Article Text

Abstract
Epithelial ovarian carcinoma (EOC) is a relatively rare malignancy but is the fifth-leading cause of cancer-related death in women, largely reflecting early, prediagnosis dissemination of malignant disease to the peritoneum. At odds with other neoplasms, EOC is virtually insensitive to immune checkpoint inhibitors, correlating with a tumor microenvironment that exhibits poor infiltration by immune cells and active immunosuppression. Here, we comparatively summarize the humoral and cellular features of primary and metastatic EOC, comparatively analyze their impact on disease outcome, and propose measures to alter them in support of treatment sensitivity and superior patient survival.
- immunologic surveillance
- immunotherapy
- tumor biomarkers
- tumor microenvironment
- genital neoplasms
- female
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Epithelial ovarian carcinoma (EOC) is among the top five causes of cancer-related death in women.1 Indeed, while EOC is relatively rare (it accounts for approximately 2% of all malignancies affecting women, basal cell and squamous cell skin cancers excluded), prognosis is particularly poor as most cases are diagnosed as late-stage invasive disease.1 EOC is traditionally classified into five histological subtypes: high-grade serous ovarian carcinoma (HGSOC), low-grade serous ovarian carcinoma (LGSOC), mucinous ovarian carcinoma (MOC), endometrioid ovarian carcinoma (EnOC), and ovarian clear cell carcinoma (OCCC), each of which has different cellular origins and molecular profiles.2 The immune contexture and density of tumor-infiltrating T lymphocytes (TILs) vary considerably among different EOCs, being highest in HGSOCs, intermediate in EnOCs, and lowest in LGSOCs, MOCs, and OCCCs.3 Consistent with a role for TIL in EOC progression, a recent study on more than 5900 advanced EOC patients demonstrated a markedly higher risk of mortality for women with MOC and OCCC subtypes as compared with patients with HGSOC and EnOC.4 HGSOC, in which epithelial ovarian cells or secretory cells are present in the mucosa of fallopian tubes, is the most common and aggressive form of EOC.2 Poor outcomes in HGSOC are largely dictated by early dissemination to the peritoneal cavity, especially the omentum, resulting in the formation of metastatic lesions and malignant ascites that ultimately resist currently approved therapeutic strategies.2 A recent study harnessing single-cell RNAseq has provided an even more in-depth resolution of HGSOC, identifying multiple subtypes with differential disease outcome.5 6
The majority of women with EOC achieve indeed complete remission after primary or interval cytoreductive surgery combined with chemotherapy based on a platinum-taxane doublet. Homologous recombination (HR) defects imposed by germline or somatic BRCA1 DNA repair-associated (BRCA1) or BRCA2 mutations are not only key determinants of platinum sensitivity in EOC patients but also provide a strong rationale for maintenance therapy based on poly(ADP-ribose) polymerase (PARP) inhibitors, which is generally associated with improved progression-free survival (PFS).7 Nonetheless, more than 50% of women affected by EOC ultimately experience recurrence with treatment-resistant disease and succumb within 5 years of diagnosis, calling for the urgent development of novel therapeutic approaches to this deadly malignancy.
Successful introduction of immune checkpoint inhibitors (ICIs) for the treatment of multiple tumor types has created enormous expectations around the possibility of harnessing the patient’s own immune system against EOC.8 9 However, compared with other neoplasms, such as non-small cell lung carcinoma and melanoma, EOC is poorly sensitive to ICIs employed as standalone immunotherapeutic agents,9 most likely due to indolent anticancer immunity and robust immunosuppression at baseline.10 In this context, a strongly immunosuppressive tumor microenvironment (TME) may considerably contribute to disease progression and metastatic dissemination, calling for the implementation of combinatorial immunotherapeutic strategies beyond immune checkpoint inhibition.11 Indeed, additional mechanisms appear to be crucial for the generation of an immunosuppressive contexture in EOC, including increased levels of immunomodulatory cytokines, enzymes and metabolites including interleukin (IL)-6, IL-10, vascular endothelial growth factor A (VEGFA), macrophage migration inhibitory factor (MIF), indoleamine 2,3-dioxygenase 1 (IDO1), arginase 1 (ARG1), and lactate.12–15 These factors increase along with disease progression, paralleling the accumulation of immunosuppressive cell types such as CD4+CD25+forkhead box P3 (FOXP3+) regulatory T (TREG) cells, tumor-associated macrophages (TAMs), tolerogenic dendritic cells (DCs), and myeloid-derived suppressor cells (MDSCs).16–19 Thus, multiple immunosuppressive factors quench anticancer immunity in the TME of EOCs, hence representing potential therapeutic targets for drug development.
Importantly, recent technological developments, including modern genomic, transcriptomic and phenotypic assays at single-cell resolution, have provided an in-depth characterization of the cellular and humoral immune contexture of EOC and its impact on disease outcomes. If validated by independent prospective clinical studies, these immunological biomarkers may not only assist in determining the response of patients with EOC to treatment but also enable the adoption of personalized treatment approaches with superior likelihood for success. Here, we present key immunological features of primary versus metastatic EOC and critically discuss their potential value as prognostic and predictive biomarkers.
The immune microenvironment in ovarian carcinoma
The immunological contexture of both primary and metastatic EOC lesions builds on a complex network of immune and non-immune cells that interact, both physically and via soluble mediators, with each other, with malignant cells and with the extracellular matrix (figure 1.20–22 Importantly, while some immune cells, such as T lymphocytes, B cells and DCs, can be found within EOC cell nests, many cellular components of the immune system, such as MDSCs, natural killer (NK) cells, mast cells and neutrophils, are primarily localized at the invasive margin.21 Moreover, it became clear that the immunological configurations of primary and metastatic EOC differ considerably from each other,20 23 as well as that the degree of primary and metastatic EOC infiltration by immune cells exhibit considerable heterogeneity across patients.24–26 In this setting, analytical approaches that go beyond the mere estimation of cellular density in diagnostic biopsies to include spatial localization and functional orientation of the immune compartment of metastatic EOC have identified components of the EOC immune contexture that are linked to improved disease outcome, as discussed here below.10 27

Principle of cancer immunosurveillance in primary and metastatic ovarian carcinoma. Primary immune cell populations, cytokines and chemokines involved in the interaction between primary and metastatic ovarian carcinoma and the host immune system. ARG1, arginase 1; CCL, chemokine (C-C motif) ligand; CXCL, chemokine (C-X-C motif) ligand; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; GITR, glucocorticoid-induced TNFR related gene; GZMB, granzyme B; IDO1, indoleamine 2,3-dioxygenase 1; IFNG, interferon gamma; IL, interleukin; LAG3, lymphocyte activation gene 3; mDCs, myeloid dendritic cells; MDSCs, myeloid-derived suppressor cells; NK, natural killer; PD-1, programmed cell death 1; PD–L1, programmed death ligand 1; pDC, plasmacytoid dendritic cell; PGE2, prostaglandin E2; PRF1, perforin 1; TAM, tumor-associated macrophage; TIM-3, coinhibitory receptor hepatitis A virus cellular receptor 2 (HAVCR2, best known as Tim-3); TGFB1, transforming growth factor beta 1; TLS, tertiary lymphoid structure; TNF, tumor necrosis factor; TREG, regulatory T cell; VEGFA, vascular endothelial growth factor A.
