Article Text

Abstract
Background Recurrent and/or metastatic (R/M) disease develops in approximately 65% of patients with squamous cell carcinoma of the head and neck (SCCHN) and is associated with a poor prognosis. Immune checkpoint inhibitors have proven effective in multiple tumor types, including R/M SCCHN. We report the efficacy and safety of avelumab (antiprogrammed death ligand 1 antibody) in an expansion cohort of patients with platinum-refractory/ineligible R/M SCCHN enrolled in the phase I JAVELIN Solid Tumor trial (NCT01772004).
Methods Eligible patients with R/M SCCHN were aged ≥18 years and had received ≥1 line of platinum-based chemotherapy with disease progression or recurrence within 6 months of the last dose or were ineligible for platinum-based chemotherapy. All patients received avelumab 10 mg/kg every 2 weeks. Tumor assessments were carried out by a blinded independent review committee (IRC) and investigators according to Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST 1.1). Key endpoints included best overall response, duration of response (DOR) and progression-free survival (PFS) assessed by IRC and investigator per RECIST 1.1, overall survival (OS), and safety.
Results Between April 24, 2015, and November 13, 2015, 153 patients were enrolled. Patients had a median of two prior lines of therapy for metastatic or locally advanced disease (range 0–6); 12 patients (7.8%) were not eligible for platinum-based chemotherapy. At data cut-off (December 31, 2017), the confirmed objective response rate was 9.2% (95% CI 5.1% to 14.9%) assessed by IRC and 13.1% (95% CI 8.2% to 19.5%) assessed by investigator. Median DOR was not reached (95% CI 4.2 to not estimable) based on IRC assessment. Median PFS was 1.4 months (95% CI 1.4 to 2.6) assessed by IRC and 1.8 months (95% CI 1.4 to 2.7) assessed by investigator; median OS was 8.0 months (95% CI 6.5 to 10.2). Any-grade treatment-related adverse events (TRAEs) occurred in 83 patients (54.2%) and were grade ≥3 in 10 patients (6.5%). The most common TRAEs were fatigue (n=19, 12.4%), fever (n=14, 9.2%), pruritus (n=12, 7.8%), and chills (n=11, 7.2%), and there were no treatment-related deaths.
Conclusion Avelumab showed clinical activity and was associated with a low rate of grade ≥3 TRAEs in heavily pretreated patients with platinum-refractory/ineligible R/M SCCHN.
- immunotherapy
- head and neck neoplasms
Data availability statement
Data are available upon reasonable request. For all new products or new indications approved in both the European Union and the United States after January 1, 2014, Merck (CrossRef Funder ID: 10.13039/100009945) will share patient-level and study-level data after deidentification, as well as redacted study protocols and clinical study reports from clinical trials in patients. These data will be shared with qualified scientific and medical researchers, upon researcher’s request, as necessary for conducting legitimate research. Such requests must be submitted in writing to the company’s data sharing portal. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. Where Merck has a co-research, co-development, or co-marketing/co-promotion agreement or where the product has been out-licensed, it is recognized that the responsibility for disclosure may be dependent on the agreement between parties. Under these circumstances, Merck will endeavor to gain agreement to share data in response to requests.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Squamous cell carcinoma of the head and neck (SCCHN) accounts for approximately 90% of all head and neck cancers.1 Potential causes of SCCHN include tobacco use, alcohol consumption, and human papillomavirus (HPV) infection.2–4 Recurrent and/or metastatic (R/M) SCCHN develops in approximately 65% of patients and is associated with poor prognosis; median overall survival (OS) is <1 year.5
Treatment options for patients with R/M SCCHN have improved in recent decades.5 The antiepidermal growth factor receptor monoclonal antibody cetuximab, in combination with platinum-based chemotherapy, was the first treatment to improve survival for the first-line treatment of R/M SCCHN compared with platinum-based chemotherapy alone.5 6 However, this treatment strategy is associated with increased toxicity.5 6 Furthermore, not all patients are eligible for platinum-based chemotherapy, and treatment options for platinum-ineligible patients include single-agent non-platinum chemotherapy (eg, paclitaxel, 5-fluorouracil, docetaxel, methotrexate, or capecitabine).7 8
