Article Text

Abstract
Background Immune checkpoint therapy (ICT) has low response rates in patients with metastatic castration-resistant prostate cancer (mCRPC), in part due to few T cells in the tumor microenvironment (TME). Anti-cytotoxic T lymphocyte-associated protein 4 (CTLA-4) promotes intratumoral T cell infiltration but induces upregulation of PD-1 and programmed death ligand-1 (PD-L1) within the prostate TME. Combined anti-CTLA-4 plus anti-PD-1 can partly overcome this adaptive resistance and was recently shown to augment responses in patients with mCRPC with measurable disease. Although bone is the most common site of metastasis in prostate cancer, patients with bone-predominant disease are frequently excluded from trials because they lack measurable disease, which limits assessment of disease progression and tissue sampling. We therefore designed this study to investigate combined ICT in mCRPC to bone.
Hypothesis Combined anti-CTLA-4 (tremelimumab) plus anti-PD-L1 (durvalumab) is safe and well tolerated in patients with chemotherapy-naïve mCRPC to bone.
Patients and methods In this single-arm pilot study, men with chemotherapy-naïve mCRPC to bone received tremelimumab (75 mg intravenous) plus durvalumab (1500 mg intravenous) every 4 weeks (up to four doses), followed by durvalumab (1500 mg intravenous) maintenance every 4 weeks (up to nine doses). The primary endpoint was incidence of adverse events. Secondary endpoints included serum prostate-specific antigen (PSA), progression-free survival (PFS), radiographic PFS (rPFS), and maximal PSA decline.
Results Twenty-six patients were treated between August 8, 2017 and March 28, 2019. Grade ≥3 treatment-related adverse events (TRAEs) occurred in 11 patients (42%), with no grade 4 or 5 events. TRAEs leading to discontinuation occurred in three patients (12%). PSA decline ≥50% occurred in three patients (12%). Six patients (24%) achieved stable disease for >6 months. At a median follow-up of 43.6 months, median rPFS was 3.7 months (95% CI: 1.9 to 5.7), and median overall survival was 28.1 months (95% CI: 14.5 to 37.3). Post-treatment evaluation of the bone microenvironment revealed transcriptional upregulation in myeloid and neutrophil immune subset signatures and increased expression of inhibitory immune checkpoints.
Conclusions Tremelimumab plus durvalumab was safe and well tolerated in patients with chemotherapy-naïve mCRPC to bone, with potential activity in a small number of patients as measured by rPFS. Combination of CTLA-4 and PD-L1 blockade with therapies targeting the myeloid compartment or other inhibitory immune receptors may be necessary to overcome mechanisms of resistance within prostate bone microenvironment.
Trial registration number NCT03204812.
- prostatic neoplasms
- tumor microenvironment
- immunotherapy
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Metastatic castration-resistant prostate cancer (mCRPC) is an immunologically ‘cold’ tumor, with a tumor microenvironment (TME) characterized in part by a relative paucity of infiltrating T cells, high proportion of suppressive myeloid cells, and immunosuppressive cytokines.1 In line with these characteristics, mCRPC has not achieved the same clinical benefit with immune checkpoint therapy (ICT) observed in many other cancers. Clinical trials of ICT targeting the programmed cell death-1/programmed death ligand-1 (PD-[L]1) axis alone have demonstrated minimal responses in patients with mCRPC.2 3 Moreover, ICT targeting cytotoxic T lymphocyte-associated protein 4 (CTLA-4) has been well studied in prostate cancer. In a phase III study, the addition of ipilimumab (anti-CTLA-4 monoclonal antibody) to radiotherapy to bone metastases in docetaxel-treated patients with mCRPC failed to meet its primary endpoint of increasing overall survival (OS), although ipilimumab did improve progression-free survival (PFS) compared with placebo.4 A subset of these patients achieved durable benefit with ipilimumab, which was associated with favorable prognostic features such as including hemoglobin >110 g/L, alkaline phosphatase <1.5 times the upper limit of normal, and absence of visceral metastases, features that may reflect disease biology, such as an absence of bone or liver involvement and overall low tumor burden.4 This durable benefit was borne out in the long-term survival analysis, in which ipilimumab was associated with two to three times higher survival rates at 3 years and beyond.5 Clinical trials of vaccines in mCRPC at the time also suggested that chemotherapy-naïve patients may derive the greatest benefit from immunotherapy, including a live poxviral-based heterologous prime-boost vaccine (PSA-TRICOM, PROSTVAC).6 A later phase III study of ipilimumab compared with placebo in chemotherapy-naïve mCRPC showed improved PFS, but did not meet its primary OS endpoint.7
Mechanistically, our group has shown that ipilimumab promoted intratumoral T cell infiltration.8 However, CTLA-4 blockade also induced upregulation of the inhibitory immune checkpoints PD-1 and PD-L1 within the prostate TME, suggesting a mechanism of adaptive resistance that could be overcome by targeting the CTLA-4 and PD-(L)1 pathways.8 Concurrent blockade of the CTLA-4 and PD-(L)1 pathways has demonstrated clinical activity in a range of tumor types, including melanoma, renal cell carcinoma, non-small cell lung cancer, hepatocellular carcinoma, and colorectal cancer.9–14 The combination of ipilimumab plus nivolumab (anti-PD-1) was subsequently reported in mCRPC in a single-institution trial of 30 patients selected for expression of the androgen receptor splice variant AR-V7 (including a 15 patient cohort that also received enzalutamide), irrespective of prior chemotherapy, and in a larger, multicenter trial of 90 patients with mCRPC stratified by chemotherapy exposure, which showed augmented responses in patients with measurable, that is, soft tissue disease.15–17 Notably, despite the fact that bone is the most common site of metastasis (in over 70% of patients) in prostate cancer,18 patients with bone-predominant disease are often excluded from clinical trials due to a lack of measurable disease, which can limit both tissue sampling as well as assessments of disease progression . We therefore designed this study to specifically enroll patients with bone metastases.