T lymphocytes
An elevated number of T lymphocytes infiltrating the tumor core or stroma has been linked to favorable prognosis in a large panel of malignancies.10 28 In line with this notion, a high density of CD3+ T cells in either primary or metastatic tumor biopsies has been attributed to independent prognostic value for improved PFS and overall survival (OS) in numerous cohorts of women with EOC (table 1).3 21 29–40 Importantly, such a positive prognostic impact is primarily associated with the expression of CD8+ T cell memory markers, markers of CD4+ TH1 polarization, and TIL localization to EOC islets rather than stromal areas.29 41 42 Moreover, although CD8+ T cell density appears to be increased in metastatic EOC samples compared with their primary counterparts,20 23 the abundance of CD8+ T cells in either compartment retains prognostic value.29 43
Prognostic relevance of T lymphocytes, dendritic cells (DCs), tertiary lymphoid structures (TLSs) and B cells in primary and metastatic ovarian carcinoma
A retrospective analysis of the immune landscape of more than 7000 EOC samples encompassing all major histological subtypes (HGSOC, LGSOC, MOC, EnOC, and OCCC) revealed that the strong positive prognostic impact of CD8+ TILs is limited to HGSOC, MOC, and EnOC, but not LGSOC and OCCC.44 Intriguingly, HGSOC generally harbors the most dense immune infiltrate as compared with other EOC subtypes.44 These findings have largely been recapitulated by independent investigators.41 Of note, abundant CD8+ T cell infiltration was linked to favorable disease outcome, regardless of residual disease, therapeutic strategy, or germline BRCA1 mutations.44 Similar results were obtained in a meta-analysis encompassing the results of 10 previously published studies, ultimately including a total of 1815 patients encompassing all EOC histologies.28
Although some studies have attributed a negative prognostic value to robust EOC infiltration by TILs expressing programmed cell death 1 (PDCD1, best known as PD-1) or cytotoxic T lymphocyte-associated protein 4 (CTLA4), potentially linked to PD-1- or CTLA-4-dependent T cell exhaustion,35 other studies have revealed a rather beneficial prognosis, possibly because an elevated number of PD-1+ cells correlates with abundant tumor infiltration by T cells altogether45 or because a subset of CD8+PD-1+ T cells expressing integrin subunit alpha E (ITGAE, best known as CD103) retain functional competence in the ovarian TME.33 In support of this possibility, a subset of CD8+CD103+ T cells that was preferentially localized at epithelial tumor regions and expressed cytotoxic molecules has been significantly correlated with improved disease outcome in HGSOC patients.33 46 47 Conversely, CD8+ cytotoxic T lymphocytes (CTLs) expressing the coinhibitory receptor hepatitis A virus cellular receptor 2 (best known as TIM-3) exhibit bona fide features of functional exhaustion, and their abundance has been associated with poor disease outcome, indicating that TIM-3 plays a prominent role in limiting immune responses against HGSOC (table 1).45 In line with this notion, recent studies have identified a key role for the coexpression of various coinhibitory receptors for T cell exhaustion/dysfunction.48 49 Specifically, IL-27 was shown to drive a transcription program that promotes the coexpression of PD-1 and TIM-3, as well as lymphocyte activating 3 (LAG3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT).48 Taken together, these findings identify a potential interaction between TIM-3 and other coinhibitory receptors that may be relevant for the establishment of robust immunosuppression in EOCs. In line with this possibility, coblockade of TIM-3 and PD-1 has been linked to tumor regression and improved anticancer T cell responses in patients with advanced solid carcinomas.50 Importantly, this combination might also circumvent some of the toxic effects observed with CTLA-4 and PD-1 coinhibition, as the expression of TIM-3 (but not CTLA-4 and PD-1) is predominantly linked to terminally differentiated T cells producing interferon gamma (IFNG).50
In summary, tumor infiltration by CD8+ CTLs, memory T cells and TH1 cells is associated with prolonged PFS and OS, particularly in patients with HGSOC. Conversely, tumor infiltration by TIM-3+ cells stands out as a negative prognostic factor. The impact of T cells expressing other activation and exhaustion markers on EOC outcome remains to be precisely elucidated.
TREG cells
TREG cells are a heterogeneous population of CD4+ T lymphocytes that express the high-affinity IL-2 receptor chain IL-2 receptor subunit alpha (best known as CD25) and the transcription factor FOXP3.51 TREG cells are essential for maintaining tolerance and preventing autoimmunity.51 However, developing malignancies, including EOC, harness TREG cells to establish local immunosuppression through a variety of mechanisms: (1) direct lysis of immune effector cells; (2) inhibition of antigen-presenting cells (APCs); (3) secretion of immunosuppressive cytokines, such as IL-10 and transforming growth factor beta 1 (TGFB1); and (4) depletion of growth factors and nutrients.52 While most circulating TREG cells stably express FOXP3 as a gene imprinted during thymic development, a tumor-infiltrating subset of TREG cells appears to retain some degree of plasticity and transdifferentiate towards a phenotype with limited immunosuppressive functions and the capacity to secrete IFNG and IL-17 under inflammatory conditions.51 52 Similarly, a subset of tumor-infiltrating TH1-polarized CD4+ T cells can transdifferentiate into FOXP3+ TREG cells in response to TGFB1.51
The majority of studies have shown that a high prevalence of TREG cells within TILs is associated with poor outcome in patients with all EOC histologies, especially when overall CTL infiltration is limited (table 1).17 30 37 38 53–56 Similar results were obtained by a meta-analysis of 869 patients encompassing all EOC histologies from four previous studies.55 Conversely, a positive prognostic value has been attributed to tumor-infiltrating TREG cells in a cohort of 270 HGSOC patients, most likely reflecting abundant TIL infiltration altogether.29 These findings suggest that the ratio of CTLs and TREG cells may constitute a superior indicator of active immunity in the ovarian TME, as validated in a number of studies.29 30 57
Tumor-infiltration by TREG cells is influenced by a variety of mechanisms, including multiple pathways driven by TAMs.58 For instance, C-C motif chemokine ligand 22 (CCL22), produced by malignant cells and TAMs, recruits TREG cells through a C-C motif chemokine receptor 4 (CCR4)-dependent mechanism.59 Moreover, miRNAs contained in TAM-derived exosomes appear to promote the interaction of TREG cells with CTLs, resulting in an increased TREG/TH17 cell ratio and disease progression.60 Conversely, TREG cells promote expression of the immunosuppressive molecule V-set domain containing T cell activation inhibitor 1 (VTCN1, best known as B7-H4) on various APCs, including TAMs.56 Of note, hypoxia-induced upregulation of CCL28 also promotes the recruitment of TREG cells to the ovarian TME through a mechanism that involves CCR10 and ultimately leads to IL-10 production in support of disease progression.61 TREG cells isolated from HGSOCs express various receptors associated with TCR engagement, including the coinhibitory receptor PD-1 and the coactivating receptors inducible T cell costimulator and tumor necrosis factor (TNF) receptor superfamily member 9 (TNFRSF9, best known as 4-1BB).62 Moreover, compared with TREG cells from other carcinomas, TREG cells from EOCs exhibit a highly activated state and increased immunosuppressive capacity, as documented in numerous studies on various histological subtypes of EOC.62
Thus, the abundance of TREG cells in primary EOC is commonly associated with poor disease outcome and metastatic progression. Conversely, the impact of TREG cell infiltration in metastatic EOCs remains relatively unknown.
