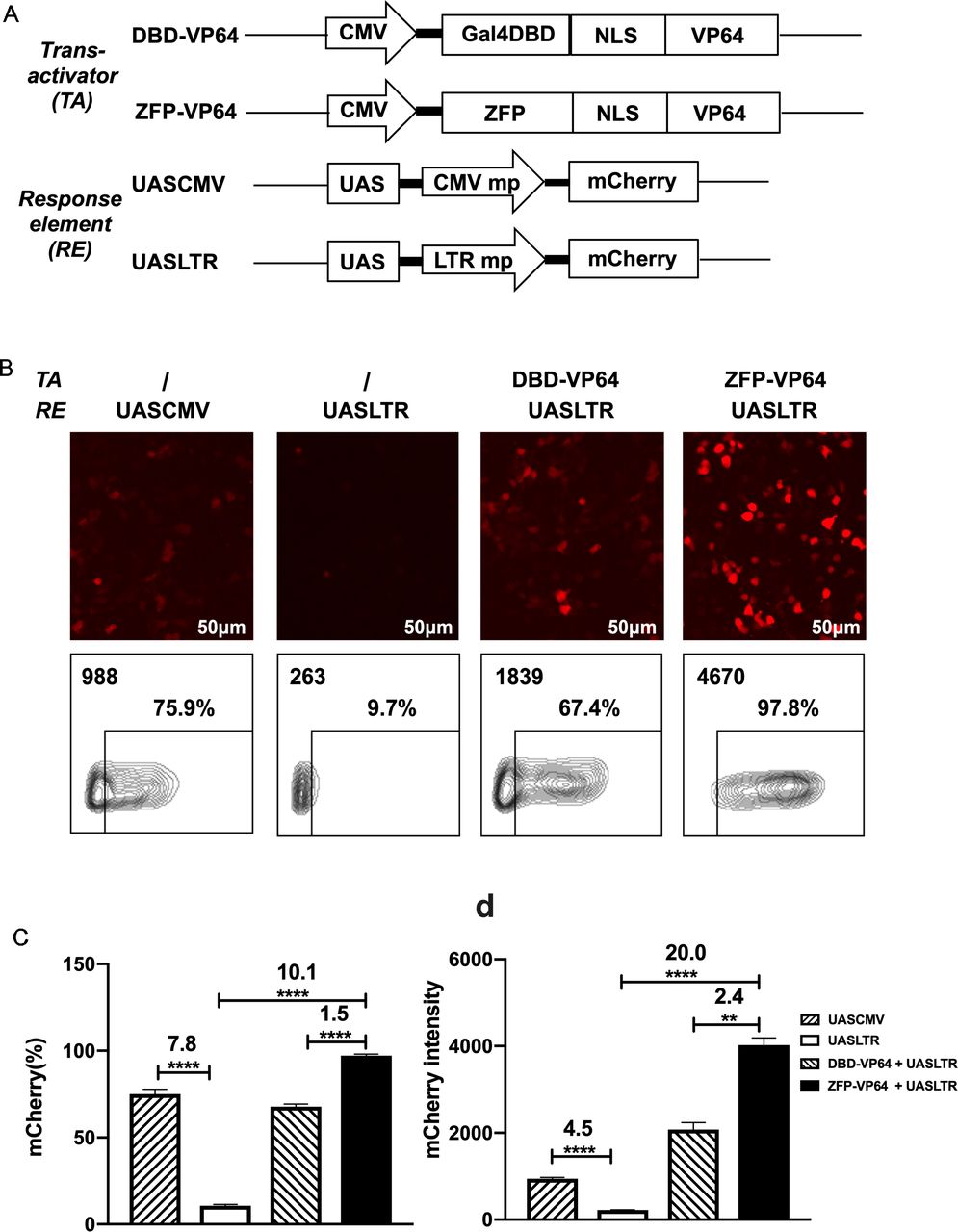
We used Zinc Finger Tools38 as an assistant platform to design and generate two ZFDBDs specific to UAS sequence, with six (6ZFDBD) and seven (7ZFDBD) zinc fingers, respectively. To construct an effective trans-activator, ZFPDBD was fused with a nuclear localization signal (NLS) and the transcriptional activation domain VP64, a tetrameric repeat of the minimal activation domain of VP16.42 As positive control, we construct the other transcriptional activator, in which the NLS-VP64 sequence is fused to the DBD of yeast Gal4 protein, which is the natural transcriptional regulator recognizing UAS element. (figure 1A). The ZFP7-VP64 transactivator could be detected by the triple-FLAG tag at its C-terminus by western blot (online supplemental figre S1A,B). To determine whether the two designed ZFP-VP64 constructs were able to effectively activate transgene expression, we cotransfected the ZFP-VP64 expressing plasmid pLV-ZFP-VP64 with the reporter vector pHR_UASCMV to HEK-293T cells and Hela cells. The reporter vector transfected alone was used as a negative control. In this transient mCherry reporter assay, ZFP7-VP64 showed effective induction in both cell lines 48 hours post transfection, with nearly all cells (98.3% in 293T and 89.5% in Hela) expressing mCherry, producing 15-fold and 3-fold intensity upregulation in 293T and Hela cells, respectively. ZFP6-VP64 exhibited significantly lower transcriptional activity compared with ZFP7-VP64, particularly justified by intensity of mCherry of mCherry positive cells (online supplemental figure S1C,D). Consequently, we choose ZFP7-VP64 for further investigation in subsequent studies.