Recently, immune checkpoint inhibitors have proven effective in patients with R/M SCCHN, leading to regulatory approvals of the antiprogrammed death 1 (PD-1) monoclonal antibodies nivolumab and pembrolizumab.9–14 Nivolumab is approved as second-line treatment of R/M SCCHN with disease progression on or after platinum-containing chemotherapy, irrespective of programmed death ligand 1 (PD-L1) status.10 Pembrolizumab is approved for first-line treatment in combination with platinum and 5-fluorouracil or as a monotherapy for patients with unresectable R/M SCCHN whose tumors express PD-L1 with a combined positive score of ≥1 in both the USA and Europe (PD-L1 with a combined positive score of ≥1 required for monotherapy in the USA only) and as a second-line treatment for patients with R/M SCCHN with disease progression on or after platinum-containing chemotherapy and whose tumors express PD-L1 with a tumor proportion score of ≥50% (PD-L1 with a tumor proportion score of ≥50% required in Europe only).13 14 Avelumab, an anti–PD-L1 monoclonal antibody, has shown clinical activity and durable responses in patients with a range of tumor types.15–18 Avelumab is approved in multiple countries worldwide as a monotherapy for the treatment of metastatic Merkel cell carcinoma and locally advanced or metastatic urothelial carcinoma (first-line maintenance and second-line treatment), and in combination with axitinib for the first-line treatment of advanced renal cell carcinoma.19
Here, we report the efficacy and safety of avelumab in the dose-expansion cohort of patients with platinum-refractory/ineligible R/M SCCHN enrolled in the phase I JAVELIN Solid Tumor trial.
Methods
Study design and patients
JAVELIN Solid Tumor (NCT01772004) is an open-label, multicenter trial in patients with various advanced solid malignancies and included several expansion cohorts enrolled after the initial dose-escalation phase. In this phase Ib dose-expansion cohort, eligible patients were aged ≥18 years; had histologically or cytologically confirmed R/M SCCHN; had received ≥1 line of platinum-based chemotherapy with disease progression or recurrence within 6 months of the last dose of platinum-based therapy given in the adjuvant, neoadjuvant, first-line, or R/M setting (or were ineligible for platinum-based chemotherapy); had measurable disease according to Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST 1.1); and had an Eastern Cooperative Oncology Group performance status (ECOG PS) score of 0 or 1. Patients were not selected based on PD-L1 or HPV status. Exclusion criteria included prior therapy with any therapy targeting T-cell coregulatory proteins (including anti-PD-L1/anti-PD-1 or anticytotoxic T lymphocyte-associated protein 4 antibodies), known autoimmune disease or hypersensitivity to monoclonal antibodies, active or history of central nervous system metastases, and other cancer diagnosis within 5 years prior to study entry. Full eligibility criteria have been published previously.15
Treatment
All patients received avelumab 10 mg/kg every 2 weeks until disease progression, unacceptable toxicity, or other protocol-specified criterion for withdrawal; dose reductions were not permitted (guidelines for treatment delay or discontinuation have been reported previously).15 Patients received premedication with antihistamine and paracetamol (acetaminophen) prior to each dose of avelumab to mitigate infusion-related reactions (IRRs).
Assessments
Clinical activity and safety were analyzed in all patients who received at least one dose of avelumab. Radiographical tumor assessments were carried out by a blinded independent review committee (IRC) and investigators according to RECIST 1.1 every 6 weeks for the first year, then every 12 weeks thereafter.
Safety was assessed every 2 weeks; adverse events (AEs) were classified according to the Medical Dictionary for Regulatory Activities (MedDRA) V.21.1 and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events V.4.0. Immune-related adverse events (irAEs) were based on a prespecified list of MedDRA preferred terms followed by comprehensive medical review. IRRs were identified using an expanded definition that included a prespecified list of MedDRA preferred terms (IRR, drug hypersensitivity, hypersensitivity, type I hypersensitivity, or anaphylactic reaction) that occurred on the day of infusion or the following day after infusion, in addition to signs and symptoms of IRR that occurred on the same day of infusion and resolved within 2 days (including AEs classified by investigators as related or unrelated to treatment).