Dendritic cells
Conventional DCs (cDCs) are commonly viewed as superior APCs, largely reflecting their capacity to efficiently process extracellular antigens and present them on MHC-II and MHC-I molecules to naïve CD4+ and CD8+ T cells, respectively, in the context of the abundant secretion of pro-inflammatory cytokines.63 Based on functional and phenotypic features, cDCs can be subdivided into at least two main subsets: type I (cDC1s) and type II cDCs (cDC2s),64 classified as CD11clowHLA-DR+DEC205+XCR1+, and CD11c+HLA-DR+CD11b+CD1a+CD14+, respectively.63 cDC1s are not only highly proficient at cross-priming tumor-targeting CD8+ CTLs in tumor draining lymph nodes but can also recruit T cells to the TME and provide them with proinflammatory cytokines.64 Unfortunately, cDC1s are very rare in the ovarian TME and exhibit features of immaturity, especially at early disease stages, implying that they might contribute to tumor progression.64 At least in part, this reflects the abundance of immunosuppressive cytokines, including IL-10, TGFB1, and VEGFA,14 65 66 and other immunosuppressive factors, including the PD-1 ligand CD274 (best known as PD-L1).67 68 In line with this notion, PD-L1 blockade enhances DC-mediated T-cell activation, correlating with IL-10 downregulation and increased secretion of IL-2 and IFNG.69
HGSOC infiltration by BAFT3-dependent CD103+ cDC1s correlates with the abundance of C-X-C motif chemokine receptor 3 (CXCR3) ligands, including C-X-C motif chemokine ligand 9 (CXCL9), CXCL10, and CXCL11, which facilitate the recruitment of clinically relevant effector T cells into the TME.43 70 CD103+ cDC1s are also dependent on the transcription factor IFN regulatory factor 8 (IRF8) and zinc finger and BTB domain containing 4,71 as well as the cytokines colony stimulating factor 2 (CSF2, best known as GM-CSF) and FMS-related receptor tyrosine kinase 3 ligand, which are associated with favorable clinical outcome in ovarian carcinoma.72 Similarly, the abundance of mature DCs expressing lysosomal-associated membrane protein 3 (LAMP3, best known as DC-LAMP) has been associated with improved prognosis in patients with various malignancies, including primary and metastatic HGSOC.21 Of note, the majority of mature DC-LAMP+ DCs are localized to the tumor stroma and are associated with tertiary lymphoid structures (TLSs) rather than in direct contact with malignant cell nests.21 73 Nonetheless, an elevated density of mature DCs in the ovarian microenvironment correlates with biomarkers of TH1 polarization and cytotoxic activity, both of which are favorable indicators in patients with all EOC histologies (table 1).20 21 42 74
At odds with cDCs, plasmacytoid DCs (pDCs), defined as CD11c-CD123+CD303+HLA-DRlow cells, are mostly involved in antiviral immune responses, reflecting their capacity to produce elevated amounts of type I IFN on activation.75 High levels of pDCs in the ovarian microenvironment are generally associated with immunosuppression and poor prognosis, as comprehensively documented in patients with various EOC histological subtypes (table 1).17 18 76–79 IL-10 and CXCL12 are the primary factors responsible for EOC infiltration by CXCR4-expressing pDC precursors, culminating in the accumulation of pDCs expressing the immunosuppressive enzyme IDO1.78 79 Consistent with this, a high density of pDCs in the EOC environment is associated with impaired TIL proliferation, decreased effector functions as well as neoangiogenesis and metastatic disease dissemination in preclinical disease models.15 Interestingly, tumor-infiltrating CD4+CD123+BDCA2+ pDCs exhibit a partially mature phenotype (indicative of activation) compared with their ascitesborne and bloodborne counterparts.77 However, these tumor-associated pDCs produce limited amounts of type I IFN, IL-6, CCL4, CCL5, and TNF on Toll-like receptor (TLR) stimulation, suggesting that local pDC dysfunction may contribute to disease progression.77
Altogether, these findings suggest that EOCs harness various mechanisms to alter DC functions to establish immunosuppressive circuitries that foster disease progression across various EOC subtypes.63 76 Thus, therapeutic interventions that restore DC functions stand out as promising approaches to initiate EOC-targeting immune responses of clinical relevance.80
B cells and TLSs
Tumor infiltration by B cells is robustly associated with improved survival in patients with EOC, especially HGSOC (table 1).3 21 36 81–85 Nevertheless, accumulating findings suggest a positive impact of B cells also in other histological EOC subtypes, including MOC, EnOC, and OCCC.36 84 Although B cells primarily reside in the tumor stroma in the context of TLSs, they can also be found within tumor cell nests.86 B cells at all stages of differentiation have been detected in EOC, including IgD+CD38+/- naïve B cells, IgD+CD38+ pregerminal and IgD−CD38+ germinal B cells; IgD−CD38+/− memory B cells as well as plasma cells (PCs) with a IgD−CD38++ phenotype.87 Similar to lymph nodes, TLSs contain prominent B-cell follicles adjoined by discrete T-cell zones containing CD4+ and CD8+ T cells, as well as follicular DCs, high endothelial venules, and lymphatic vessels.73 TLSs are documented in only approximately 30% of all EOC patients, but their presence is strongly associated with favorable clinical outcome (table 1).21 83
Interestingly, in the ovarian setting, TLSs are frequently surrounded by dense infiltrates of mature PCs.83 85 PCs are generally associated with a high density of CD8+ and CD4+ T cells, as well as CD20+ B cells, which stands out as an immunological configuration compatible with the induction of clinically relevant tumor-targeting immunity.83 CD20+ B cells are found in more than 40% of HGSOCs, and their abundance also correlates with tumor infiltration by CD4+ and CD8+ T cells, as well as with the abundance of transcripts encoding various T cell markers, such as TIA1 cytotoxic granule associated RNA binding protein (TIA1), granzyme B (GZMB) and FOXP3.3 21 Importantly, abundant EOC infiltration by both CD8+ CTLs and CD20+ B cells is associated with a more favorable disease outcome than infiltration by either cell population alone, suggesting the existence of cooperative interactions between CD8+ CTLs and CD20+ B cells in the ovarian microenvironment.21 81 83 In line with this notion, the majority of EOC-infiltrating CD20+ B cells express high levels of costimulatory molecules, including CD80 and CD86, as well as MHC Class I and Class II molecules, as they display a CD27- memory phenotype linked to markers of somatic hypermutation, oligoclonality and IgG class switching.88
As recently shown by us and others, both omental and peritoneal HGSOC metastases are highly infiltrated by CD20+ B cells with a memory phenotype.20 23 89 As in primary EOCs, transcript levels of CD20 correlate with markers of cytotoxic responses, suggesting that B cells infiltrating metastatic EOC promote anticancer immune responses, a notion that has been mechanistically validated on B-cell depletion in syngeneic mouse models of peritoneal metastasis.89 Although the density of CD20+ B cells is significantly increased in peritoneal metastases compared with primary EOC lesions,23 the abundance of metastasis-infiltrating CD20+ B cells is not associated with disease outcome (table 1).20
Taken together, these observations suggest that TLSs represent key sites for the induction and maintenance of clinically relevant EOC-targeting immunity and that B cells mediate a central function in this context. An improved understanding of the biology of tumor-infiltrating B cells is highly anticipated to harness this lymphocyte subset for therapeutic purposes.