Supplemental material
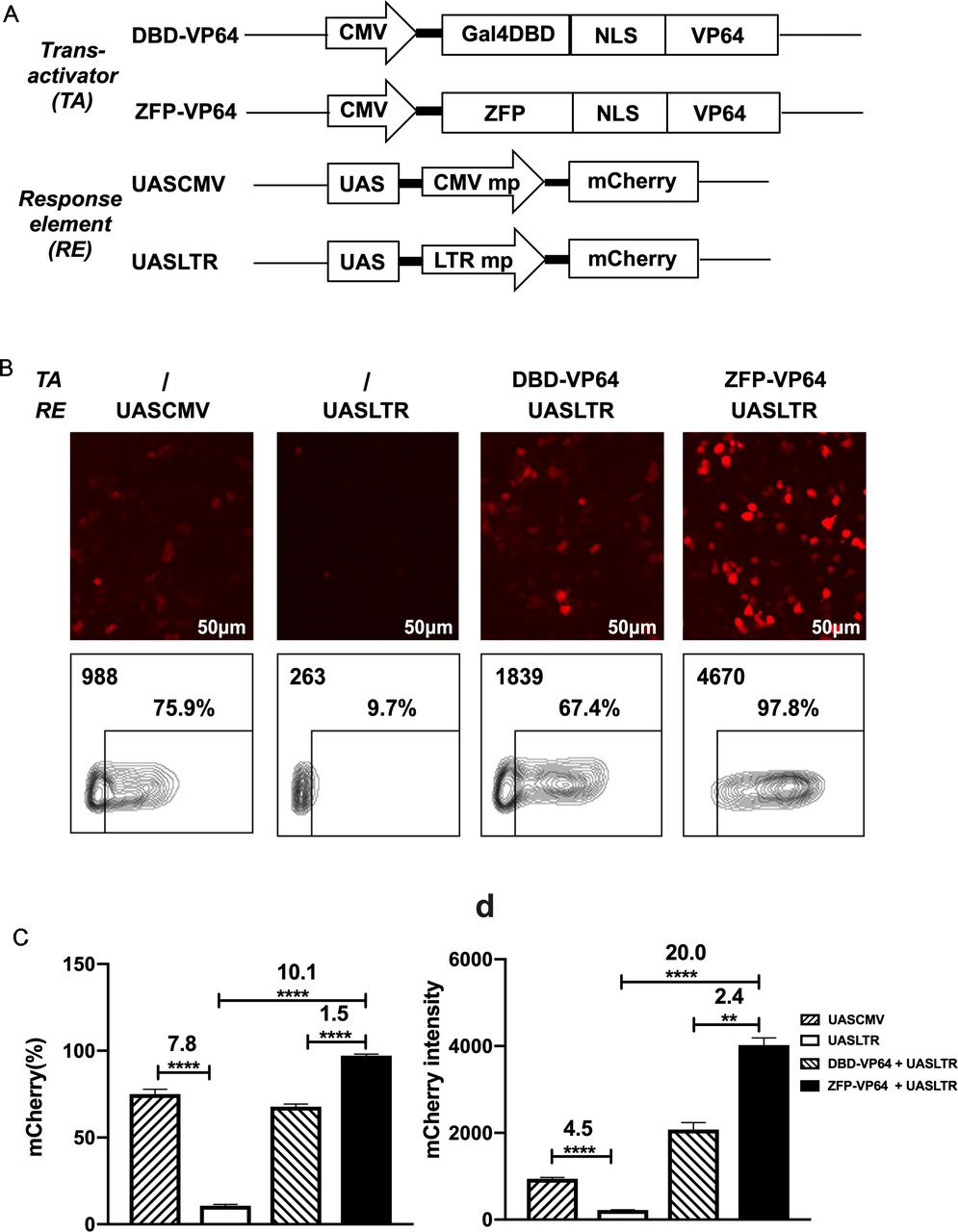
Design of a novel zinc-finger protein-based artificial transcription system for strict and effective gene control. (A) Diagram of transactivators and response elements used in the regulatory system. ZFP, a novel zinc-finger protein specifically targeting UAS sequence; VP64, activation domain; UASLTR, a novel response element consisting of UAS sequence inserted into upstream of minimal LTR promoter; UASCMV, UAS sequence linked to upstream of minimal CMV promoter. (B–D) Basal expression of mCherry driven by different hybrid promoters and inducible expression of mCherry by different transactivators were evaluated by fluorescence microscopy (B) and FACS (B–D) in 293 T cell transfection assay. (n=3, mean±SD is shown, **p<0.01, ****p<0.0001, analyzed using Student’s t-test). CMV, cytomegalovirus; DBD, DNA binding domain; NLS, nuclear localization signal; UAS, upstream activating sequence; ZFP, zinc finger proteins.
However, the basal expression of minimal CMV minimal promoter was as high as ~50% in 293T and ~30% in Hela, which is undesirable for precise transgenes regulation (online supplemental figure S1C). To create an optimal minimal promoter with low leaky expression, we cut 5’ LTR promoter of HIV-1 sequence to retain only three sp1 binding sites and TATA box sequence (PLTRSp1-TATA mp). We further deleted the three Sp1 binding sites and construct a new minimal promoter containing only TATA box (termed PLTRTATA mp), which presented the lowest leaky expression in 293T transfection assay compared with promoters described above (online supplemental figure S2). So, we replaced the CMV mp with LTR mp and constructed the new response element: UASLTR (figure 1A). In transient transfection assay, fluorescence microscopy and flow cytometry demonstrated that the basal activity of this reporter was significantly lower than that of UASCMV (figure 1B–D). When pLV-ZFP-VP64 plasmid was cotransfected, a 20-fold increase of mCherry intensity was observed (figure 1D). When compared with Gal4-VP64, ZFP7-VP64 showed a higher induction rate and intensity of mCherry expression (figure 1B–D). Based on these data, ZFP7-VP64 and UASLTR were the optimal trans-activator and the corresponding response element to generate a new inducible transcription system.
Supplemental material
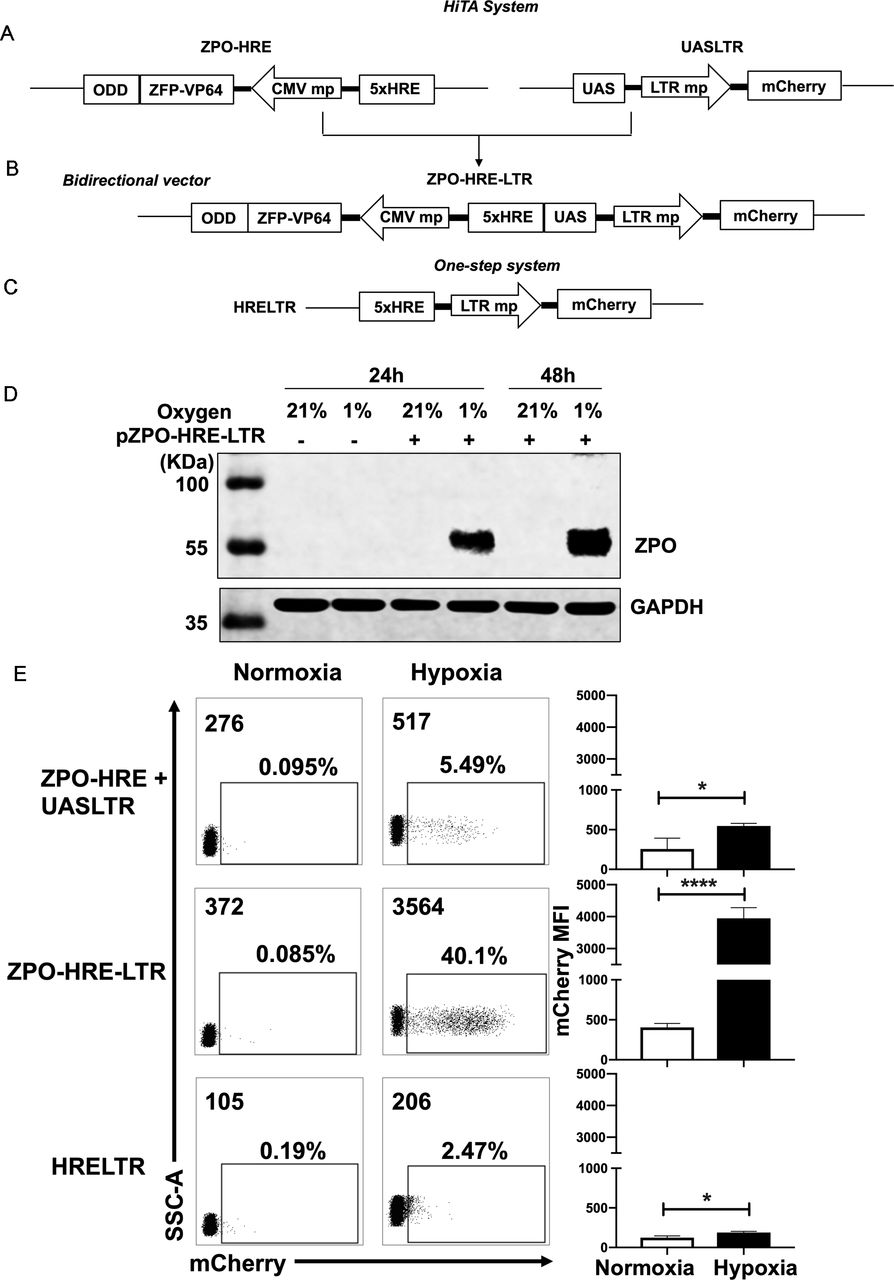
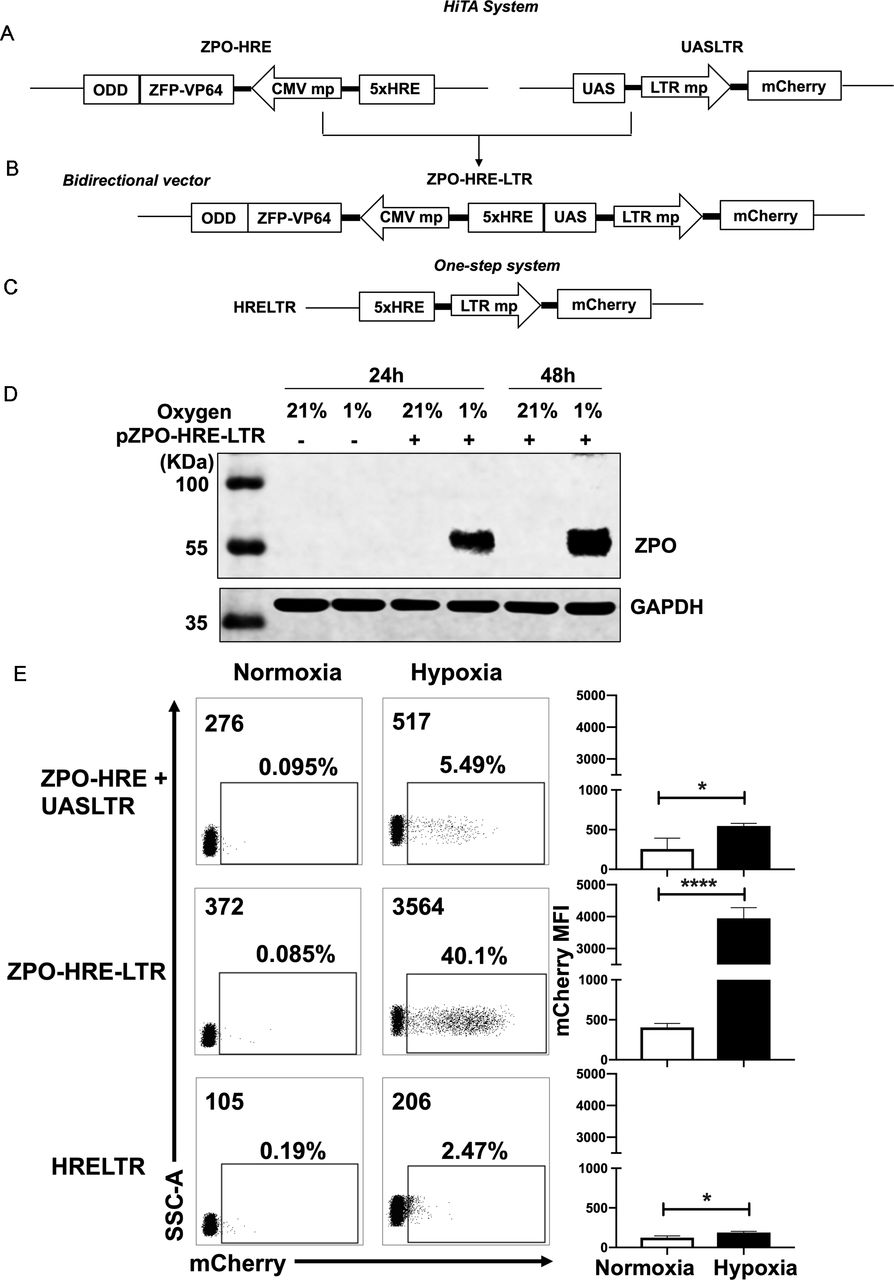