We hypothesized that the combination of anti-CTLA-4 and anti-PD-L1 would be safe, tolerable and yield clinical responses in an mCRPC population selected for bone metastases and no prior chemotherapy for castration-resistant disease. Herein, we report the clinical results and exploratory immunologic analysis of the bone microenvironment in a pilot study of tremelimumab (anti-CTLA-4) in combination with durvalumab (anti-PD-L1) in patients with chemotherapy-naïve mCRPC with bone metastases. This study represents the first published report of this combination in this patient population.
Methods
Study design and patients
This trial was an open-label, investigator-initiated, single-arm pilot study conducted at the University of Texas M.D. Anderson Cancer Center (M.D. Anderson, Houston, Texas, USA).
Key inclusion criteria included age 18 years or older; histologically or cytologically confirmed adenocarcinoma of the prostate with evidence of metastatic disease to the bone on radionuclide bone scan, CT scan or MRI scan; tumor progression on gonadotropin-releasing hormone analog with castrate levels serum testosterone (≤1.7 nmol/L or 50 ng/dL) with prostate-specific antigen (PSA) and/or radiographic progression according to Prostate Cancer Working Group 3 (PCWG3) criteria; asymptomatic or minimally symptomatic disease; Eastern Cooperative Oncology Group performance status of 0 or 1, and adequate hematologic (including hemoglobin ≥110 g/L), renal, and hepatic function. Key exclusion criteria included evidence of visceral metastases to the liver, prior treatment with taxane-based chemotherapy for CRPC, and prior treatment with anti-PD-(L)1 or anti-CTLA-4 therapy. Docetaxel was permitted in the castration-sensitive setting per CHAARTED (ChemoHormonal Therapy Versus Androgen Ablation Randomized Trial for Extensive Disease in Prostate Cancer) and STAMPEDE (Systemic Therapy in Advancing or Metastatic Prostate Cancer: Evaluation of Drug Efficacy) trials.19–21 Full inclusion and exclusion criteria are provided in the clinical protocol (online supplemental appendix 1).
Supplemental material
Treatment
Patients were treated with the combination of tremelimumab (75 mg intravenously every 4 weeks for up to four doses) and durvalumab (1500 mg intravenously every 4 weeks). Four weeks after the last dose of combination therapy, patients received maintenance durvalumab (1500 mg intravenously every 4 weeks) for up to nine doses or until disease progression, intolerable toxicity, physician decision, or withdrawal of consent. Patients who experienced adverse events (AEs) related to combination dose therapy but not meeting dose discontinuation criteria were permitted to proceed to durvalumab monotherapy without completing all four combination doses provided a minimum of two combination cycles were administered. Complete discontinuation criteria and AE management guidelines are provided in the protocol (online supplemental appendix 1). Retreatment was permitted once per protocol in two clinical scenarios: (1) patients who achieved disease control (stable disease, partial response, or complete response) through the end of the 12-month treatment period were allowed to restart treatment with the combination on disease progression; (2) patients who received at least two doses of tremelimumab and durvalumab who had evidence of disease progression during the durvalumab monotherapy portion were permitted to restart treatment with the combination.
Assessments and endpoints
PSA assessment was obtained at baseline and every 4 weeks (with each cycle). Radiographic tumor assessment by CT and technetium-99m-MDP bone scintigraphy was performed at baseline, every 8 weeks for 48 weeks and then every 12 weeks until confirmed progression. AEs were collected throughout treatment and graded per the Common Terminology Criteria for Adverse Events (CTCAEs) V.4.03. Survival was assessed during follow-up at 30 days, 3 months, and 12 months. The primary endpoint was incidence of AEs. Secondary endpoints were PSA PFS, radiographic PFS (rPFS) per RECIST V.1.1 with PCWG3 modifications assessed by investigator review, and maximal PSA decline. PSA PFS was defined as per PCWG3 criteria: time from start of therapy to first PSA increase of 25% and ≥2 ng/mL above the nadir, and which is confirmed by a second value ≥3 weeks later. Peripheral blood mononuclear cells (PBMCs) and bone marrow collections (aspirate and core biopsy from the posterior iliac crest) were collected at baseline, after two doses and after four doses of tremelimumab and durvalumab. Pre-treatment and post-treatment bone marrow collections were obtained from the same anatomic site (posterior iliac crest). Detailed endpoint definitions are provided in the clinical protocol (online supplemental appendix 1).