Tumor-associated macrophages
TAMs, which constitute the largest fraction of the myeloid infiltrate in the majority of solid malignancies, including all EOCs,90 can be found within tumor cell nests, at the tumor invasive margin and in the stroma. A high degree of TAM heterogeneity has been observed not only across different histological subtypes of EOC, with HGSOCs and MOCs being the EOCs most abundantly infiltrated by TAMs, but also in women with the same EOC subtype and even different EOC lesions in the same patient.90 91 Moreover, TAMs display a high degree of functional plasticity and can rapidly adapt to changing microenvironmental conditions to acquire different phenotypic, metabolic, and functional profiles.92 In particular, exposure of tumor-infiltrating monocytes and macrophages to cytokines, such as IL-4, IL-5, IL-10, IL-13, CCL2, TGFB1, and CSF1 (best known as M-CSF), as well as to prostaglandin E2 (PGE2), which is abundantly produced by dying cancer cells, promotes the acquisition of anti-inflammatory and protumoral (so-called M2-like) properties.93
In ovarian carcinoma, M2-like TAMs robustly promote neo-angiogenesis and disease progression in the context of largely immunosuppressive rewiring of the TME.94 In line with this notion, high levels of CD206+CD163+CD204+ M2-like TAMs within primary and metastatic EOCs of all histologies are generally associated with reduced sensitivity to treatment and poor prognosis (table 2).65 94–100 Conversely, M1-like TAMs, defined as CD68+CD86+HLA-DR+iNOS+ cells, constitute a good prognostic factor in women with EOC, largely reflecting their ability to promote robust inflammatory responses that limit disease progression, although their presence is significantly decreased in the TME of patients with advanced EOC (table 2).91 98 101 102
Prognostic relevance of tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), natural killer (NK) cells and cancer associated fibroblasts (CAFs) in primary and metastatic ovarian carcinoma
The immunosuppressive functions of M2-like TAMs involve a variety of global anti-inflammatory cytokines (eg, IL-10 and TGFB1) and chemokines (eg, CCL17, CCL18, CCL22) that facilitate the following functions: (1) inhibiting antigen presentation to T cells, (2) subverting DC maturation, (3) blocking CTL effector functions, and (4) driving the recruitment of TREG cells.103 M2-like TAMs in the ovarian TME also limit immune effector functions by producing exosomes.60 Specifically, TAM-derived exosomes contain high amounts of proteins, as well as DNA, mRNA and miRNA molecules, which together suppress T cell activity and promote an imbalance between TREG cells and TH17 cells by directly targeting signal transducer and activator of transcription 3 (STAT3) in CD4+ T cells.60
Multiple studies have identified a crucial role for TAMs, especially CD163+TIM-4+ omental TAMs, in the metastatic dissemination of ovarian cancer cells to the peritoneal cavity.19 Indeed, the specific depletion of this TAM population prevents the development of metastatic disease in mouse models of ovarian cancer, and the molecular circuitries that underlie these functions may represent a novel therapeutic target in the ovarian setting.19 Moreover, TAMs are important for the formation of spheroids during transcoelomic EOC metastasis. In particular, TAMs can produce large amounts of epidermal growth factor (EGF) to activate EGF receptor (EGFR) and VEGFC signaling in surrounding cells, ultimately leading to upregulation of multiple integrins and intercellular adhesion molecule 1 (ICAM1) and hence promoting cancer cell proliferation, migration, adhesion, spheroid formation and implantation into the peritoneal cavity.94 In line with this notion, EGFR-blocking and ICAM1-blocking strategies inhibit spheroid formation and metastatic disease progression in mouse models of EOC,94 standing out as potential targets for the development of novel approaches to the management of EOC patients.
Finally, TAMs support tumor progression by increasing the availability of selected nutrients in the primary, and even more so metastatic, ovarian TME. Specific TAM subsets can indeed accumulate lipids in support of their immunomodulatory properties, ultimately leading to deregulation of multiple factors involved in lipid metabolism, including the lipid chaperones fatty acid binding protein 4 (FABP4) and FABP5.104 In advanced EOC, TAMs preferentially express FABP4, which supports tumor progression by favoring IL-6-driven STAT3 signaling.105 FABP4 also plays a key role in the interaction between ovarian cancer cells and adipocytes.104 In line with these observations, FABP4 deficiency impairs metastatic tumor growth in mouse models of EOC.106 Intriguingly, EOC cells actively promote cholesterol efflux by TAMs, culminating in depletion of lipid rafts and increased IL-4 signaling.107 Thus, genetic deletion of the ABC transporters that mediate cholesterol efflux limits EOC progression in mice.107
Altogether, these findings indicate that EOCs harness macrophage polarization to an M2-like phenotype as a mean to establish immunosuppression in support of local and distant disease dissemination. The therapeutic potential of TAM-targeting or TAM-repolarizing agents, such as CSF 1 receptor (CSF1R) inhibitors in patients with EOC, however, remains to be elucidated.
Myeloid-derived suppressor cells
MDSCs are a heterogeneous population of relatively immature myeloid cells that differ in morphology and function from terminally differentiated myeloid cells, such as DCs, macrophages and neutrophils.108 There are two major groups of MDSCs in humans, namely, granulocytic/polymorphonuclear MDSCs (PMN-MDSCs), which generally display a CD11b+CD33+CD14−CD15+ surface phenotype, and monocytic MDSCs (M-MDSCs), which are most often CD11b+CD33+CD14+CD15− MDSCs.109
An increased number of circulating or tumor-infiltrating MDSCs has been detected in patients with various malignancies,109 including women with primary and metastatic EOC.16 110 111 Nevertheless, association between histological subtypes of EOC and MDSC abundance has never been investigated. Various tumor-derived cytokines (eg, IL-6, IL-10, IL-18, TNF, and VEGFA), growth factors (eg, M-CSF, GM-CSF) and other mitogens (eg, PGE2) promote the formation of MDSCs from myeloid progenitors in the bone marrow.14 111–113 This largely reflects the activation of signaling transduction cascades culminating with STAT3 signaling, which also promotes MDSC immunosuppression by downregulating IRF8 while upregulating CCAAT enhancer binding protein beta.114 Conversely, the accumulation of MDSCs within neoplastic lesions is driven by a variety of cytokines and chemokines, including CXCL1, CXCL8, CXCL12, CCL1, CCL2, CCL3, CCL5 and CCL7, which primarily operate via CCR4 and CCR5.113 115
Accumulating preclinical and clinical evidence indicates that PMN-MDSCs and M-MDSCs suppress both innate and adaptive immune responses driven by ovarian cancer cells.16 116–120 While the majority of such studies focused on HGSOCs, data from a limited number of patients with MOC and EnOC also support the protumoral role of MDCSs.110 Of note, PMN-MDSCs preferentially use reactive oxygen species, peroxynitrite, ARG1 and PGE2 to mediate immune suppression.112 119 Conversely, M-MDSCs predominantly harness nitric oxide, immunosuppressive cytokine such as IL-10 and TGFB1, and membrane-bound molecules, such as PD-L1 to impair CTL and NK cell functions.121 MDSCs also drive tumor progression by favoring epithelial-to-mesenchymal transition (EMT), invasiveness and metastatic dissemination in malignant cells and by promoting neoangiogenesis.114 116 Recent data suggest that MDSCs are also involved in the establishment of the premetastatic niche.122 Consistent with these findings, elevated numbers of circulating or intratumoral MDSCs correlate with poor disease outcome in women with various EOC subtypes (table 2).14 110 112 113 123 124
Altogether, these observations suggest that both M-MDSCs and PMN-MDSCs establish immunosuppression and support metastatic dissemination in EOC. Thus, targeting MDSCs stands out as a promising approach to promote EOC-directed immune responses. Potential approaches to this objective include (1) blocking the formation of MDSCs in the bone marrow, (2) impeding MDSC recruitment to neoplastic lesions, and/or (3) reprogramming MDSCs to terminally differentiate and lose their immunosuppressive potential.
