Article Text

Abstract
Background The human tumor microenvironment (TME) is a complex and dynamic milieu of diverse acellular and cellular components, creating an immunosuppressive environment, which contributes to tumor progression. We have previously shown that phosphatidylserine (PS) expressed on the surface of exosomes isolated from human TMEs is causally linked to T-cell immunosuppression, representing a potential immunotherapeutic target. In this study, we investigated the effect of ExoBlock, a novel PS-binding molecule, on T-cell responses in the TME.
Methods We designed and synthesized a new compound, (ZnDPA)6-DP-15K, a multivalent PS binder named ExoBlock. The PS-binding avidity of ExoBlock was tested using an in vitro competition assay. The ability of this molecule to reverse exosome-mediated immunosuppression in vitro was tested using human T-cell activation assays. The in vivo therapeutic efficacy of ExoBlock was then tested in two different human tumor xenograft models, the melanoma-based xenomimetic (X-)mouse model, and the ovarian tumor-based omental tumor xenograft (OTX) model.
Results ExoBlock was able to bind PS with high avidity and was found to consistently and significantly block the immunosuppressive activity of human ovarian tumor and melanoma-associated exosomes in vitro. ExoBlock was also able to significantly enhance T cell-mediated tumor suppression in vivo in both the X-mouse and the OTX model. In the X-mouse model, ExoBlock suppressed tumor recurrence in a T cell-dependent manner. In the OTX model, ExoBlock treatment resulted in an increase in the number as well as function of CD4 and CD8 T cells in the TME, which was associated with a reduction in tumor burden and metastasis, as well as in the number of circulating PS+ exosomes in tumor-bearing mice.
Conclusion Our results establish that targeting exosomal PS in TMEs with ExoBlock represents a promising strategy to enhance antitumor T-cell responses.
- immunotherapy
- melanoma
- drug evaluation
- preclinical
- T-lymphocytes
- tumor microenvironment
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Conventional cancer treatment strategies such as surgery, radiotherapy and chemotherapy often lead to tumor recurrence and metastasis. The use of immunotherapy to eradicate tumor and eliminate tumor metastasis by inducing antitumor immunological responses has met with resounding success, revolutionizing oncology in the last decade. Targeting immune checkpoints using monoclonal antibodies, termed checkpoint blockade therapy, has shown promise against multiple cancers. However, the response rate among patients is highly variable, leaving room for improvement.1 Identification of novel therapeutic targets and designing stand-alone or combination targeting strategies can potentially improve outcomes and patient survival.
Phosphatidylserine (PS) is a phospholipid that is normally present on the inner leaflet of normal cells. However, apoptotic as well as non-apoptotic cancer cells such as malignant melanoma, leukemia, neuroblastoma, and gastric carcinoma have been shown to widely express PS on their surfaces.2 PS exposed on the surface of tumor cells contributes to suppression of T-cell activity and blocks tumor clearance.3 4 PS blockade in a mouse model of melanoma has been shown to significantly reduce tumor size and tumor necrosis area.5
Multiple studies have documented the presence of immunosuppressive nanosized extracellular vesicles (EVs), called exosomes, in the microenvironment of many different types of human tumors.6–15 These reports have strongly suggested that these exosomes contribute to the arrest of antitumor T-cell responses. We and others have predicted that blockade of immunosuppressive exosomes would enhance the strength and durability of T cell-mediated antitumor responses. PD-L1+ exosomes released from melanomas in patients have been shown to suppress the function of CD8 T cells and to facilitate tumor growth, while the elimination of tumor-derived exosomes suppresses tumor growth and increases survival in mice.16 17 Multiple studies have established a causal link between exosome-mediated immune suppression and PS on the surface of exosomes.6–8 Based on these studies, we predicted that PS represents a potential therapeutic target. Molecules that bind with high avidity to PS on tumor cells, other apoptotic cells, and exosomes could potentially block PS-mediated immune suppression.