Statistical analyses
The full statistical analysis plan is available in the protocol in online supplemental appendix 1. All patients who received at least one dose of study drug were included in safety reporting and interim safety monitoring. Anti-tumor activity was analyzed for all patients who received at least two doses of study drug and had at least one follow-up for clinical outcome measures. All AEs were tabulated by their CTCAE V.4.03 name, counted once at the worst grade and the ultimate attribution within the patient. PSA PFS and rPFS were calculated from the first day of treatment and summarized by Kaplan-Meier methods. The numeric PSA value at maximal decline was summarized. No hypothesis testing was planned. The planned sample size was 20 evaluable patients as a pilot sample size with sufficient numbers to establish safety and for analysis of secondary endpoints. Up to 27 patients were permitted to be enrolled to achieve 20 evaluable patients. Exploratory subgroup comparisons used the log-rank test and are reported with median time and 95% CIs. All clinical analyses were performed in SAS V.9.4 (SAS Institute) and Kaplan-Meier figures were generated in Stata V.16 (StataCorp LLC).
Genetic testing
Data were extracted from patients who underwent standard of care germline genetic testing (Invitae or GeneDx) or somatic tumor mutational testing using next generation sequencing (NGS). All patients who underwent somatic tumor mutational testing used an in-house genomic platform except for one patient who had testing via Caris. The MD Anderson (MDA) Solid Tumor Genomic Assay (MDA STGA Oncomine) is an internal NGS-based analysis for the detection of somatic mutations in the coding sequence of 128 genes (134 genes in updated 2018 panel) and selected copy number variations (amplifications) in 47 genes (49 genes in updated 2018 panel) with overlap in 134 genes (146 genes total in updated 2018 panel) performed on DNA extracted from the sample in a CLIA-certified molecular diagnostics laboratory. For patients in whom inadequate tumor tissue was available for the MDA STGA Oncomine panel, an alternative NGS-based analysis for detection of somatic mutations in the coding sequence of 50 genes was available (MDA CM50). The genomic reference sequence used is GRCh37/hg19. Detailed information on signal-processing, base calling, alignment, variant calling, and copy number calling algorithms is available on request. DNA damage repair (DDR) defects were defined as ≥1 gene mutation in the relevant gene panel (eg, ATM, BRCA1, BRCA2, CDK12, CHEK2, FANCA, PALB2). This method did not permit calculation of tumor mutational burden.
NanoString
Bone marrow aspirates (BMAs) from patients were collected in Vacutainer or Cell Preparation Tubes (CPTs) containing sodium heparin (BD Vacutainer, Franklin Lakes, New Jersey, USA). Mononuclear cells were isolated from BMA by centrifuging the CPTs at 2000rpm (863g) for 15 min at room temperature. Diluted sample (1:5) with phosphate buffered saline (PBS) was layered over 10 mL of Ficoll. The mixture was centrifuged at 2000 rpm (863g) for 20 min at room temperature with no brakes. The interface cells were harvested and washed twice with PBS containing 10% fetal calf serum at 500g and 450g for 10 min, respectively, and cryopreserved. At the time of RNA isolation, stored cells were processed with RiboPure RNA Purification Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Extracted RNA was quantified by ND Nanodrop1000 spectrometer (Thermo Scientific, Wilmington, Massachusetts, USA). For NanoString assay, 100 ng of RNA was used to detect immune gene expression using nCounter PanCancer Immune Profiling panel along with custom CodeSet. Counts of the reporter probes were tabulated for each sample by the nCounter Digital Analyzer and raw data output was imported into nSolver (http://www.nanostring.com/products/nSolver, V.4.0). nSolver data analysis package was used for normalization, cell type analysis, and differential gene expression analysis and gene set enrichment analysis (GSEA) were performed with Qlucore Omics Explorer V.3.5 software (Qlucore, New York, USA).22 23 Data were plotted using GraphPad Prism 8 (GraphPad Software V.8.4.3). Statistical analysis was performed using the two-tailed Student’s t-test to compare two groups. P values less than 0.05 denote significant differences.