Generation of a hypoxia-inducible transcription amplification (HiTA) system for hypoxia-regulated gene control. (A–C) Schematics of lentiviral vector constructs of hypoxia-regulated system. Target gene is driven by 5× HRE-LTR mp in one-step system (A). HiTA system involved a hypoxia-regulated artificial transcription factor ZFP-VP64-ODD, which is driven by 5× HRE-CMVmp. The ZFP-VP64-ODD is degraded under normoxia via ubiquitin–proteasome pathway while accumulated and activated the transcription of target genes under hypoxia (B). A novel bidirectional lentiviral vector that incorporated transactivator and response element into one vector (C). (D) Wide type 293 T cells (plasmid -) or ZPO-HRE-UASLTR plasmid transfected 293 T cells (plasmid +) were placed under hypoxic (-) or normoxic (+) conditions for 24 hours or 48 hours. The expression of zinc finger protein regulated by both HRE and ODD (ZPO) under normoxia and hypoxia was measured by western blot. (E) Flow cytometry analyses of the mCherry expression in (A–C) hypoxia-regulated system. (n=3, mean±SD is shown, *p<0.05,****p<0.0001, analyzed using Student’s t-test). CMVmp, cytomegalovirus minimal promoter; ODD, oxygen-dependent degradation domain; UAS, upstream activating sequence; ZFP, zinc finger proteins.
Generation of a HiTA system for the control of gene expression
We next sought to develop a HiTA system for oxygen-regulation of target genes. Hypoxia-induced gene changes are mainly executed through HREs, which can be recognized by HIF-1α, a master transcriptional activator only stabilized in hypoxic cells. In normoxic cells, HIF-1α is degraded via the ubiquitin-proteasome pathway by possession of an ODD. We thus envision that HRE and ODD can be used in combination to achieve hypoxia-inducible expression of ZFP-VP64. We first generated a hypoxia-inducible promoter by placing five tandem copies of HRE (5×HRE) upstream of minimal CMV. The HRE sequence was derived from VEGF gene promoter, shown previously to confer bidirectional gene activation under hypoxic condition.39 Given the potential bidirectionality of 5×HRE, we generated four constructs to examine whether orientation impacts the performance of 5×HRE, and by using mCherry and mCherry-ODD fusion as parallel reporters, to assess the oxygen-sensing efficacy of ODD (online supplemental figure S3A). Only the ODD-modified mCherry reporter exhibited oxygen sensitivity, which, when driven by 5×HRE in 3’ orientation, allowed the lowest basal expression under 21% O2 condition (online supplemental figure S3B,C). Guided by above analyses, coupling to the observation made in cotransfection of ZFP-VP64 expression vector and UASLTR reporter plasmid that the transcriptional activity of ZFP-VP64 can be fulfilled at lower expression level (online supplemental figure S4), we subsequently constructed a hypoxia-inducible ZFP-VP64 expression unit (figure 2A).