PD-L1+ status was assessed by the central laboratory using the PD-L1 immunohistochemistry 73–10 pharmDx assay (Dako, Carpinteria, California, USA). PD-L1+ status was defined as PD-L1 expression on ≥1% of tumor cells; expression cut-offs of ≥50% and ≥80% were also analyzed. HPV status was assessed by the central laboratory in all patients using p16 immunohistochemistry (CINtec Histology, Ventana Medical Systems; Roche, Indianapolis, Indiana, USA). HPV+ status was defined as a histo score of ≥210 or expression on >70% of tumor cells with 3+ staining intensity.
Endpoints
The primary endpoint for the expansion cohorts was the best overall response as assessed by IRC per RECIST 1.1. Secondary endpoints were confirmed and unconfirmed response best overall response, duration of response (DOR) and progression-free survival (PFS, defined as the time from first administration of study treatment until the date of the first documentation of progressive disease or death by any cause (whichever occurred first)) assessed by IRC and investigator per RECIST 1.1, immune-related best overall response according to modified immune-related response criteria (irRECIST)20 assessed by investigator, DOR and immune-related PFS assessed by IRC and investigator per modified irRECIST, OS (defined as the time from the first dose to death due to any cause), and safety.
Statistical analysis
A sample size of 150 patients was planned to provide 95% Clopper-Pearson CIs for an objective response rate (ORR, proportion of patients with a complete response (CR) or partial response (PR)) of 10% (95% CI 5.7% to 16.0%) in the case of 15 responders and 20% (95% CI 13.9% to 27.3%) in the case of 30 responders. The sample size was based on an assumed ORR of 20% to provide approximately 91% power to reject the null hypothesis of ORR of ≤10%. Time-to-event endpoints were estimated using the Kaplan-Meier method, and 95% CIs of medians were calculated using the Brookmeyer-Crowley method.
Results
Patients and treatment
Between April 24, 2015, and November 13, 2015, 153 patients were enrolled at 69 sites in nine countries and were treated with avelumab. The median age was 63 years (range 37–91), and most patients had metastatic disease (n=122, 79.7%) versus locally advanced disease (n=25, 16.3%) at baseline. Patients had a median of two prior lines of systemic therapy for metastatic or locally advanced disease (range 0–6). A total of 12 patients (7.8%) were not eligible for platinum-based therapy due to impaired renal function (n=1, 0.7%), hearing loss (n=8, 5.2%), peripheral neuropathy (n=2, 1.3%), and other reasons (n=1, 0.7%). Additional baseline characteristics are shown in table 1.
Patient demographics and baseline characteristics
At data cut-off (December 31, 2017), the median follow-up was 27.9 months (range 25–32), and 10 patients (6.5%) remained on treatment. Median duration of treatment was 3 months (range 0.5–29.0). Reasons for treatment discontinuation included disease progression (n=110, 71.9%), death (n=9, 5.9%), withdrawal of consent (n=9, 5.9%), AE (n=8, 5.2%), others (n=5, 3.3%), and protocol non-compliance (n=2, 1.3%).
Antitumor activity
At data cut-off, the confirmed ORR according to RECIST 1.1 was 9.2% (95% CI 5.1% to 14.9%) assessed by IRC (including two patients (1.3%) with CR and 12 (7.8%) with PR) and 13.1% (95% CI 8.2% to 19.5%) assessed by investigator (including 5 patients (3.3%) with CR and 15 (9.8%) with PR) (table 2). In total, 105 and 48 patients did and did not receive prior cetuximab treatment. According to IRC, eight patients (7.6%) who received prior cetuximab had a response, while six patients (12.5%) who did not receive prior cetuximab responded. Per investigator assessment, 10 patients (9.5%) who received prior cetuximab responded and 10 patients (20.8%) who did not receive prior cetuximab responded.