Immunohistochemistry
Pre-treatment and post-treatment bone biopsy cores were decalcified in a working solution of equal parts 8% hydrochloric acid and 8% formic acid at 20× volume, with daily solution changes until decalcification was completed. The specimens were rinsed in water, neutralized with a concentrated ammonia solution, and washed. The bone biopsy cores and clots were fixed in 10% formalin, embedded in paraffin and cut into transverse 4 µm serial sections. H&E and immunohistochemical staining was performed using the CD8 (Thermo Scientific, cat# MS457S), CD45RO (Leica Microsystems, cat# PA0146), FOXP3 (Bio Legend, cat# 320108), and CD68 (Agilent-Dako, cat# M0876) antibodies. For quantification, slides were scanned and digitalized using the Scan ScopeXT system (Leica Technologies, Buffalo Grove, Illinois, USA). Quantification analysis was done using the HALO 3.0 software (Indica Labs, Albuquerque, New Mexico, USA) and percent positive cells (number of positive cells/mm2) were plotted. Statistical analysis was performed using the Graphpad Prism software V.8. P values of <0.05 denote statistical significance.
Results
Patients
Between August 8, 2017 and March 28, 2019, 26 patients were treated with at least one dose of the combination of tremelimumab and durvalumab (figure 1). Baseline characteristics of these patients are listed in table 1 and treatment exposure and patient disposition are listed in online supplemental table 1. The median age was 68 years old (range: 48–89), and baseline laboratory values are listed in online supplemental table 2. The median baseline PSA was 29.6 ng/mL (IQR: 12.2–77.3). Most patients (77%) had Gleason 8 or greater disease. All patients had bone metastases as shown in online supplemental figure 1. Visceral metastases were present in three patients (11%; lung-only, n=1; adrenal-only, n=1; adrenal and soft tissue (base of penis), n=1). Six patients (23%) had received docetaxel in the castration-sensitive setting. Nineteen patients (73%) had received prior next-generation hormonal therapy, including abiraterone, enzalutamide, or apalutamide; and 13 patients (50%) had received prior sipuleucel-T. A complete listing of prior therapies received is provided in online supplemental table 3. As of the June 30, 2021 data cut-off, the median follow-up was 43.6 months (online supplemental table 1).
Supplemental material

Study schema
Baseline patient characteristics
Safety
All patients except one received at least two combination doses of tremelimumab plus durvalumab (n=25, 96%); this patient was followed for safety but excluded from subsequent efficacy analyses as pre-specified in the protocol. The median number of combination doses administered was 3 (IQR: 2–4). Ten patients received all four combination doses (38%) (online supplemental table 1). One patient (#11) was retreated per protocol and received a total of eight combination doses. Treatment-related adverse events (TRAEs) of any grade occurred in 24 patients (92%), with grade ≥3 TRAEs in 11 patients (42%) (table 2). It should be noted that there were no grade 4 or 5 AEs observed. The most common AEs of any grade included rash (38%), anemia (31%), and increases in amylase (27%) or cortisol (27%), consistent with previous reports,24–27 and the most frequent grade 3 events were increased lipase (15%), increased amylase (12%), and colitis/diarrhea (8%) (table 2). Immune-related adverse events (irAEs) ≥grade 3 included myositis (4%), colitis/diarrhea (8%), and type 1 diabetes mellitus (4%). AEs leading to discontinuation of treatment occurred in three patients (12%): colitis (Patient #14), peripheral sensory neuropathy (Patient #19), and myositis (Patient #27).
Treatment-related adverse events (TRAEs)
Efficacy
Twenty-five patients were included in the final efficacy analysis with a median follow-up time of 43.6 months. One patient (#18) received only one dose of combination therapy and was excluded from efficacy analysis as pre-specified in the clinical protocol. The median PSA PFS was 0.9 months (95% CI: 0.9–1.8) (figure 2A), median rPFS was 3.7 months (95% CI: 1.9 to 5.7) (figure 2B) and median OS was 28.1 months (95% CI: 14.5 to 37.3) (figure 2C). The 1-year, 2-year, and 3-year OS were 96%, 55%, and 35%, respectively (table 3). PSA decline of any magnitude was observed in six patients (24%). A PSA decline ≥50% (PSA50) occurred in three patients (12%), with a PSA decline ≥90% (PSA90) in one patient (4%) (figure 2D). Among the six patients with measurable disease, no radiographic partial or complete responses were observed. Stable disease greater than or equal to 6 months was achieved in six patients, yielding a disease control rate of 24% (figure 2E and table 3). One patient (#11) who experienced radiographic progression during the monotherapy phase was re-treated with the combination per protocol and received a total of eight combination doses. For this patient, the time to first radiographic progression (rPFS1) was 13.8 months and time to second radiographic progression (rPFS2) was 19.8 months.

Secondary efficacy outcomes. A) PSA progression free survival (PSA PFS). B) Radiographic progression free survival (rPFS). Tick marks denote censored events. C) Overall survival (OS). D) Waterfall plot of best percentage change from baseline in PSA (top) and changes in PSA over time (bottom). Changes +100% were capped at 100%. E) Swimmer’s plot of radiographic progression free survival (rPFS) in months. *Denotes patient who was re-treated per-protocol.