Supplemental material
Supplemental material
We first separated ZFP-VP64-ODD (ZPO) transactivator and the response element UASLTR into two different lentiviral vectors and stably cotransduced them into the Jurkat cell line to evaluate hypoxia-driven transcription amplification effect (figure 2A). We also construct a one-step vector where the reporter mCherry gene was driven directly by HRE and LTR minimal chimeric promoter (figure 2C) for comparison of these two methods. Jurkat cells were divided into two groups after lentiviral transduction of two separated plasmids, the first group was placed in a hypoxic chamber (1% O2, 5% CO2 and 94% N2) and the second group of control cells were kept in standard normoxic condition (5% CO2, 21% O2, 74% N2), all of them were maintained in a humidified incubator at 37°C. After 48 hours, we assessed the transcriptional activity by mCherry expression under normoxia and hypoxia. Both groups had very low basal mCherry expression under normoxia but with minimal induction (less than 10%) under hypoxia (figure 2E). We suspected whether the limited transcription amplification was attributed to separated plasmids of the two components, resulting in incoherent expression. Therefore, we construct a bidirectional vector where ZPO gene was placed in 3’ orientation of HRE concatemers and UASLTR response element inserted in in 3’ orientation (figure 2B). This single bidirectional vector allowed for a more coherent expression of transcription factor and transgenes regulation. With this design, western blot showed that the HRE and ODD regulated ZPO was hypoxia-dependent expression, only retained and tested in hypoxia (figure 2D). Furthermore, flow cytometry showed that the majority of transduced cells demonstrated a marked improvement in hypoxia-induced mCherry expression with retaining low background. A comparison of mCherry expression reveals a~20-fold gain with the HiTA system when compared with the one-step system (HRELTR) (figure 2E).
HiTA system mediates ‘on’ and ‘off’ switch of CAR expression in an O2-dependent manner
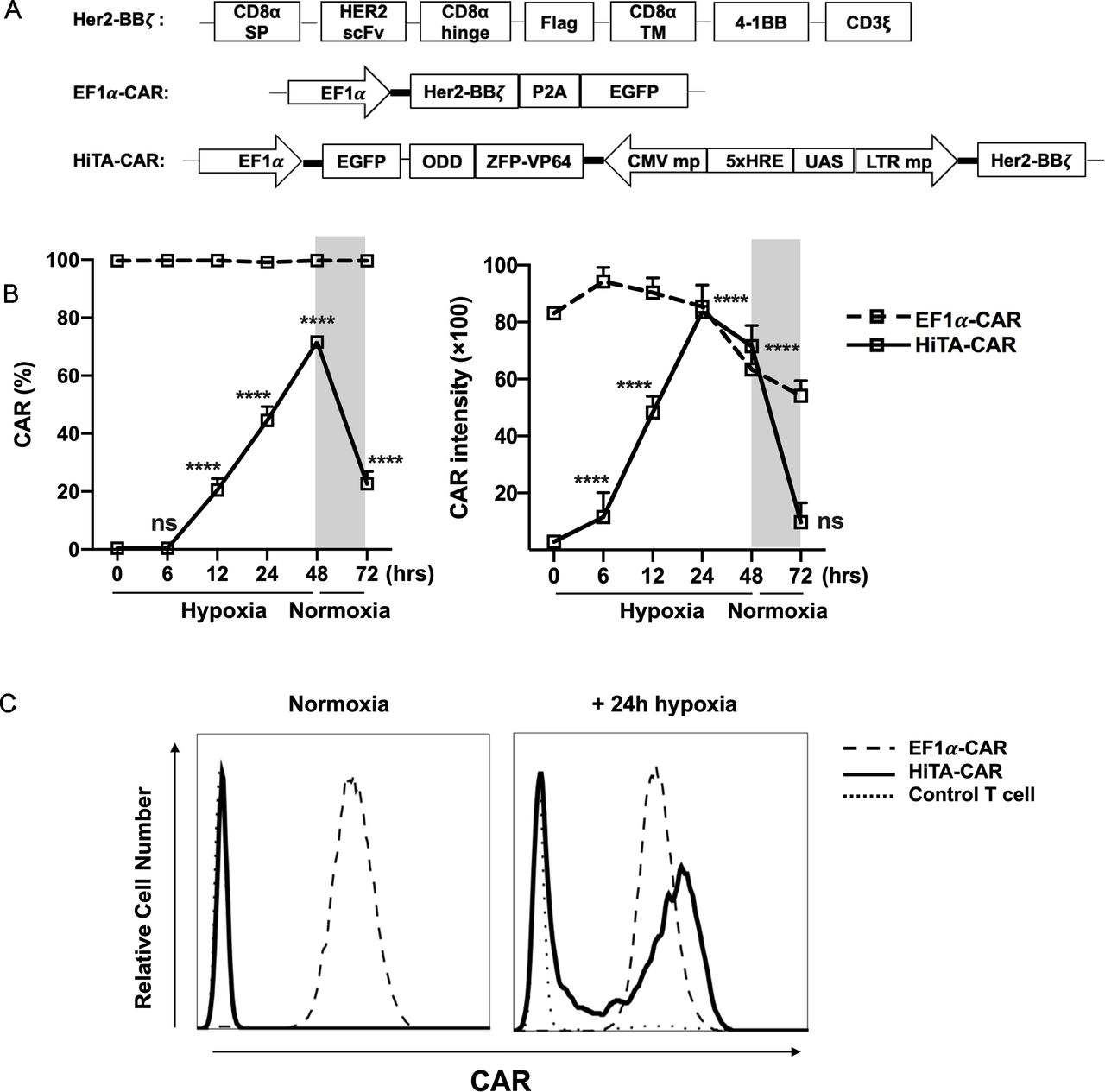
Considering the very low basal expression and transcription amplification of HiTA system, it has significant advantages in designing hypoxia-sensing CAR-T cells. We placed α-Her2 CAR (Her2-BBz) under control of HiTA system and generated a HiTA-CAR (figure 3A). To assess the oxygen sensitivity, we prepared engineered T cells stably transduced with HiTA-CAR, and EF1α-CAR as a positive control. The surface CAR expression of HiTA-CAR was hypoxia-dependent, showing upregulation in 1% O2 and downregulation when returned to normoxia condition. Our results confirmed a highly dynamic and stringent on-off switch by oxygen concentration, with hypoxia-inducible 18.2-fold CAR expression rate and 16.8-fold CAR intensity upregulation. Although ~20% CAR expression was detectable 24 hours after returning to normoxia, the CAR intensity has already returned to the basal level. When EF1α-CAR was compared with the HiTA-CAR, the CAR intensity in the HiTA system was observed similar expression level as that driven by the EF1α promoter after 24 hours hypoxia treatment (figure 3B–C). Besides, the reduced oxygen level significantly increased CAR induction frequency and expression intensity of HiTA-CAR, confirming its excellent oxygen sensitivity (online supplemental figure S5).
Supplemental material

Hypoxia-dependent ON-OFF kinetic characteristic of CAR expression in HiTA-CAR-T cells. (A) Diagram of EF1α-CAR and HiTA-CAR constructs. The Her2-BBξ construct contained an internal Flag tag for the detection of CAR expression. Both EF1α-CAR and HiTA-CAR constructs included an EGFP reporter gene under the control of the constitutively active EF1α promoter for marking the transduced cells. (B) EF1α-CAR, HiTA-CAR and untransduced control T cells were cultured in normoxic or hypoxic (1% O2) conditions for 24 hours. (C) Example histograms of surface CAR expression on CD3 +T cells were assessed using flow cytometry. (C) Surface CAR expression on EF1α-CAR and HiTA-CAR at the indicated time under hypoxia (1% O2) or normoxia were assessed using flow cytometry. (n=3 donors, mean±SD of each time point compared with 0 hour in HiTA-CAR is shown, ****p<0.0001, ns: not significant, analyzed using one-way ANOVA). ANOVA, analysis of variance; CAR, chimeric antigen receptor; CMV, cytomegalovirus; EF1α, eukaryotic elongation factor 1-α; EGFP, enhanced green fluorescent protein; HiTA, hypoxia-inducible transcription amplification; ODD, oxygen-dependent degradation domain; UAS, upstream activating sequence; ZFP, zinc finger protein.