Confirmed best overall response per Response Evaluation Criteria in Solid Tumors V.1.1 assessed by IRC and investigator
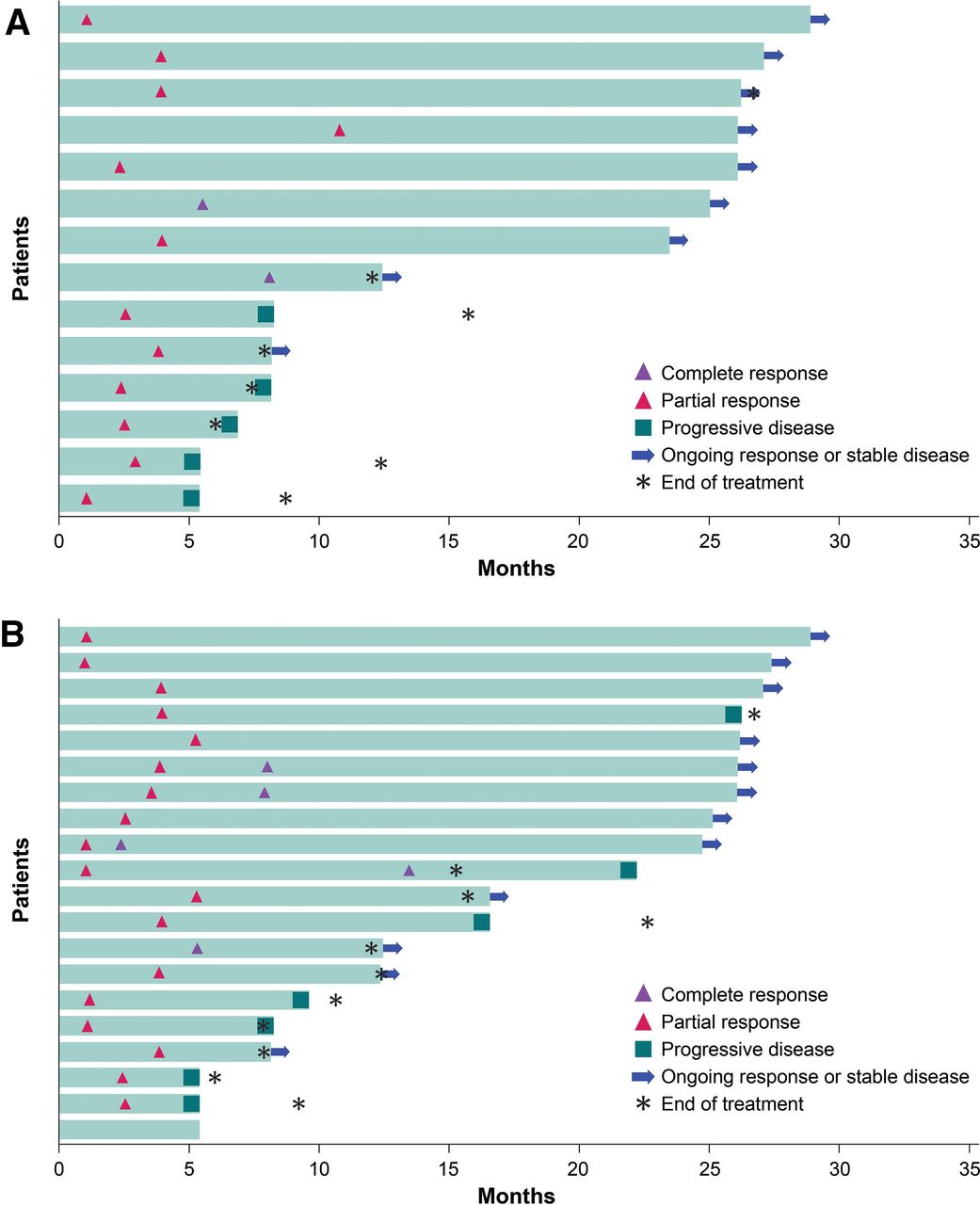
ORRs by subgroup are given in online supplemental figures 1 and 2. Immune-related ORR assessed by investigator according to modified irRECIST was 13.7% (95% CI 8.7% to 20.2%). Based on IRC and investigator assessment, the median time to response was 2.8 months (range 1.3–11.0) and 3.3 months (range 1.2–5.5), respectively (figure 1). Median DOR was not reached (95% CI 4.2 months to not estimable) based on IRC assessment. Of the 14 patients who had a best overall response of CR or PR assessed by IRC, nine patients (64.3%) had an ongoing response at data cut-off. Patients were followed up beyond data cut-off for DOR assessed by investigator; as of February 3, 2020, median DOR was 30.4 months (95% CI 8.3 to not estimable).