NK cells
NK cells are a subset of innate lymphoid cells that play a central role in defending the organism from viral infection, early malignant transformation and metastatic tumor dissemination.125 126 NK cell effector functions encompass potent cytotoxicity against target cells, as well as the secretion of immunomodulatory cytokines that orchestrate innate and adaptive immune responses.126 Such functions do not involve the recognition of specific antigens, as they do in the case of CTLs, but are controlled by the balance between inhibitory and stimulatory signals that are conveyed to NK cells on interaction with potential targets.125
Results on EOC infiltration by NK cells are rather inconsistent, at least in part due to the use of rather heterogeneous markers for NK cell detection (table 2).13 20 21 33 39 127 128 Thus, high levels of NK cells in the TME of all EOC subtypes have been positively associated with improved prognosis when beta-1,3-glucuronyltransferase 1 (best known as CD57) and CD103 were used as phenotypic markers, although these molecules are also expressed by activated CD8+ T cells.33 39 In contrast, when NK cells were identified using natural cytotoxicity triggering receptor 1 (NCR1, best known as NKp46), their abundance in primary and metastatic HGSOC lesions did not correlate with clinical outcome,20 21 perhaps due to functional impairments imposed by local immunosuppression.129 Indeed, NK cell effector functions in peritoneal ascites are inhibited on MIF-driven downregulation of killer cell lectin-like receptor K1 (KLRK1, best known as NKG2D).13 130 Additionally, NK cell cytotoxicity in the TME of metastatic EOC is limited on the downregulation of NCR2 (best known as NKp30), as induced by soluble and surface-exposed NK cell cytotoxicity receptor 3 ligand 1 (NCR3LG1, best known as B7-H6).131 In line with these findings, increased B7-H6 expression has been associated with metastatic disease progression and poor clinical outcome in patients with various EOC histological subtypes.100
In addition to mediating direct cytotoxic effects against neoplastic cells, NK cells can also exert anticancer activity by engaging the adaptive arm of the immune system. Specifically, NK cells can recruit cDCs to the TME on secretion of CCL5, X-C motif chemokine ligand 1 (XCL1) and XCL2.132 Moreover, IL-18-primed NK cells can favor tumor infiltration through immature DCs via CCL3 and CCL4, a process that culminates in the secretion of CTL chemoattractants, including CXCR3 and CCR5 ligands.133
Altogether, these observations indicate that NK cells dynamically interact with malignant and immune components of the ovarian TME most often in support of anticancer immunity. However, available data fail to elucidate a robust prognostic value for EOC infiltration by NK cells, potentially linked to an elevated degree of methodological heterogeneity and/or to the functional impairment of EOC-infiltrating NK cells downstream of local immunosuppression. Efforts aimed at homogenizing the quantification of EOC-infiltrating NK cells and obtaining further insights into their functional rewiring on tumor infiltration are urgently needed to clarify the therapeutic potential of NK cell-targeting agents in women with EOC.126
Cancer-associated fibroblasts
Cancer-associated fibroblasts (CAFs) are key components of the ovarian TME with diverse biological functions, including matrix remodeling as well as reciprocal interactions with TILs and cancer cells.134 135 Tissue-resident quiescent fibroblasts, which are predominant in the normal stroma, and mesenchymal stem cells transform into CAFs on interaction with cancer cells.136 137 CAFs found in EOC lesions generally express actin alpha 1, skeletal muscle (ACTA1, best known as SMA), fibroblast activation protein alpha, S100 calcium binding protein A (S100A4, best known as FSP1) and fibroblast growth factor 1 (FGF1).138 139 However, due to the continuous reciprocal interactions between CAFs with cancer cells, the former tend to undergo dynamic changes that enable high degrees of phenotypical and functional heterogeneity.139 140 Indeed, neoplastic cells secrete various cytokines and soluble factors such as IL-6, IL-8, IL-1β, TGFB1, platelet-derived growth factor, FGF and EGF to activate fibroblasts.141 142 Moreover, ovarian cancer cells reprogram fibroblasts to become CAFs via alternations in the levels of 3 miRNAs, namely downregulation of miR-214 and miR-31, coupled to upregulation of miR-155.143
On reprogramming, CAFs promote tumor growth and invasion through increased secretion of multiple cytokines, chemokines and growth factors such as CCL5, IL-6, IL-8, TGFB1, VEGFA among others.144–146 Moreover, CAFs promote EOC progression by favoring the EMT141; angiogenesis,147; altered cancer metabolism144 148; chemoresistance;32 149 150 and immune modulation.151 152 However, an extensive description of all the mechanisms through which CAFs drive EOC progression goes largely beyond the scope to of this review, and can be found elsewhere.134 135 Importantly, CAF abundance positively correlate with disease progression and poor disease outcome in women with primary138 139 142 150 153 and metastatic EOC143 144 154 (table 2).