Summary of efficacy outcomes
Exploratory analysis
To characterize subsets of patients who benefited from treatment, we performed post-hoc exploratory subgroup analyses of rPFS and OS by baseline characteristics that have previously been hypothesized to confer benefit with treatment with ICT (eg, baseline PSA, alkaline phosphatase, volume of disease) and did not observe an association with outcomes (online supplemental table 4).4 19 28 29
Immunogenetics
Immunogenetics have recently been shown to identify subsets of patients who may respond to ICT.30 In particular, as prior studies have shown that DDR defects may sensitize to ICT, we examined the subset of patients who had undergone germline (n=13) and/or somatic (n=16) tumor mutational testing.31 Among the six patients with rPFS >6 months, four underwent germline or somatic mutational testing, and DDR alterations were identified in three patients: #23, somatic PALB2 and germline BRCA2; #21, somatic ATM; and #7, germline ATM (online supplemental table 5). Notably, two patients who carried somatic CDK12 alterations (#3 and #16) had rPFS less than 6 months, although our method did not permit identification of the biallelic CDK12 loss that has previously been reported as a distinct subset of patients that may benefit from ICT (online supplemental table 5).32 We observed a prolonged response in one patient (#11) with a somatic mutation in SMARCB1 (figure 3). Finally, as we have previously demonstrated that two doses of anti-CTLA-4 therapy with ipilimumab induced T cell infiltration into prostate tumors, we examined the association between number of combination doses received and rPFS and observed numerically prolonged rPFS with increasing doses of the combination (online supplemental figure 2).
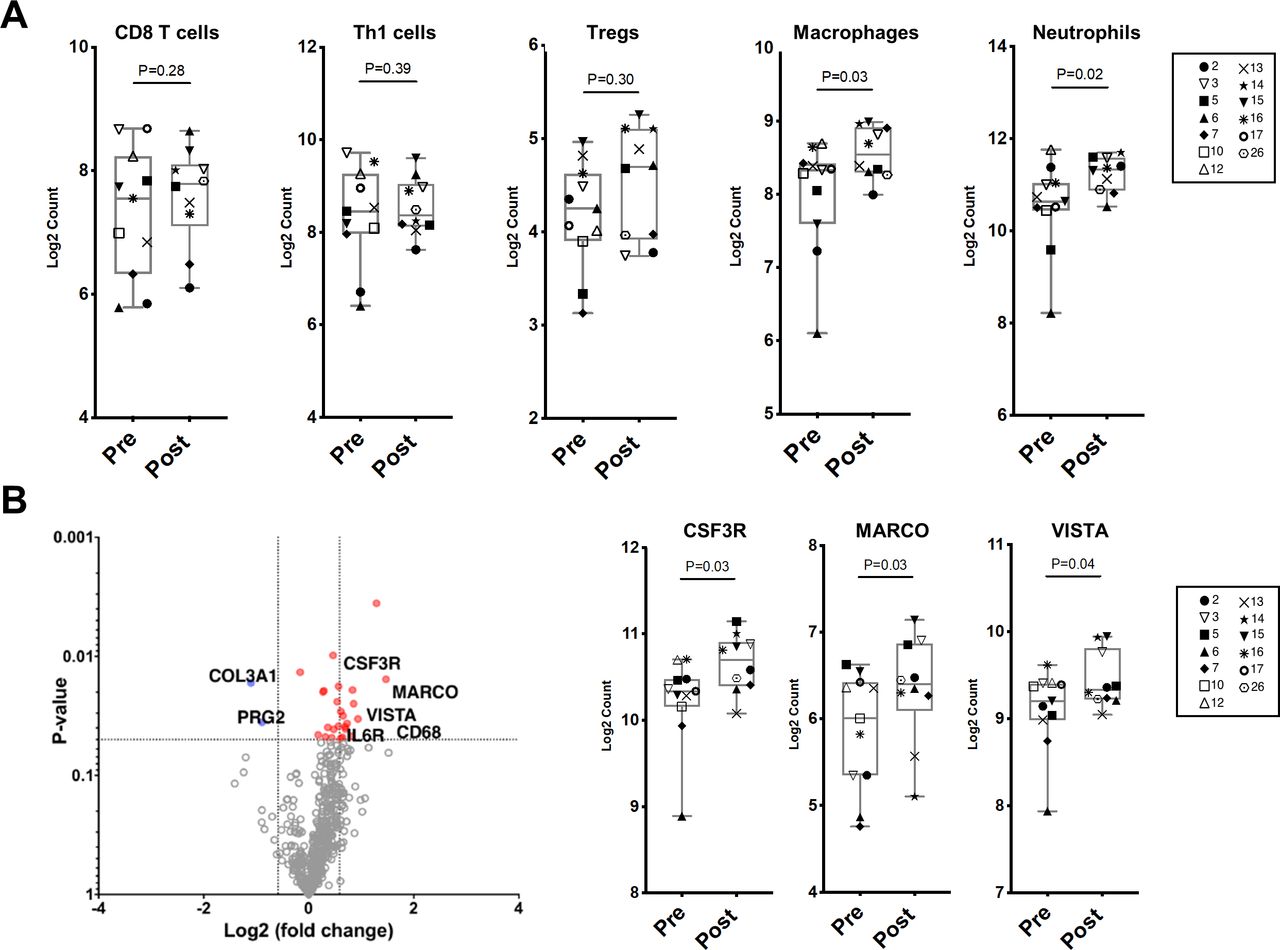
Changes in immune cell subsets and specific immune markers in bone marrow aspirates by Nanostring RNA expression profiling after tremelimumab and durvalumab. A) Scatter plots of immune cell phenotypes in bone marrow aspirates pre- and post-treatment with tremelimumab and durvalumab. B) Left: Volcano plot of differentially expressed genes (Log2 FC>1.2 and p-value <0.05) pre-and post-treatment. (Pre in blue, n=11; post in red, n=10). Right: Scatter plots showing expression of specific immune markers pre- and post-treatment.