HiTA-CAR-T cells exhibited hypoxia-restricted effector functions
To determine whether HiTA-CAR-T cells effector functions were oxygen-regulated, SKOV3 ovarian cancer cells were seeded into culture plates and coincubated with EF1α-CAR or HiTA-CAR-T cells from five donors under normoxic and hypoxic conditions. After 24 hours incubation, we tested surface CAR expression and cytokines secretion of CAR-T cells. The results demonstrated a very stringent hypoxia-restricted CAR expression with nearly no detectable CAR expression under normoxia, but different induction levels observed in different donors because of individual variation under hypoxia (figure 4A). The magnitude of induction by hypoxia was ~300-fold in CAR frequency and ~30-fold in CAR intensity (figure 4B,C). Consistently, IL-2 and IFN-γ, the two functional cytokines related to T cell response,43 44 were found little secretion under normoxia and hypoxia-inducible secretion to different degree (figure 4D). Besides, when coincubated with SKOV3, HiTA-CAR-T cells displayed efficient hypoxia-dependent cytotoxicity against the SKOV3 cells, almost equivalent to EF1α-CAR-T cells at the E/T ratio of 2:1, with no significant killing observed in normoxic conditions. In contrast, the control EF1α-CAR-T cells presented similar cytolytic activities under hypoxia and normoxia (figure 4E). These results confirmed oxygen-controlled function of HiTA-CAR-T cells in vitro.

Stringent oxygen control of effector functions of HiTA-CAR-T cells. HiTA-CAR T cells were cocultured with SKOV3 cells in hypoxic and normoxic conditions for 24 hours at E/T ratio of 3:1 (n=5 donors) (A–D). (A) The Surface CAR expression on HiTA-CAR T cells were assessed using flow cytometry. (B, C) Statistical results of Her2 expression intensity of HiTA-CAR-T cells. (D) Quantification of supernatant IL-2 and IFN-γ releases from HiTA-CAR-T and untransduced control T cell(n=3, mean±SD is shown, ***p<0.001, ****p<0.0001, analyzed using Student’s t-test). (E) In vitro cytotoxicity of EF1α-CAR and HiTA-CAR-T cells to SKOV3 tumor cells in indicated E/T ratios in normoxic and hypoxic conditions after 48 hours coincubation. (Bar on charts shows mean±SD of technical triplicates. In line charts, the dots mark means of three independent experiments (n=3 donors) and error bars displayed as the mean±SD, *p<0.05, analyzed using two-way ANOVA). ANOVA, analysis of variance; CAR, chimeric antigen receptor; EF1α, eukaryotic elongation factor 1-α; E/T, effector/target; IFN-γ, interferon γ; IL-2, interleukin 2; HiTA, hypoxia-inducible transcription amplification.
HiTA-CAR-T cells exhibited enhanced safety without on-target liver toxicity
Having validated the hypoxia-restricted activation and cytotoxicity in vitro, we next sought to evaluate safety of HiTA-CAR-T cells in vivo. We have established a humanized mouse model where human target antigens (eg, Her2) were selectively expressed in mouse liver (unpublished data). The murine liver was transduced to coexpress human Her2 antigen and luciferase by i.p. injection of Ad5-Her2. The expression of luciferase in murine hepatocytes was detected using bioluminescent imaging (BLI) at day 8 post-infection, indicating the distribution of Her2, and then mice with similar luciferase expression level were randomly divided into four groups (figure 5A).

HiTA-CAR-T-cells demonstrated no systemic toxicity to mice with human Her2 antigen on hepatocytes. (A) Overview of the experimental design for evaluating safety of HiTA-CAR-T cells in human Her2-expressing mice. All mice (except mice in Her2-, EF1α-CAR group) were injected (i.p.) with 1×109 PFU Ad5-Her2 and received 5×106 transduced (GPF+) T cells (intravenous) at day 8. Mice that had hepatic Her2 expression (+Ad5-Her2) but infused with vehicle, control T cells (untransduced T cells), or mice that received EF1α-CAR-T cells but lacked hepatic Her2 expression (- Ad5-Her2) served as negative control. Mice that had hepatic Her2 expression but infused with EF1α-CAR T cells served as positive control. (B) Liver damage was determined by serum ALT and AST levels collected at indicated time points. Weight change shown by percent change from initial weight (n=5–8, mean±SD is shown, a one-way ANOVA test was performed, and comparisons are shown between the HiTA-CAR and EF1α-CAR groups, *p<0.5, **p<0.01, ***p<0.001). (C) Detection of the kinetic expression of Her2 in hepatocytes. Her2 expression was monitored by anti-Her2 immunofluorescence staining of paraffin-embedded liver section slides at day 0/3/7 post T cells transfer separately. (D) The statistical results of Her2 +hepatocytes in different groups (n=3, mean±SD is shown, ***p<0.001, ****p<0.0001, analyzed using Student’s t-test). (E) Systemic cytokine release by T cells 7 days post T cells transfer was detected in mouse serum by CBA assay (left) (n=5–8, mean±SD is shown, a one-way ANOVA test was performed, and comparisons are shown between the HiTA-CAR and EF1α-CAR groups, ***p<0.001). Representative hepatic tissue slides by HE staining. Black arrows indicate inflammatory lesions (right). (F) The statistical results of liver inflammatory lesioned proportion of mice in different groups (n=5, mean±SD is shown, ****p<0.0001, analyzed using Student’s t-test). ALT, alanine aminotransferase; ANOVA, analysis of variance; AST, aspartate aminotransferase; CAR, chimeric antigen receptor; EF1α, eukaryotic elongation factor 1-α; HiTA, hypoxia-inducible transcription amplification; i.p., intraperitoneally; i.v., intravenous; CBA, cytometric bead array.
To compare the on-target toxicity to normal liver, EF1α-CAR-T cells were included and served as a positive control, which resulted in fatal on-target normal tissues toxicity.19 We postulated that our HiTA-CAR-T cells would exhibit less toxicity to mice compared with EF1α-CAR-T cells, since there is no CAR induction in normoxic condition although in the presence of antigens (figure 4A). We also included a Her2-negative control group that received EF1α-CAR-T cells, and a negative control group that expressed Her2 but received UTD to detect a Her2-independent T cell response. As predicted, mice infused with EF1α-CAR-T cells resulted in severe liver damage, evident by elevated serum ALT and AST levels, and a rapid loss of body weight. In contrast, mouse ALT and AST levels and weight change in HiTA-CAR-T group were similar to other negative control groups after T cells infusion (figure 5B). we monitored the expression of Her2 in hepatocytes at day 0, 3, and 7 post T cell transfer, equivalent to day 7, 10, and 14 after adenovirus inoculation, by immunofluorescence. The result indicated similar kinetics of Her2 expression in the hepatocytes among vehicle, control T and HiTA-CAR groups: the number of Her2-positive cells were gradually decreased during the 7-day observation period, starting with approximately 60% while ending with about 40%. In sharp contrast, the declining of Her2-positive hepatocytes was significantly more dramatic for EF1α-CAR group, consistent with effective targeted killing by EF1α-CAR T cells (figure 5C,D). Besides, the result of BLI kinetics revealed that the HiTA-CAR group, like the control T group, retained a strong bioluminescent signal by day 7 while the signal in the EF1α-CAR group was substantially reduced by day 3, and could not be detected by day 7, in line with the elimination of luciferase+ (surrogate of Her2+) hepatocytes by EF1α-CAR-T cells (online supplemental figure S6). Besides, only Her2-expressing mice received EF1α-CAR-T cells had elevated T cell activation-associated cytokines, including human IL-2, IFN-γ and TNF-α (figure 5E, left) in serum collected at day 7. Nine days after T cells infusion, the mice were sacrificed, and the H&E staining of hepatic tissue slides showed an obvious infiltration of myeloid cells distributed throughout liver, especially around blood vessels in EF1α-CAR-T cells treated group compared with other groups, which revealed the CAR-mediated inflammation in the liver expressing target antigens (figure 5E, right; Figure 5F). T cells from enzyme-digested spleen, lung or liver tissues and blood were isolated and analyzed for surface CAR expression by flow cytometry. Importantly, there was no detectable surface CAR expression on HiTA-CAR-T cells in blood and tissues, compared with nearly 100% CAR expression on EF1α-CAR T cells (figure 6A; online supplemental figure S7). Notably, the ratio of CAR+ (namely GFP+) EF1α-CAR-T preparation increased from 50% to about 100% 9 days after infusion, mainly due to antigen-specific expansion. However, CAR +HiTA-CAR-T cells retained similar ratio (figure 6A). In addition, immunofluorescence staining of liver tissues demonstrated that robust CAR-T cells only detected in Her2-expressing murine liver of EF1α-CAR-T cells treated group (figure 6B). Overall, compared with the conventional EF1α CAR-T cells, our HiTA-CAR-T cells exhibited enhanced safety without systemic toxicity and on-target toxicity to normoxic liver in vivo.