Supplemental material

Time to and duration of response of patients with confirmed objective response per Response Evaluation Criteria in Solid Tumors V.1.1 assessed by (A) an independent review committee and (B) an investigator.
Median PFS according to RECIST 1.1 was 1.4 months (95% CI 1.4 to 2.6) assessed by IRC (figure 2A) and 1.8 months (95% CI 1.4 to 2.7) assessed by investigator (figure 2B). PFS rates of 6 and 12 months were 16.3% (95% CI 10.5% to 23.1%) and 10.7% (95% CI 6.0% to 17.0%), respectively, assessed by IRC, and 22.3% (95% CI 15.9% to 29.4%) and 13.5% (95% CI 8.4% to 19.7%) assessed by investigator. Median immune-related PFS assessed by investigator according to modified immune-response criteria was 2.8 months (95% CI 2.7 to 4.1). Median OS was 8 months (95% CI 6.5 to 10.2); 1-year and 2-year OS rates were 35.9% (95% CI 28.3% to 43.6%) and 17.1% (95% CI 11.5% to 23.7%), respectively (figure 3A).

Kaplan-Meier estimates of progression-free survival per Response Evaluation Criteria in Solid Tumors V.1.1 assessed by (A) IRC, (B) investigator, (C) IRC by PD-L1 status (≥1% cut-off), and (D) investigator by PD-L1 status (≥1% cut-off). IRC, independent review committee; PD-L1, programmed death ligand 1.

{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier estimates of (A) OS and (B) OS by PD-L1 status (≥1% cut-off). OS, overall survival; PD-L1, programmed death ligand 1.
Biomarker analysis
In the HPV+ SCCHN (n=39, 25.5%) and HPV− SCCHN (n=99, 64.7%) subgroups, ORR by IRC assessment was 15.4% (95% CI 5.9% to 30.5%) and 5.1% (95% CI 1.7% to 11.4%), respectively (online supplemental figure 1); ORR by investigator assessment was 17.9% (95% CI 7.5% to 33.5%) and 11.1% (95% CI 5.7% to 19.0%) (online supplemental figure 2). The median PFS was 2.7 months (95% CI 1.4 to 3.9) vs 1.4 months (95% CI 1.4 to 1.4) by IRC assessment and 3.3 months (95% CI 1.4 to 5.0) vs 1.4 months (95% CI 1.4 to 2.2) by investigator assessment. Median OS was 11. 8 months (95% CI 7.8 to 16.3) vs 7.4 months (95% CI 5.0 to 8.7).
In patients with PD-L1+ (n=107, 69.9%) and PD-L1− (n=30, 19.6%) tumors (≥1% cut-off), ORR by IRC assessment was 10.3% (95% CI 5.2% to 17.7%) and 3.3% (95% CI 0.1% to 17.2%), respectively; ORR by investigator assessment was 15.0% (95% CI 8.8% to 23.1%) and 6.7% (95% CI 0.8% to 22.1%) (online supplemental figures 1 and 2; online supplemental table 1). Median PFS was 1.4 months (95% CI 1.4 to 2.8) vs 1.4 months (95% CI 1.3 to 2.7) by IRC assessment (figure 2C) and 1.8 months (95% CI 1.4 to 3.5) vs 1.5 months (95% CI 1.3 to 4.0) by investigator assessment (figure 2D). Median OS was 7.9 months (95% CI 6.1 to 10.6) vs 8.9 months (95% CI 6.7 to 15.0) (figure 3B). ORRs by PD-L1 status at ≥1%, ≥50%, and ≥80% cut-offs are given in online supplemental table 1, and median PFS and OS values by PD-L1 status at ≥50% and ≥80% cut-offs are given in online supplemental table 2.