In summary, CAFs may also constitute valuable target to limit immunosuppression in the TME of patients with EOC. So far, this strategy has been mostly been investigated in other tumors with a large CAF component, such as pancreatic carcinoma.134
Cytokine and chemokine profile
Accumulating preclinical and clinical evidence indicates that the cytokine and chemokine milieu of EOC plays a key role in the establishment of local and systemic immune contexture (table 3).155 156 Thus, the intratumoral or circulating abundance of multiple cytokines and chemokines impacts disease outcome in patients with EOC. For instance, elevated IL-6 levels in the ovarian TME have been associated with disease progression, resistance to treatment and poor clinical outcome in patients with various EOC subtypes.156–159 At least in part, this reflects the ability of IL-6 to promote EOC cell invasion through the basal membrane, as well as to (1) mediate mitogenic effects linked to chemoresistance and (2) promote IL-10 secretion.160 Moreover, IL-6 reportedly activates protumorigenic signal transducers, including JAK1 (Janus kinase 1) and STAT3.161 162 In metastatic EOC, TAMs are the primary producers of IL-6, and their presence, as well as high bloodborne and peritoneal IL-6 levels, correlate with poor disease outcome.65 111 159 160 Similar findings have been obtained with IL-8, IL-10, VEGFA, TGFB1 and TNF, all of which appear to condition the ovarian TME in favor of disease progression and escape from immunosurveillance (figure 2 and table 3).14 65 66 156 163–168

{kind=link}
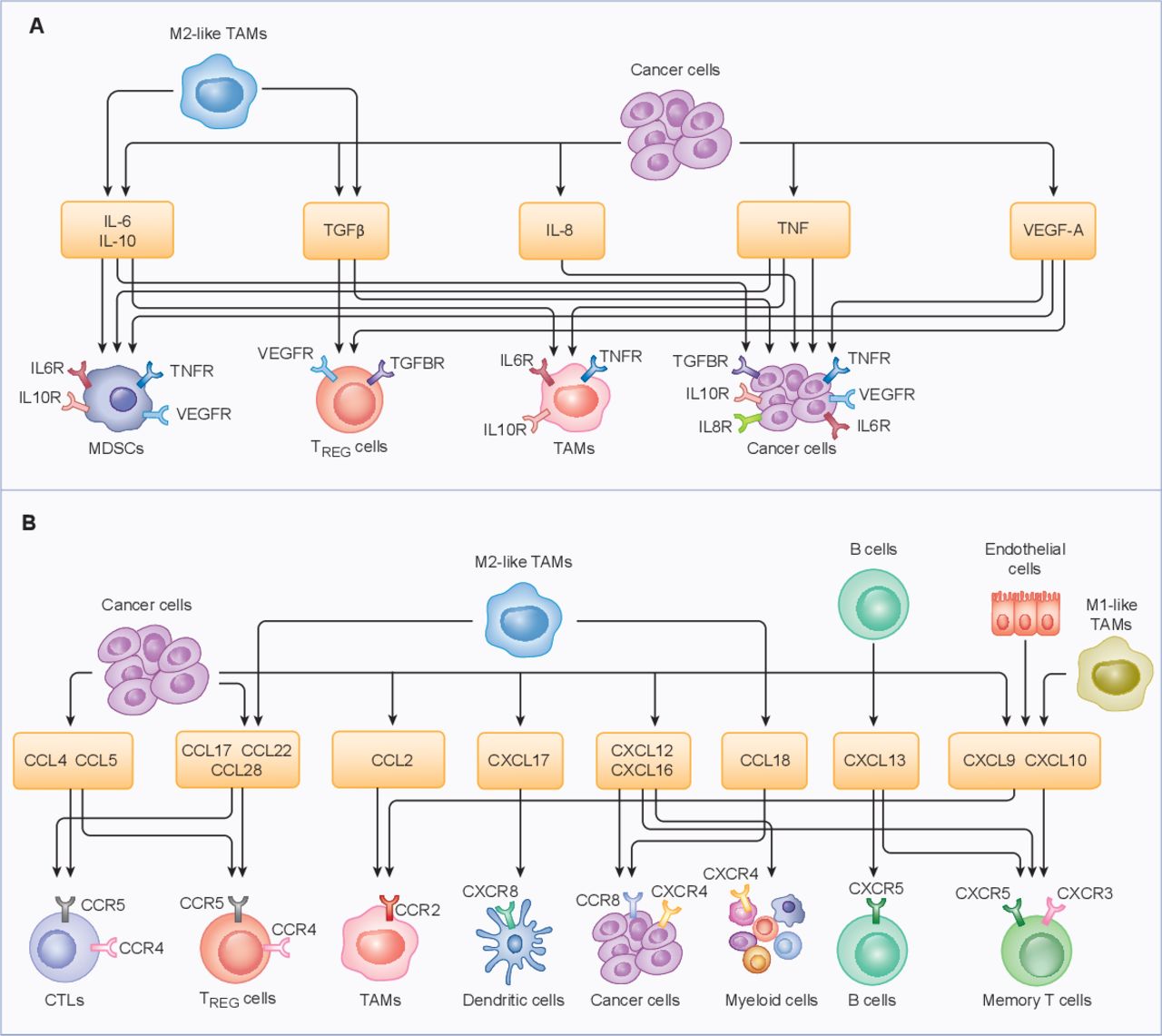{kind=link}
Cytokines and chemokines that coordinate the tumor microenvironment. Primary cytokines (A) and chemokines (B) produced in the ovarian tumor microenvironment. The most prominent sources and major receptors are depicted. CCL, chemokine (C-C motif) ligand; CCR, C-C chemokine receptor; CXCL, chemokine (C-X-C motif) ligand; CXCR, C-X-C chemokine receptor; CTL, cytotoxic T lymphocyte; IL, interleukin; IL6R, interleukin 6 receptor; MDSC, myeloid-derived suppressor cell; NK, natural killer; pDC, plasmacytoid dendritic cell; TAM, tumor-associated macrophage; TGFB1, transforming growth factor beta 1; TGFBR, transforming growth factor beta receptor; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; TREG, regulatory T; VEGFA, vascular endothelial growth factor A; VEGFR, vascular endothelial growth factor receptor.
Pro-tumoral and anti-tumoral roles of cytokines and chemokines in primary and metastatic ovarian carcinoma
The overall chemokine landscape of EOC is heterogeneous, with CCL2, CCL5, CXCL12 and CXCL16 being the most predominant molecules.155 Importantly, the high levels of CXCR6 and CXCL16 in serous papillary carcinoma tissues suggests an association with aggressive histological subtype as compared with EnOC.169 CCL2 is mostly produced by malignant cells and contributes to TAM accumulation.170 Conversely, while CCL4 and CCL5 expression is mostly associated with CTL recruitment,133 171 CCL22 and CCL28 levels positively correlate with an increased abundance of TREG cells (at least in primary EOCs).54 61 155 The expression of genes associated with T cell recruitment is restricted to the epithelial tumor component and preserved across metastatic sites, suggesting that T cells might easily home to metastatic lesions.155 However, the impact of some cytokines and chemokines on EOC progression and clinical outcome is controversial. For instance, although EOC-derived CXCL12 is associated with T cell recruitment,172 as are CXCL9 and CXCL10,70 it also drives tumor progression by activating the MAPK cascade in EOC cells,173 and promotes tumor infiltration by myeloid cells.113 156 Consistent with this notion, high levels of CXCL12 or its receptor (CXCR3), as well as CXCL16, CXCR6 and CCL8, have been associated with metastatic dissemination to the peritoneum and ascites formation (table 3).169 173–175
In summary, the net effect of cytokine and chemokine signaling on EOC progression depends on the balance between their ability to recruit and activate specific immune cell populations and their capacity to drive mitogenic signaling in EOC cells.