Supplemental material
Supplemental material

Analysis of CAR-T cells from different tissues. (A) Representative contour plot of CAR-T preparation in vitro and live nucleated cells (Pacific Blue), CD45+ (PerCP-Cy5-5) and CD8+ (APC) CAR-T cells from the indicated enzyme-dispersed tissues and blood, the GFP+ CAR-T cells and surface CAR expression (PE) were analyzed by using flow cytometry (left). The statistical data of CAR expression rate of HiTA and EF1α- CAR-T cells in vitro and from indicated mice tissues (right) (n=3, mean±SD is shown, ****p<0.0001, analyzed using Student’s t-test). (B) Immunofluorescence staining of CD3 and CAR molecules expressed on T cells in liver tissue. White arrows indicate CD3+ CAR+ T cells. CAR, chimeric antigen receptor; EF1α, eukaryotic elongation factor 1-α; HiTA, hypoxia-inducible transcription amplification.
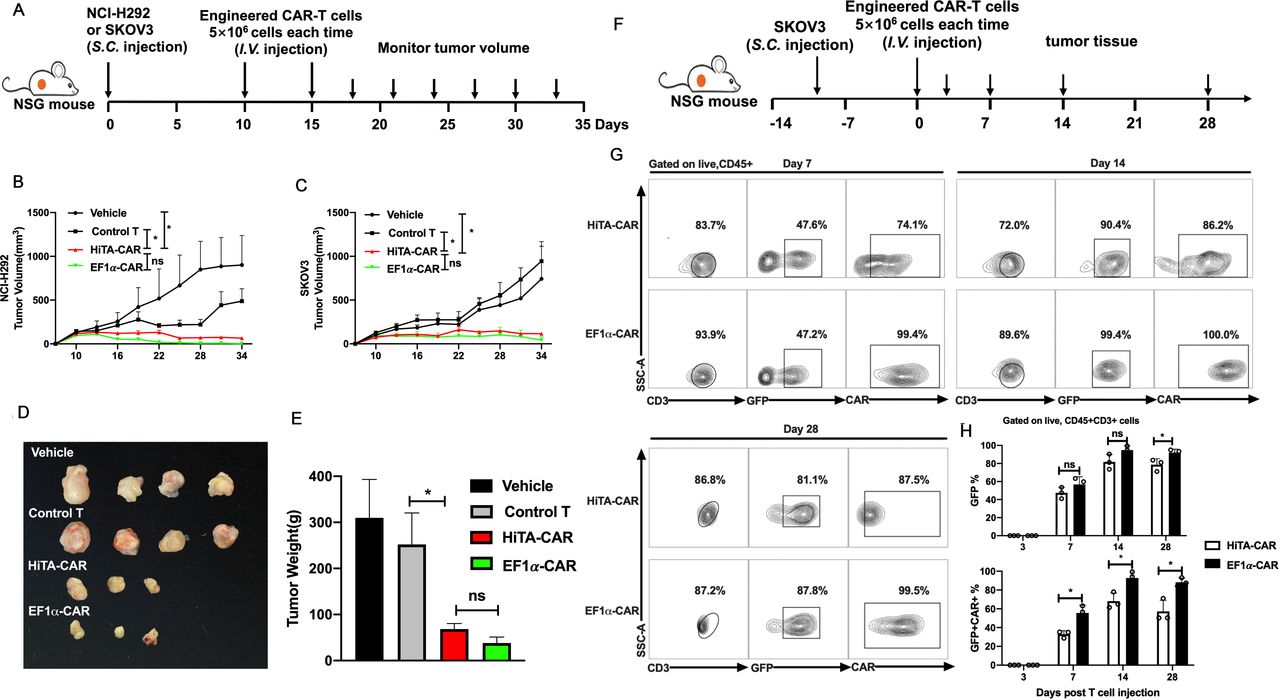
HiTA-CAR-T-cells demonstrated antitumor efficacy in vivo. (A) Overview of the experimental design for confirming the tumoricidal effect of HiTA-CAR- T cells to Her2+ tumor in vivo. Mice were implanted with 2×106 Her2 +SKOV3 tumor cells or 5×106 HER2 +NCI-H292 tumor cells. Ten and fifteen days post subcutaneous tumor cells inoculation, mice were infused (intravenous) with either vehicle or 5×106 transduced (GFP+) HiTA-CAR or non-transduced control T cells. The NCI-H292 (B) and SKOV3 (C) tumor growth curves at different time points are presented and the significant differences between HiTA-CAR-T cells and control T cells or vehicle were analyzed (n=4, mean±SEM is shown, *p<0.05, ns: not significant, analyzed using two-way ANOVA). SKOV3 tumors were isolated from mice, observed for size (D) and weighted (E) at day 34. (F) Overview of the experimental design for confirming the migration and accumulation of HiTA-CAR- T cells to SKOV3 tumor in vivo. Mice were implanted with 2×106 HER2 +SKOV3 tumor cells. Ten days postsubcutaneous tumor cells inoculation, mice were infused (intravenous) with either vehicle or 5×106 transduced (GFP+) HiTA-CAR-T cells. (G) Representative contour plot of tumor-infiltrating CAR-T cells. (H) The statistical data of GFP+ and GFP+CAR+ tumor-infiltrating CAR-T cells at different time point were analyzed using flow cytometry (n=3, mean±SD is shown, *p<0.05, ns: not significant, analyzed using Student’s t-test). ANOVA, analysis of variance; CAR, chimeric antigen receptor; HiTA, hypoxia-inducible transcription amplification.