Supplemental material
Safety
AEs of any grade occurred in 149 patients (97.4%); grade ≥3 AEs occurred in 91 patients (59.5%). Treatment-related adverse events (TRAEs) of any grade occurred in 83 patients (54.2%); grade ≥3 TRAEs occurred in 10 patients (6.5%, table 3). The most common TRAEs were fatigue (n=19, 12.4%), fever (n=14, 9.2%), pruritus (n=12, 7.8%), and chills (n=11, 7.2%). Serious TRAEs occurred in six patients (3.9%) and led to permanent treatment discontinuation in four patients due to hepatocellular injury (elevated alanine aminotransferase and aspartate aminotransferase; n=1, 0.7%), hyperbilirubinemia (n=1, 0.7%), diarrhea (n=1, 0.7%), and hypophosphatemia (n=1, 0.7%). IRRs (based on an expanded definition) occurred in 23 patients (15.0%, all were grade 1/2), and irAEs occurred in 23 patients (15.0%, online supplemental table 3); the most common irAEs were hypothyroidism (n=11, 7.2%), rash (n=3, 2.0%), pruritus (n=2, 1.3%), diarrhea (n=2, 1.3%), and hepatocellular injury (n=2, 1.3%). Grade 3 irAEs occurred in three patients due to hepatocellular injury (n=2, 1.3%) and psoriasis (n=1, 0.7%) and led to permanent treatment discontinuation in one patient (0.7%) due to hepatocellular injury; no grade 4 or 5 irAEs occurred. Twenty-five patients (16.3%) had an AE leading to death (online supplemental table 4), and none were treatment related.
TRAEs (any grade in ≥5% of patients or grade 3/4 in all patients) and IRRs
Discussion
Avelumab showed clinical activity, including durable responses (median DOR was 30.4 months, assessed by investigator), in heavily pretreated patients with platinum-refractory/ineligible R/M SCCHN. TRAEs were grades 1 and 2 in the majority of cases (73/83 TRAEs, 88.0%), and a low proportion of patients had grade ≥3 TRAEs (10/83, 12.0%).
In this cohort, patients were not selected based on HPV or PD-L1 status. A higher proportion of patients had HPV− tumors than HPV+ tumors (64.7% (n=99) vs 25.5% (n=39)); the proportion of patients with PD-L1+ tumors (≥1% cut-off) vs PD-L1− tumors was 69.9% (n=107) vs 19.6% (n=30). Patients with HPV− disease are known to have poorer prognosis than those with HPV+ disease,5 and ORRs were lower in patients with HPV− tumors compared with HPV+ tumors. ORRs were higher in patients with PD-L1+ tumors (≥1% cut-off) compared with those with PD-L1− tumors; however, responses were seen in a small number of patients with PD-L1− tumors. Increased ORRs and median OS values were also observed with higher PD-L1 expression cut-offs of ≥50% and ≥80% compared with 1%. Furthermore, ORRs were higher in patients with PD-L1 +tumors using the ≥50% cut-off compared with the ≥80% cut-off, whereas OS values were similar using the two cut-offs. PFS values were similar in both PD-L1+ and PD-L1− subgroups and regardless of PD-L1 cut-off.
Historically, trials of single-agent non-platinum chemotherapy in patients with R/M SCCHN have reported varied ORRs (range 10%–43.3%) and median OS of <10 months.21–24 The results of this phase Ib cohort were similar to those reported with nivolumab monotherapy in the randomized, open-label, phase III CheckMate 141 trial of nivolumab (n=240) versus the investigator’s choice of chemotherapy or cetuximab (n=121) in patients with R/M SCCHN and disease progression after platinum-based chemotherapy.25 After 2 years of follow-up of the intention-to-treat population (all randomized patients, n=361), the ORR was 13.3% with nivolumab vs 5.8% with investigator’s choice, and median OS was 7.7 months vs 5.1 months, respectively.25 The results reported here are also similar to those reported in the randomized, open-label, phase III KEYNOTE-040 trial, in which patients with R/M SCCHN who had disease progression with platinum-containing therapy received pembrolizumab (n=247) or investigator’s choice of chemotherapy or cetuximab (n=248).12 In the intention-to-treat population (all randomized patients, n=495), the ORR was 14.6% with pembrolizumab vs 10.1% with investigator’s choice, and the median OS was 8.4 months vs 6.9 months, respectively.12 In the randomized, open-label, phase III EAGLE trial, which compared durvalumab with or