Modulating the ovarian TME
EOC was one of the first malignancies in which a positive association between TIL density and OS was identified.40 However, EOC-infiltrating lymphocytes are often suppressed and/or functionally exhausted by a variety of mechanisms, including (but not limited to) (1) abundant secretion of immunosuppressive cytokines, such as TGFB1, IL-6 and IL-10, by EOC cells65 66 156 163; (2) expression of metabolic immunosuppressors, such as IDO178; (3) robust tumor infiltration by immunosuppressive TREG cells,38 46 54 M2-like TAMs and MDSCs94 95 109; and (4) activation of coinhibitory receptors, such as PD-1 and TIM-3.45 176 In line with such multipronged immunosuppression, ICIs are not very effective in women with EOC.177
Chemotherapy
Some anticancer agents, including conventional chemotherapeutics, targeted drugs and radiation therapy (RT), can be harnessed to stimulate anticancer immunity, as they can increase the antigenicity of malignant cells, boost their adjuvanticity or repolarize the TME in support of immunological disease control.178 At least in principle, genotoxic chemotherapies and RT can favor the formation and/or expression of mutated neoepitopes.179 However, expression levels may remain low and hence be incompatible with robust immune recognition. Chemotherapeutic agents used for the clinical management of platinum-resistant EOC, including doxorubicin, oxaliplatin and paclitaxel, are known to drive immunogenic cell death,180 which is associated with the abundant emission of immunostimulatory molecules commonly known as damage-associated molecular patterns (DAMPs) and thus might synergize with immunotherapy.178 181 In line with this notion, pegylated doxorubicin has been shown to boost the uptake of dying EOC cells by cDCs and ultimately promote the cross-priming of T cells specific to EOC-associated antigens.182 However, the combination of pegylated doxorubicin with an ICI specific for PD-L1 (avelumab) failed to demonstrate superior activity compared with standard of care in a recent phase III clinical trial.183 184 Similar findings have been documented in a randomized phase II study testing the combination of pegylated doxorubicin with a TLR 8 (TLR8) agonist (motolimod).185 In addition to promoting polyploidization and hence boosting the immunogenicity of EOC cells,186 paclitaxel promotes the repolarization of M2-like TAMs into their M1-like counterparts, as well as the depletion of TREG cells and MDSCs from the ovarian TME.187 However, available preclinical data are insufficient to support the initiation of clinical trials testing paclitaxel in combination with ICIs in women with advanced EOC. Metronomic cyclophosphamide has also been shown to deplete TREG cells from the ovarian TME, suggesting some potential for synergy with immunotherapeutic regimens.188 In line with this notion, metronomic cyclophosphamide combined with an angiogenesis inhibitor and a PD-1-targeting ICI (pembrolizumab) is well tolerated and mediates clinical benefits in 95.0% and durable treatment responses (>12 months) in 25.0% of women with recurrent EOC.189
Immune checkpoint inhibitors.
PD-1, CTLA-4, LAG3, TIM-3, and other coinhibitory receptors are widely expressed by EOC-infiltrating cells and mediate robust immunosuppressive effects.45 176 Thus, ICIs a priori represent a valid strategy to reverse local immunosuppression in women with EOC. However, comprehensive phenotypic and functional analyses of EOC-infiltrating T cells and the ovarian TME have revealed the existence of a multilayered immunosuppressive network,176 190 potentially explaining the poor clinical activity of ICIs documented so far in patients with EOC. Indeed, in the first phase II study evaluating the efficacy of a PD-1-targeting ICI (nivolumab) for recurrent EOC, the overall response rate (ORR) in 20 assessable patients was only 15%, with a 10% durable complete response rate.11 Similarly, the use of pembrolizumab as a single therapeutic agent for EOC has been linked to an ORR of 9%, which was primarily associated with increased expression of PD-L1.191
One potential approach to improving the efficacy of ICIs in patients with EOC relies on the use of multiple nonredundant ICIs as a combinatorial regimen.176 Supporting the validity of this approach, nivolumab combined with a CTLA-4-specific ICI (ipilimumab) has been associated with an ORR of 31.4% (vs 12.2% for nivolumab alone) in phase II clinical trials enrolling 100 patients with persistent or recurrent EOC.192 Based on these clinical findings and the preclinical data discussed above, current efforts are being refocused on targeting coinhibitory receptors other than CTLA-4 and PD-1, including (but not limited to) TIM-3, LAG3 and TIGIT (source https://www.clinicaltrials.gov). The results of these trials are urgently awaited.
Angiogenesis inhibitors
The anti-angiogenic drug bevacizumab, a humanized monoclonal antibody targeting VEGFA, has now been employed for first-line management of advanced EOC for more than 7 years.193 Based on preclinical findings from mouse models of EOC, bevacizumab is expected to synergize with ICIs, largely reflecting its ability to promote tumor infiltration by T cells.194 Consistent with this notion, bevacizumab in combination with nivolumab has been associated with improved ORR (28.9%) and PFS (median 9.4 months) in women with relapsed EOC, an activity that was even more pronounced in patients with platinum-sensitive lesions.195 Similarly, an ICI specific to PD-L1 (durvalumab) combined with a PARP inhibitor (olaparib) and cediranib, a tyrosine kinase inhibitor with anti-angiogenic activity, achieved an ORR of 50% and a disease control rate of 75% in 12 randomized patients.196 However, the results from a recent phase III study evaluating the addition of atezolizumab (an ICI specific for PD-L1) to platinum-based chemotherapy and bevacizumab failed to support the use of ICIs for newly diagnosed stage III or IV EOCs.197
PARP inhibitors
PARP inhibitors have emerged as key therapeutic interventions for patients with EOC.198 Indeed, rather common germline mutations in BRCA1 and BRCA2, resulting in HR defects, make EOCs highly sensitive to PARP inhibitors.198 Thus, no less than three distinct PARP inhibitors (ie, niraparib, olaparib, and rucaparib) are currently approved for the treatment of recurrent, platinum-sensitive EOC as maintenance on platinum chemotherapy.199 Of note, PARP inhibitors have been shown to mediate multipronged immunostimulatory effects, largely reflecting their ability to inhibit DNA repair in malignant cells.200 and indicating the possibility for synergy with ICIs.201 Thus, PARP inhibitors might enhance the mutational load of EOCs as consequence of unrepaired DNA damage, favoring T cell infiltration, but also appear to drive robust type I IFN secretion downstream of cyclic GMP-AMP synthase and stimulator of IFN response cGAMP interactor 1 activation.202 203 Based on these preclinical findings, PARP inhibitors are currently being tested in combination with ICIs in more than 10 ongoing clinical trials.12 The results of this wave of investigation are highly anticipated.
Tumor vaccines
A variety of tumor-associated antigens (TAAs) that can be specifically targeted by vaccination strategies have been identified in EOC.204 Cancer/testis antigen 1B (CTAG1B, best known as NY-ESO-1) is one such antigen, and several NY-ESO-1-based vaccines have been shown to provide NY-ESO-1+ EOC patients with an OS advantage.205 However, vaccine-driven immunoediting may ultimately promote the selection of NY-ESO-1− EOC cell clones and hence enable clinical relapse. An alternative approach for vaccination involves the use of mutated TAAs as targets.206 Although advantageous in some aspects, this approach does not circumvent the possible emergence of antigen-negative malignant cell clones, indicating an advantage for vaccination strategies targeting multiple TAAs at the same time, such as DC-based vaccines.63 In this context, DCs from a patient with EOC must be provided either ex vivo or in vivo with a source of TAAs in the context of activation cues in the former setting followed by DC reinfusion into the patient.63 Such sources can be as diverse as recombinant full-length TAAs or epitopes thereof, TAA-encoding nucleic acids, autologous tumor lysates, and allogeneic cancer cell lysates. Results from a number of clinical trials testing ex vivo DC-based vaccines in women with EOC demonstrate that this approach is well tolerated and associated with at least some activity.63 207 Moreover, in consideration of their mechanism of action, DC-based vaccines are expected to synergize with other immunotherapeutic interventions, such as ICI-based immunotherapy and adoptive T cell transfer (ACT).208 Nonetheless, no clinical trial is currently investigating a DC-based vaccine combined with ICIs or ACT in patients with EOC, an entire line of clinical investigation that is urgently awaited.