HiTA-CAR-T cells exhibit vigorous antitumor efficacy
To test the antitumor efficacy of HiTA-CAR-T cells, two tumor cell lines with different Her2 antigen expression levels-high densities of antigen-expressing SKOV3 and NCI-H292 expressing low levels of antigen, were selected as representative Her2-expressing tumors and were injected s.c. into NSG mice. HiTA-CAR-T cells, EF1α-CAR-T cells, untransduced control cells or vehicle were administered when tumor volumes reached approximately 150 mm3 in volume and were proved to be hypoxic (online supplemental figure S8). These tumor-bearing mice were injected (intravenous) with 5×106 CAR-T cells on day 10 and day 15 and serial tumor size assessments were conducted (figure 7A). The progressive tumor growth of NCI-H292 and SKOV3 were observed in the mice treated with untransduced control cells or vehicle. In contrast, mice treated with HiTA-CAR-T cells in both xenograft models exhibited excellent anti-tumor effect, showing no significant difference with EF1α-CAR-T cells. (figure 7B,C). At day 34, mice were sacrificed and the SKOV3 tumors were harvested, weighted and analyzed for the infiltration and CAR expression of T cells. Tumors from HiTA-CAR-T and EF1α-CAR-T cells treated mice showed significantly smaller size and lighter weight than that from mice received control T cells (figure 7D,E). To explore the kinetic accumulation of CAR-T at tumor site, mice with SKOV3 tumors were sacrificed 3/7/14/28 days post T cells infusion.(figure 7F). CAR +HiTA-CAR-T and EF1α-CAR-T were undetectable on day 3. CAR frequency and intensity of HiTA-CAR-T cells increased from day 7 to day 14 by the hypoxic tumor microenvironment (figure 7G). The proportion of tumor-infiltrated CAR +T cells rose from day 7 to day 14 due to migration and proliferation, and then felled from day 14 to day 28, which may result from tumor shrinkage and reduced CAR-T expansion (figure 7H). All together, these results indicated HiTA-CAR-T cells provided excellent antitumor effect in vivo.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
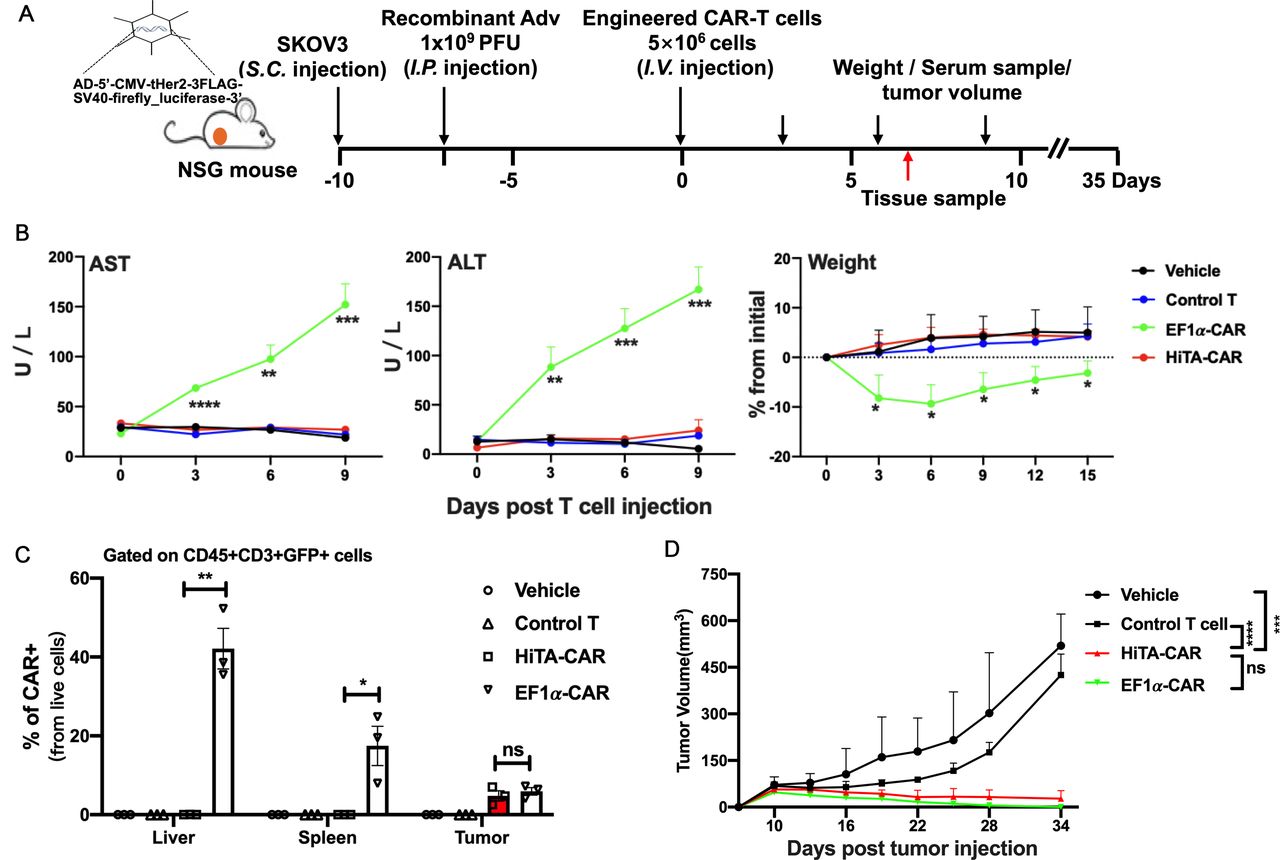
HiTA-CAR T-cells demonstrated anti-tumor efficacy without on-target off-tumor toxicity in vivo. (A) Overview of the experimental design for confirming the tumoricidal effect as well as reduced toxicity of HiTA-CAR- T cells in vivo. Mice were implanted with 1.5×106 HER2 +SKOV3 tumor cells. 1×109 PFU Ad5-Her2 was injected (i.p.) 7 days before 5×106 transduced (GPF+) T cells infusion (intravenous) (B) Liver damage was determined by serum ALT and AST levels collected at indicated time points. Weight change shown by percent change from initial weight (n=5, mean±SD is shown, a one-way ANOVA test was performed, and comparisons are shown between the HiTA-CAR and EF1α-CAR groups, *p<0.5, **p<0.01, ***p<0.001, ****p<0.0001). (C) The statistical results of proportion of CAR+ T cells in liver, spleen, and tumor tissues on day 7. (n=3, mean±SD is shown, *p<0.05, **p<0.01, ns: not significant analyzed using Student’s t-test). (D) The SKOV3 tumor growth curves are presented and the significant differences between HiTA-CAR group with other groups were analyzed (n=5, mean±SEM is shown, ***p<0.001, ****p<0.0001, ns: not significant, analyzed using two-way ANOVA). ALT, alanine aminotransferase; ANOVA, analysis of variance; AST, aspartate aminotransferase; CAR, chimeric antigen receptor; EF1α, eukaryotic elongation factor 1-α; HiTA, hypoxia-inducible transcription amplification; i.p., intraperitoneally; i.v., intravenous; s.c., subcutaneously.
HiTA-CAR-T suppressed tumor growth without on-target off-tumor toxicity
We established a mouse model that is more correlative to clinical cancer patients by injecting Adv-Her2 to SKOV3 tumor bearing mice, to explored both anti-tumor efficacy and safety of HiTA-CAR-T cells (figure 8A). Mice in HiTA-CAR-T group revealed no severe liver damage or weight loss (figure 8B), which was consistent with the result in figure 5B. Quantification of CAR +T cells in liver, spleen, and tumor at day 7 post infusion by flow cytometry clearly indicated that the induction of CAR expression in HiTA-CAR-T cells was restricted to tumor site, with the frequency of resultant CAR+ cells similar to that detected in tumors of EF1α-CAR group (figure 8C). Importantly, as expected, HiTA-CAR-T cells demonstrated excellent antitumor effect comparable to EF1α-CAR-T cells (figure 8D). Collectively, these data strengthened the notion that HiTA-CAR-T specifically target hypoxic tumor with minimal off-tumor toxicity.