Immune monitoring of the bone microenvironment
We have previously shown that anti-CTLA-4 with ipilimumab induces T cell infiltration within the soft tissue TME, with adaptive upregulation of the inhibitory checkpoints PD-L1 and V-domain Ig suppressor of T cell activation (VISTA) on immune cells, including subsets of macrophages.8 We therefore investigated the impact of combined anti-CTLA-4 and anti-PD-L1 therapy on the bone microenvironment. We analyzed BMAs collected from patients pretreatment and post-treatment by NanoString RNA expression profiling. Patients #11 and #27 had the longest rPFS, which allowed us to view them as having a clinical response. We had available samples for immune monitoring studies for patient #11 but not patient #27. Across all evaluable patients except patient #11, we did not observe a difference in transcriptional profiles associated with T cell populations pretreatment and post-treatment (CD8 T cells, Th1 cells, and Tregs) (figure 3A). We did identify a statistically significant increase in macrophage and neutrophil transcriptional signatures following treatment with tremelimumab and durvalumab (figure 3A). In keeping with this increase in macrophage and neutrophil transcriptional profiles, specific immune markers that are highly expressed on myeloid cells were found to be upregulated post-treatment. These markers included CSF3R (encodes for granulocyte colony-stimulating factor receptor and promotes differentiation into neutrophils and macrophage), MARCO (encodes for macrophage receptor with collagenous structure, a scavenger receptor on macrophages), VISTA (inhibitory immune checkpoint strongly expressed on myeloid cells), and leukocyte immunoglobulin-like receptor subfamily B member 4 (LILRB4), an inhibitory receptor prominently expressed on tumor-associated macrophages (figure 3B and online supplemental figure 3).33 Finally, to confirm these findings, we performed quantitative immunohistochemistry (IHC) on bone marrow cores from patients at baseline and post-treatment. There was no change in T cell subsets (CD8, CD45RO, and FoxP3), however, there was a trend toward increased CD68 macrophages following treatment (online supplemental figure 4).
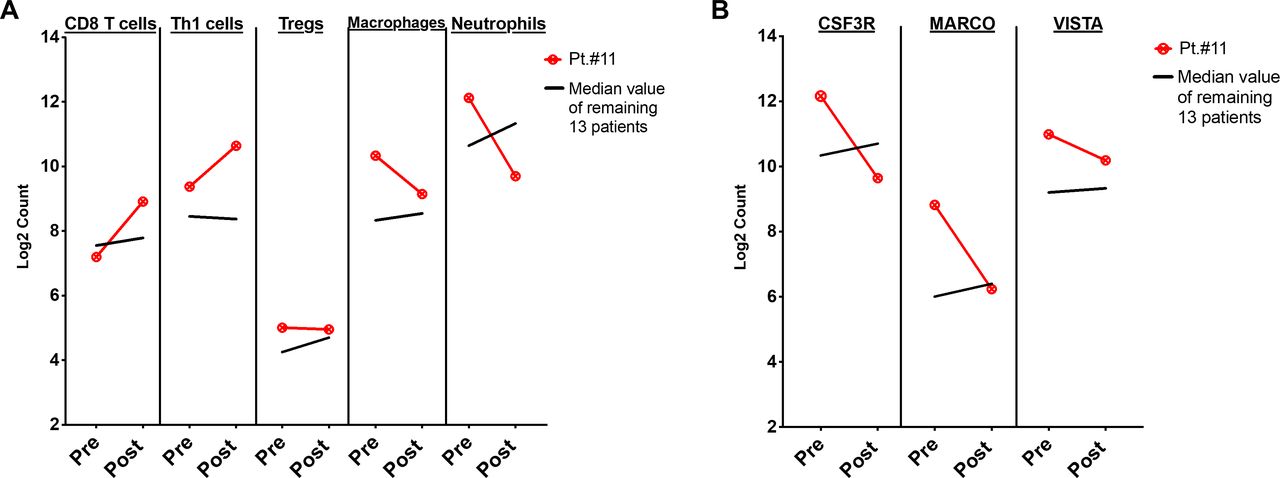
To further investigate correlates of response in the bone microenvironment, we next analyzed patient #11, who had prolonged PFS and had been retreated with the combination per protocol (bone marrow samples for this patient were collected prior to retreatment with the combination). Compared with all other patients, this patient had increases in transcriptional profiles associated with CD8 T cells and Th1 cells, without increases in regulatory T cells (Tregs), macrophages, or neutrophils (figure 4A). Similarly, this patient did not have increased CSF3R, MARCO, or LILRB4 expression (figure 4B and online supplemental figure 5). To confirm these transcriptional findings, quantitative IHC was performed on bone marrow cores for this patient, which demonstrated increased CD8 T cells and CD45RO memory T cells following treatment, without an increase in FoxP3+ Tregs or CD68 macrophages (online supplemental figure 6). Finally, to evaluate if these changes were exclusive to patient #11, we analyzed transcriptional profiles for patient #7 (rPFS 12.8 months). We observed no significant changes in immune cell subsets in this patient (online supplemental figure 7), which may have been attributable to his history of chronic lymphocytic leukemia (CLL, Rai Stage 0) and the impact of low-grade CLL on the immune response.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.Changes in immune cell subsets and specific immune markers in a patient with prolonged response (#11). A) Line graphs showing immune cell phenotypes by Nanostring at baseline (Pre), and after treatment with tremelimumab and durvalumab (Post) in patient #11. B) Line graphs showing expression of immune markers by gene expression at baseline (Pre), and after treatment with tremelimumab and durvalumab (Post) for patient #11. Red line denotes patient #11; black line denotes median values of each cell type/immune marker for remaining 13 patients.