Adoptive T cell transfer
ACT represents a personalized immunotherapy based on autologous TILs expanded ex vivo and reintroduced into patients together with high-dose IL-2 on lymphodepletion.209 ACT has demonstrated considerable potency in patients with metastatic melanoma but limited success in women with EOC, potentially due to the strong immunosuppressive networks at play in the ovarian TME or suboptimal TIL expansion ex vivo.210 Several pilot and phase I/II clinical studies are currently open to investigating the therapeutic profile of ACT in women with advanced EOC.9 That said, available results from a pilot study enrolling six patients with advanced EOC suggest that ACT is primary active on existing target lesions but fails to control distant progression,211 potentially linked to TIL exhaustion, insufficient expansion or intralesion/interlesion heterogeneity.212
Chimeric antigens receptor-T cells and TCR therapy
Chimeric antigens receptors (CARs) are fusion proteins engineered into T cells for them to recognize specific antigens independent on MHC presentation.213 CAR-T cell therapy has achieved unprecedented success in the treatment of hematological malignancies such as relapsed/refractory B-cell leukemia and lymphoma.213 However, a similar success has not been witnessed in patients with solid tumors, due to variety of obstacles.214 In line with this notion, a phase I study evaluating the safety and efficacy of first-generation of CAR-T cells targeting the folate receptor alpha (FOLR1) in patients with metastatic EOC documented limited efficacy.215 Current efforts are focusing on increasing CAR-T cell potency, with a particular interest around promoting CAR-T cells infiltration and intratumoral persistence.216 217 Despite these and other limitations, numerous early phase clinical trials are currently testing CAR-T cells with a variety of specificities in women with EOC (source https://www.clinicaltrials.gov). In this setting, a recent case report documented some therapeutic benefit (partial response and inhibition of hepatic progression) in a patient with metastatic EOC receiving CAR-T cells targeting mesothelin (MSLN) and engineered to secrete a PD-1-blocking single-chain fragment in combination with apatinib.218 These results suggest a novel therapeutic strategy for EOC and a Phase I study investigating this possibility is ongoing (NCT04503980). In similar line, T cells engineered by viral vectors to express the TCR gene with defined specificity (TCR-T cells) targeting NY-ESO-1 (NCT01567891, NCT03017131), MUC16 (NCT02498912), MAGE-A4 (NCT03132922) and neoantigens (NCT03412877) are tested in early phase clinical studies in EOC patients.
Oncolytic virus therapy
An alternative strategy to resolve immunosuppression is administer oncolytic viruses (OVs) directly into the TME.219 OVs preferentially infect and replicate in malignant cells, culminating with cell lysis accompanied by the release of various cytokines and DAMPs in support of tumor-targeting immunity.219 Indeed, talimogene laherparepvec (T-VEC)—the first OV approved by the US Food and Drug Administration—mediated multiple immunomodulatory functions, including the GM-CSF-dependent recruitment, maturation, and activation of APCs culminating with the initiation of robust T cell responses with systemic outreach.220 Along similar lines, the oncolytic adenovirus AD5/3 has recently been shown to restore immunostimulation in the EOC microenvironment along with increased infiltration by CTLs.221 Therefore, OVs represent a promising combinatorial partner for other immunotherapeutic regimens in the management of solid tumors including EOC. For instance, T-VEC in combination with ICIs showed promising results in early phase clinical trials enrolling melanoma patients.222
Concluding remarks
Immunotherapy with ICIs has revolutionized the management of multiple tumor types, creating enormous expectations around the possibility of harnessing the patient immune system against EOC. However, the clinical benefit of ICIs as standalone immunotherapeutic interventions in women EOC is limited. This may reflect limited pre-existing immunity and/or the existence of robust immunosuppressive pathways in the EOC microenvironment.
In line with this notion, the findings discussed herein demonstrate that pre-existing immunity in the ovarian TME has a major impact on the sensitivity of EOC to (immuno)therapy,40 calling for the identification of immune biomarkers to integrate into common diagnostic assessments and guide treatment selection (table 4). For instance, women with highly infiltrated EOCs (so-called ‘hot’ tumors with an elevated immunoscore) may benefit from ICI-based immunotherapy or ACT, whereas individuals with an intermediate degree of immune infiltration are expected to respond to agents that stimulate CD8+ T cell infiltration (table 4). So-called ‘cold’ tumors which are characterized by a low immunoscore, remain the most challenging to eradicate and hence are associated with poor prognosis.
Potential immunotherapeutic strategies against ovarian carcinoma
A potential strategy to overcome the lack of pre-existing immunity in EOC is to combine a priming therapy that enhances T cell responses, such as DC-based vaccination, or strategies that turn the tumor into an in situ vaccine, such as RT, using an approach that either removes immunosuppressive cues (eg, ICI-based immunotherapy, TAM depletion) or provides immunostimulatory signals.10 Moreover, accumulating preclinical and clinical evidence indicates that epigenetic modifiers, including DNA demethylating agents and some chemotherapeutics, can stimulate anticancer immunity by various mechanisms, including the (1) selective depletion of immunosuppressive cells; (2) lymphodepletion associated with renovation of the patient immunological repertoire and (3) activation of immune effector cells and hence may be beneficial in patients with low or absent TILs (table 4). Similarly, immunogenic chemotherapeutics such as doxorubicin and paclitaxel, antiangiogenic drugs and PARP inhibitors stand out as promising partners for ICIs in the management of patients with ‘cold’ EOC.
We surmise that rationally designed combinations of conventional and immunotherapeutic agents will be critical to unlock immunosuppression in the EOC microenvironment in support of clinical efficacy. Preclinical studies in immunocompetent EOC identifying not only the agents to be used in such combinations, but also their optimal administration schedule223 are urgently required to translate an expanding literature on the immune contexture of EOC into clinically relevant therapeutic strategies.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors All authors listed have made substantial, direct and intellectual contributions to the work and approved it for publication.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests JF and RS are employees of Sotio Biotech a.s.; IV declares consulting for Astra Zeneca, Clovis Oncology, Carrick Therapeutics, Deciphera Pharmaceuticals, Elevar Therapeutics, F. Hoggmann-La Roche, Genmab, GSK, Immunogen, Jazzpharma, Mersana, Millennium Pharmaceuticals, MSD, Novocure, Octimet Oncology NV, Oncoinvent AS, Sotio a.s., Verastem Oncology, Zentalis; contracted research for Oncoinvent AS, Genmab; research funding from Amgen, Roche; LG declares research funding from Lytix and Phosplatin (completed) and speaker and/or advisory honoraria from Boehringer Ingelheim, Astra Zeneca, OmniSEQ, The Longevity Labs, Inzen, Onxeo, the Luke Heller TECPR2 Foundation; SO has patents for molecular signatures in ovarian cancer (US10253368 and EU2908913) and is funded by NCI (R01 CA208753) and VA (VA-ORD BX004974) grants. AC declares consulting for SOTIO; contracted research for Novocure and Oncoinvent.
Provenance and peer review Not commissioned; externally peer reviewed.