Discussion
Here, we developed a HiTA system, which could be used for hypoxia-dependent control of CAR expression in T cells and thereby of their effector functions. Our developed HiTA system has several advantages: First, it incorporated an optimized artificial zinc finger transcriptional system. Compared with the most widely used eukaryotic transcriptional regulatory system-yeast Gal4/UAS system,31 our refined minimal LTR promoter and designed ZFP7-VP64 transactivator demonstrated lower basal activity and higher transcriptional induction level compared with minimal CMV promoter and Gal4-VP64 transactivator, respectively. Second, HRE concatemers together with ODD provided a double-sensing effect of hypoxia. ZFP7-VP64 regulated by both HRE and ODD confirmed excellent oxygen sensitivity and transcriptional amplification of target genes, compared with that driven by HRE promoter alone. Third, many researchers constructed transgenes fused with ODD domain to achieve hypoxia regulation. However, the function of transgenes, especially for small molecules, may be affected by the large ODD molecules. Target gene in our HiTA system was regulated by transcriptional process but not the protein level, thereby retains function. In addition, we constructed a refined bidirectional lentiviral vector to incorporate trans-activator and response element. Although many studies have relied on cotransduction of cells with separate plasmids, gene expression mediated by the dual vector system is difficult to operate because it strictly depends on two vectors entering one cell at once and requires precise control of the ratio of two vectors. Our results showed a limited induction when the ZPO-HRE trans-activator and response element were split into two vectors and cotransduced into Jurkat cells. Moreover, in the practical application for gene therapy, the dose of virus administrated to patients should be as low as possible, in order to reduce the risk of insertion mutation and oncogene activation caused by retrovirus/lentivirus integration. Therefore, our development of HiTA system with one bidirectional vector possesses advantages in applications for its cost-effective preparation and for its expression efficacy. However, this two-in-one vector system also had difficulty in lentivirus packaging due to the limited capacity of cargo size.45 This problem may be solved by using transposon system with larger transgene cargo size in the future.46 47
Adoptive CAR-T cell therapy can cause fatal on-target off-tumor toxicity to normal tissues,19 besides, off-tumor targeting can cause CAR-T cells to be sequestered and delayed from reaching their intended tumor targets, thus weaken the antitumor effect, as demonstrated in a recent study.48 Recent years, the hypoxia-regulated CAR fused with an ODD showed high background under normoxia and limited induction in hypoxic conditions,24 25 thus demonstrating the inability of ODD alone to achieve precise hypoxia-regulation as a single oxygen sensor. Our previous work improved the ODD-regulated HiCAR by adding HREs into upstream of EF1α promoter to generate a chimeric HRE-EF1α promoter (chHE).25 The improved chHE-HiCAR demonstrated enhanced CAR induction level, but also observed leaky CAR expression and residual cytotoxicity under normoxia. To further improve the design of HiCAR, we constructed HiTA-CAR-T cells for selective tumor targeting as this system showed stringent hypoxia-regulation of target genes by HiTA system and by the hypoxic tumor environment. As expected, our HiTA-CAR-T cells showed highly hypoxia-restricted CAR expression, cytokines secretion and cytotoxicity. Importantly, the adoptively transferred HiTA-CAR-T cells suppressed the growth of NCI-H292 and SKOV3 tumor xenografts, implicating its antitumor efficacy in vivo.
To rapidly evaluate on-target off-tumor toxicity of HiTA-CAR-T cells, we generated a mouse model that expresses human Her2 antigen on hepatocytes by i.p injection of Her2-expressing type 5 adenovirus (Ad5-Her2) (unpublished data). We considered it as a reasonable tool to assess off-tumor toxicity because it has CAR-targeted human expression on normal liver tissues, which is overlapped expressed on tumor cells. Recently, Castellarin et al have developed a similar mouse model to test on-target off-tumor toxicity of affinity-tuned CAR-T cells that recognize human Her2 antigens in normal murine livers by recombinant adenovirus-associated virus and transposon system.48 As expected, EF1α-CAR-T cells caused severe liver damage, as evidenced by elevated ALT and AST in mouse serum, and an obvious weight loss, suggesting on-target liver toxicity. In contrast, mice injected with HiTA-CAR-T cells did not show any significant toxicity as shown in mice received EF1α-CAR-T cells, which is further proved by the absence of surface CAR expression on HiTA-CAR-T cells from different tissues, including spleen, blood, lung and Her2 expressing liver. Overall, these data demonstrated that HiTA-CAR-T cells are highly conditioned by hypoxia and may provide a safe platform for CAR-T cell-mediated immunotherapy.
Our HiTA system could be adapted to drive additional immunomodulators, such as IL-12 which may cause side effect when expressed in normal tissues, in hypoxia tumor microenvironment or incorporated into oncolytic virus to express hypoxia-induced effectors to kill tumor cells.49 50 In addition, we may integrate cancer-specific promoters with HRE to create dual specific expression systems for increased tumor specificity, as demonstrated by a combination of cancer-specific promoter with HREs in a previous study.51 Moreover, the combination with other promoters of hypoxia up-regulated gene, like glyceraldehyde-3-phosphate dehydrogenase (GAPDH)29 and VEGF,52 as an enhancer for gene expression in the future.
In summary, we established a highly effective HiTA system for stringent gene control to specific hypoxic conditions in this study. HiTA-CAR-T cells provided tumoricidal efficacy both in vitro and in vivo in the absence of on-target toxicity to mouse normal liver expressing human antigen. Hypoxia is one of the most common characteristics of solid tumors, our HiTA system could be applied to regulate CARs specific to diverse tumor antigens or any immunomodulators in tumor lesions. Our study provided a new promising strategy to balance the safety and efficacy of cancer immunotherapy or other diseases that require precise hypoxia-targeted delivery as a supplement to the existing therapy approaches.
Supplemental material
Data availability statement
All supporting data are included in the manuscript and the online supplemental materials. Additional data are available on reasonable request to corresponding author.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the Ethics Committee of Shanghai Public health Clinical Center. All animal experiments are conducted in accordance with the principles and procedures.
Acknowledgments
We thank Chenli Qiu, Peng Sun for their help in flow cytometry. We thank Linxia Zhang, Lingyan Zhu for their advice in performing experiment. We also thank Animal Center of Shanghai Public health Clinical Center and its staff for their help in animal feeding and animal model construction.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
HH and QL contributed equally.
Contributors JX and XZ conceived and designed this project and supervised the experiments. MY supervised the experiments. HH and QL performed the analysis. HH, MF performed the in vitro experiments. HH, QL performed the in vivo experiments with the help of LJ, ZL, LZ and XD. C.Zhao helped writing and revising the manuscript. C.Zhu performed the IHC & IF experiments. All authors read and approved the final manuscript.
Funding This work was supported by the National Key Research and Development Program (2016YFC1303402), National 13th Grand Program on Key Infectious Disease Control(2017ZX10202102-006),Clinical Research Plan of SHDC (SHDC2020CR3002A & SHDC12018112) and the Intramural Funding from Shanghai Public Health Clinical Center (KY-GW-2019-16).
Competing interests The authors declare that they have no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.