Discussion
The prostate TME is ‘immunologically cold’, with few infiltrating T cells, high myeloid suppressive cells,34 35 and immunosuppressive cytokines,36 37 among other features, which may help explain the poor responses observed in trials of ICT with PD-(L)1 inhibition, including a phase II study of pembrolizumab (anti-PD-1) that reported response rates of 3%–5%, and a phase III study that showed the addition of atezolizumab (anti-PD-L1) to enzalutamide (androgen receptor antagonist) did not improve OS.1–3 Our group has previously shown that CTLA-4 inhibition with ipilimumab promoted T cell infiltration into prostate tumors.8 However, CTLA-4 blockade also induced upregulation of the inhibitory immune checkpoints PD-1 and PD-L1 within the prostate TME, suggesting a mechanism of adaptive resistance that could be overcome through combination immune checkpoint blockade.8
In this single-arm pilot study of durvalumab plus tremelimumab in chemotherapy-naïve mCRPC to bone, the combination was safe and well tolerated, with activity observed in a subset of patients. Post-treatment evaluation of the prostate bone microenvironment revealed a transcriptional upregulation in myeloid and neutrophil immune subset signatures, and increased expression of inhibitory immune checkpoints.
Combination blockade of the CTLA-4 and PD-L1 pathways has been evaluated in three clinical trials to date, of which two are ongoing. The first trial examined durvalumab with or without tremelimumab following enzalutamide or abiraterone and no more than one taxane chemotherapy (NCT02788773), and has reported preliminary results, which have not been published.38 The second trial was a phase II study of ipilimumab 1 mg/kg plus nivolumab 3 mg/kg (IPI1+NIVO3) with or without enzalutamide (n=15 in each cohort) in AR-V7+ mCRPC irrespective of prior chemotherapy (NCT02601014).15 17 Among patients with measurable disease, this trial reported objective responses in two patients, leading the authors to conclude that the combination was ineffective in the overall AR-V7+ study population, though higher response rates were observed among DDR+ patients.15 The heterogeneous study population, small sample size, and restriction to the small fraction of patients with AR-V7 positivity limits the generalizability of this trial. The third study, a multicenter phase II trial of ipilimumab 3 mg/kg plus nivolumab 1 mg/kg (IPI3 +NIVO1) (NCT02985957, CheckMate 650), reported response rates of 10%–25% in patients with measurable (ie, soft tissue) disease, at the expense of greater toxicity.16 We observed a relatively low degree of toxicity in our durvalumab plus tremelimumab study, which may be related to the dose intensity of anti-CTLA-4—an expansion phase of CheckMate 650 exploring alternative dosing strategies of ipilimumab is currently ongoing. Furthermore, unlike CheckMate 650, we did not observe radiographic responses in this study, which may be attributable to the fact that all patients had bone metastases.16 Therefore, strategies accounting for mechanisms of resistance to ICT in patients with bone metastases are essential.