without tremelimumab versus the investigator’s choice of single-agent standard of care (cetuximab, a taxane, methotrexate, or a fluoropyrimidine) in patients with R/M SCCHN and disease progression or recurrence following platinum-containing therapy, the primary endpoint of improved OS was not met in either durvalumab arm (median OS of 7.6 and 6.5 months vs 8.3 months with standard of care). ORRs were 17.9% with durvalumab, 18.2% with durvalumab plus tremelimumab, and 17.3% with standard of care.26 It must be noted that in our cohort reported here, patients were heavily pretreated (median of two prior lines of therapy for metastatic or locally advanced disease), and this may have contributed to the slightly lower ORR of 9.2% by IRC and 13.1% by investigator compared with the results observed in the phase III studies. Furthermore, the phase III JAVELIN Head and Neck 100 trial of avelumab plus chemoradiation followed by avelumab maintenance in patients with previously untreated locally advanced SCCHN was stopped for not meeting the primary endpoint of prolonging PFS. The lack of improvement in PFS with the addition of avelumab to chemoradiotherapy was unexpected, and there was no obvious explanation for these findings.27
In summary, in this phase 1b expansion cohort of heavily pretreated patients with platinum-refractory/ineligible R/M SCCHN, avelumab showed similar clinical activity to that reported in phase III trials of immune checkpoint inhibitors and a manageable safety profile.
Data availability statement
Data are available upon reasonable request. For all new products or new indications approved in both the European Union and the United States after January 1, 2014, Merck (CrossRef Funder ID: 10.13039/100009945) will share patient-level and study-level data after deidentification, as well as redacted study protocols and clinical study reports from clinical trials in patients. These data will be shared with qualified scientific and medical researchers, upon researcher’s request, as necessary for conducting legitimate research. Such requests must be submitted in writing to the company’s data sharing portal. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. Where Merck has a co-research, co-development, or co-marketing/co-promotion agreement or where the product has been out-licensed, it is recognized that the responsibility for disclosure may be dependent on the agreement between parties. Under these circumstances, Merck will endeavor to gain agreement to share data in response to requests.
Ethics statements
Patient consent for publication
Ethics approval
The trial was conducted in accordance with the Declaration of Helsinki and the International Council for Harmonisation Guideline for Good Clinical Practice. The protocol was approved by the institutional review board or independent ethics committee of each center, and all patients provided written informed consent before enrollment.
Acknowledgments
The authors thank the patients and their families, investigators, coinvestigators, and study teams at each of the participating centers, and Eleanor Green of ClinicalThinking for medical writing support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Provision of study materials or patients: JG, K-WL, MRP, AD, DJW, SG, MSG, MG, AB, CLT, AM, DV, UK, PS, and NP. Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of the manuscript and accountability for all aspects of the work: all authors.
Funding This trial was funded by Merck (CrossRef Funder ID: 10.13039/100009945), as part of an alliance between Merck and Pfizer.