Our group has recently demonstrated that the prostate bone microenvironment may serve as a site of primary immune resistance, a relevant consideration given that over 70% of patients with mCRPC harbor bone metastases.18 39 We have shown that whereas ipilimumab increases Th1 subsets in the soft tissue TME (associated with anti-tumor responses), the bone microenvironment is enriched in transforming growth factor beta (TGF-β), which favors Th17 development over Th1 responses and is associated with primary resistance to ICT.39 Due to the difficulty of obtaining adequate samples from sclerotic bone lesions, we were unable to perform the same CyTOF and cytokine protein analyses in this study. Nevertheless, our evaluation of the immune compartment within the BMAs in this trial demonstrated upregulation in myeloid and neutrophil immune subset transcriptional signatures, and increased expression of inhibitory immune checkpoints. Based on these results, future trials of ICT in patients with bone-predominant disease should consider concurrent targeting of the myeloid compartment (either through direct targeting, such as via CSF-1R,40 41 or through the addition of specific tyrosine kinase inhibitors42), immunosuppressive cytokines (such as IL-843 44 or TGF-β39) or inhibitory immune checkpoints such as VISTA or LILRB4.
Limitations of this trial included its single-center patient population and small sample size, having been designed as a pilot study to investigate safety and explore efficacy. Additionally, the selection criteria were relatively narrow, enrolling patients with chemotherapy-naïve mCRPC with bone metastases, with or without visceral metastases (excluding those with liver metastases) and with excellent bone marrow reserve (including hemoglobin ≥110 g/L).
In conclusion, the combination of tremelimumab plus durvalumab is safe and well tolerated in chemotherapy-naïve patients with mCRPC to the bone, with anti-tumor activity in a subset of patients. The path forward for combination ICT in mCRPC will rely on: (1) optimal dosing strategies to balance clinical benefit with toxicity, (2) improved patient selection for treatment, accounting for both clinical factors and immunogenetics, and (3) rational combination strategies to overcome mechanisms of immunologic resistance.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
The trial was conducted in accordance with the Declaration of Helsinki Good Clinical Practice guidelines. The protocol and its amendments were approved by the Institutional Review Board (IRB) at MD Anderson. Written informed consent for participation in the study was obtained from all participants. Patients also provided written informed consent to M.D. Anderson IRB-approved laboratory protocols, PA13-0291 and LAB02-152.
Acknowledgments
We are grateful to our patients and their families for their participation in this study. We thank AstraZeneca for their support of this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @BeckyTidwell1
SKS and BAS contributed equally.
Contributors The first draft of the manuscript was written by BAS, AV, and SKS. All authors contributed to the final manuscript and approved the decision to submit the manuscript for publication. The corresponding author and principal investigator had access to all data in the study and had final responsibility for the decision to submit for publication.
Funding These studies were supported in part by MedImmune/AstraZeneca. The research work was also supported by The University of Texas MD Anderson Cancer Center Prostate Cancer Moon Shot Program; The V Foundation for Cancer Research’s Lloyd Family Clinical Oncology Scholar Award D2018-014 (to SKS); NIH Training Grant T32 CA009666 (BAS); and NIH/NCI Award P30CA016672. The Genitourinary Cancers Program of the Cancer Center support grant shared resources at The University of Texas M.D. Anderson Cancer Center. JPA and PS are members of the Parker Institute for Cancer Immunotherapy at The University of Texas M.D. Anderson Cancer Center.
Competing interests SS reports Consulting or Advisory Role: Amgen, Apricity Health, AstraZeneca, Bayer, Bristol-Myers Squibb, Cancer Expert Now, Dava Oncology, Dendreon, Exelixis, Kahr Bio, Janssen Oncology, Javelin Oncology, and MD Education Limited; Research Funding: AstraZeneca, Bristol-Myers Squibb, and Janssen Oncology; Other (Joint Scientific Committee): Janssen Oncology, Polaris. AA reports Consulting or Advisory Role: Astellas Pharma US, Bayer HealthCare Pharmaceuticals, Janssen Research & Development; Other (Joint Scientific Committee): American Cancer Society. CJL reports Grants or Contracts: Janssen, Bristol-Myers Squibb, Pfizer, ORIC Pharmaceuticals; Payment or Honoraria: Merck, Sharp & Dohme, Bayer, Amgen. JA reports Consulting or Stock Ownership or Advisory Board: Achelois, Adaptive Biotechnologies, Apricity, BioAtla, BioNTech, Candel Therapeutics, Codiak, Dragonfly, Earli, Enable Medicine, Hummingbird, ImaginAb, Jounce, Lava Therapeutics, Lytix, Marker, PBM Capital, Phenomic AI, Polaris Pharma, Time Bioventures, Trained Therapeutix, Two Bear Capital, Venn Biosciences. PS reports Consulting or Stock Ownership or Advisory Board: Achelois, Adaptive Biotechnologies, Affini-T, Apricity, BioAtla, BioNTech, Candel Therapeutics, Catalio, Codiak, Constellation, Dragonfly, Earli, Enable Medicine, Glympse, Hummingbird, ImaginAb, Infinity Pharma, Jounce, JSL Health, Lava Therapeutics, Lytix, Marker, Oncolytics, PBM Capital, Phenomic AI, Polaris Pharma, Sporos, Time Bioventures, Trained Therapeutix, Two Bear Capital, Venn Biosciences.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.