Competing interests JG reports providing a consulting or advisory role for AstraZeneca, Bristol Myers Squibb, Merck, MSD, Nanobiotix, and Innate Pharma; and has received research funding from Bristol Myers Squibb, Chugai Pharmaceutical Co., Merck and GlaxoSmithKline. K-WL reports providing a consulting or advisory role for Bayer and ISU ABXIS; has received honoraria from Bristol Myers Squibb, Eli Lilly, and Genexine; and received research funding from ABL Bio, ALX Oncology, AstraZeneca/MedImmune, BeiGene, Daiichi Sankyo, Five Prime Therapeutics, GC Pharma, Genexine, LSK BioPharma, MacroGenics, MSD, Merck, Oncologie, Ono Pharmaceutical, Pfizer, Pharmacyclics, Taiho Pharmaceutical, Y-BIOLOGICS, and Zymeworks (to his institution for conducting clinical trials). MRP has received honoraria from AbbVie, Bayer, Genentech, Janssen Pharmaceutical, Pharmacyclics, and Pfizer; provided speakers services for Celgene, Merck, and Exelixis; and research funding from Acerta Pharma, ADC Therapeutics, Agenus, Aileron Therapeutics, AstraZeneca, Bicycle Therapeutics, BioNTech, Boehringer Ingelheim, Calithera Biosciences, Celgene, Checkpoint Therapeutics, Ciclomed, Covis Pharma, Curis, Cyteir Therapeutics, Daiichi Sankyo, eFFECTOR Therapeutics, Eli Lilly, Merck, Evelo Biosciences, Forma Therapeutics, Genentech/Roche, Gilead, GlaxoSmithKline, H3 Biomedicine, Hengrui Therapeutics, Hutchinson MediPharma, Ignyta, Incyte, Jacobio Pharmaceuticals, Janssen Pharmaceutical, Jounce Therapeutics, KLUS Pharma, Kymab, Loxo Oncology, LSK Biopartners, Lycera, MacroGenics, Merck, Millennium Pharmaceuticals, Mirati Therapeutics, Moderna, Pfizer, Phoenix Molecular Designs, Placon Therapeutics, Portola Pharmaceuticals, Prelude Therapeutics, Qilu Puget Sound Biotherapeutics, Revolution Medicines, Ribon Therapeutics, Seven and Eight Biopharmaceuticals, Stemline Therapeutics, Syndax, Synthorx, Taiho Pharmaceutical, Takeda, Tesaro, TopAlliance, Vedanta Biosciences, Verastem Oncology, Vigeo Therapeutics, and Xencor. AD reports providing a consulting or advisory role for and receiving reimbursement for travel and accommodation expenses from Bristol Myers Squibb and Merck. DW reports providing a consulting or advisory role for Bristol Myers Squibb and Sanofi-Aventis, and has received research funding from Astellas Pharma, AstraZeneca, Bristol Myers Squibb, Eli Lilly, Enzychem Lifesciences, F-star, Genentech, Iovance Biotherapeutics, Kura Oncology, MSD, Merck, Pfizer, Regeneron, and Sanofi. MSG reports providing a consulting or advisory role for Deciphera Pharmaceuticals, ImaginAb, Redhill Biopharma, and TRACON Pharmaceuticals; and owns stock in Caremission and Medilus. MG reports providing a consulting or advisory role for Guardant360; speakers services for Merck; has received research funding from Array Biopharma, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Care Progress, Celgene, Compass Therapeutics, Constellation Pharmaceuticals, CytRx, Eisai, Esanex, Genentech, Incyte, Infinity Pharmaceuticals, Janssen, Karyopharm Therapeutics, MedImmune, Merck, Mirati Therapeutics, Moderna, Pfizer, Regeneron, Sanofi, Seattle Genetics, Tesaro, and Vendata Biosciences; owns stock in Cota Healthcare; and is an employee of Reginal Cancer Care Associates. AB reports providing speakers services for AstraZeneca, Bristol Myers Squibb, Eli Lilly, Genentech, and Merck; and has received research funding from Arcus Biosciences, Bristol Myers Squibb, Genentech, GlaxoSmithKline, MedImmune, and Merck. CLT has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, Merck, GlaxoSmithKline, MSD, Nanobiotix, Rakuten, Roche, and Seattle Genetics; provided a consulting or advisory role for Idem Healthcare; and reimbursement for travel and accommodation expenses from AstraZeneca, Bristol Myers Squibb, Merck, and MSD. DV reports providing a consulting or advisory role for Nestlé and Nutricia. PS has received honoraria from Blueprint Medicines, Boehringer Ingelheim, and Deciphera Pharmaceuticals; provided a consulting or advisory role for Adaptimmune, Advanced Medical, Blueprint Medicines, Boehringer Ingelheim, Deciphera, Ellipses Pharma, Exelixis, Guided Clarity, Intellisphere, Medscape, and Transgene; research funding from CoBioRes, Eisai, G1 Therapeutics, Novartis, and PharmaMar; and reimbursement for travel and accommodation expenses from Boehringer Ingelheim, Ipsen, and MSD. HJG is an employee of Merck Healthcare KGaA, Darmstadt, Germany. DZ is an employee of Merck Serono (Beijing) Pharmaceutical R&D Co., Ltd. Beijing, China, an affiliate of Merck KGaA. MB is an employee of Merck Healthcare KGaA, Darmstadt, Germany. All other authors have nothing to disclose.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.