We have designed and synthesized a new molecule, (ZnDPA)6-DP-15K, that binds PS with high avidity. We report here that this molecule, which we have named ExoBlock, consistently and effectively blocks immunosuppressive exosomes. We first demonstrate that ExoBlock has the ability to significantly reverse T-cell immune suppression mediated by exosomes derived from human ovarian and melanoma tumor tissues in vitro. The in vivo therapeutic efficacy of ExoBlock was then tested using two tumor xenograft models: (1) the melanoma-based xenomimetic (X-)mouse model that was developed and validated to evaluate the therapeutic efficacy of strategies that are designed to enhance T-cell antitumor responses using patient-derived T cells and patient-derived neoantigen expressing tumor target cells18; and (2) the ovarian tumor-based omental tumor xenograft (OTX) model that represents a more complete microenvironment, established using ovarian tumor aggregates derived from patients.19 Using these models, we establish here that treatment of tumor xenograft-bearing mice with ExoBlock results in a significant suppression of tumor growth and dissemination, with concomitant enhancement of antiumor T-cell responses. We conclude that targeting PS using ExoBlock represents a viable strategy to target immunosuppressive exosomes and thereby enhances antitumor T-cell immune responses in tumor microenvironments (TME).
Materials and methods
Specimens
Ascites fluids and solid tumors from patients with stage III or stage IV ovarian canccers were received anonymously under an approved institutional review board protocol from the Roswell Park Comprehensive Cancer Center (RPCCC) Tissue Procurement Facility. All the tumors obtained were high-grade serous epithelial ovarian carcinomas. Experiments were done using cell-free ascites fluids that had been stored at −80°C. Normal donor peripheral blood was provided by the Flow and Image Cytometry Facility at RPCCC. Normal donor peripheral blood lymphocytes (NDPBL) were obtained by monocyte depletion and Ficoll–Hypaque density separation. Cells were cryopreserved in liquid nitrogen until use. Specimens from 12 different patients with ovarian cancer and NDPBL from 10 different donors were used in this study.
Cell culture
DM6-Mut cells and TKT R438W cells were generated and authenticated as described previously.18 20 DM6-Mut cells and Jurkat cells were maintained in RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 20 U/mL penicillin, 20 µg of streptomycin, and 50 µM 2-mercaptoethanol (complete medium) at 37°C under 5% CO2. DM6-Mut cells are adherent and were grown in 75 or 175 cm3 flasks until they were 90% confluent. Cells were harvested with 0.25% trypsin-EDTA for 3–4 min at 37°C following removal of the medium and washed with phosphate-buffered saline (PBS). Complete medium was added to stop the reaction, and cells were centrifuged at 300×g for 10 min.
Mice
NOD.Cg-Prkdcscid Il2rgtm1Wjl (NSG) mice (young adult female mice), raised in a research colony at The Jackson Laboratory, were used in this study as they have been shown to support the growth of a variety of human tumors.18 19 21 Age-matched and sex-matched mice were randomly assigned to control (untreated) and test (ExoBlock-treated) groups.
ExoBlock
ExoBlock was synthesized at Molecular Targeting Technologies Inc. (MTTI) at the 0.5 g scale. Structural integrity and consistency between different batches was confirmed using nuclear magnetic resonance studies as well as amino group analysis.
In vitro competition study
Apoptosis was induced in Jurkat cells by culture (16–20 hours) with 10 µM etoposide (Sigma Aldrich) at 1×106/mL. Cells were then washed, resuspended in N-tris-(hydroxymethyl)-methyl-2-aminoethane sulfonic acid (TES) buffer (5 mM TES, 145 mM NaCl, pH=7.4). The cells were then stained with PSVue 499 (MTTI, Cat# P-1009) at 10 µM in the presence or absence of equimolar or titrating amounts of ExoBlock using the protocol provided by the manufacturer. Sytox Red (Life Technologies) was used to exclude dead cells. The samples were acquired using an LSRFortessa (BD Biosciences) flow cytometer and data analyzed using FlowJo (Tree Star).
In vitro efficacy studies
Tumor-associated exosomes were isolated by ultracentrifugation as described previously7 8 (online supplemental methods). T cells from NDPBL were stimulated using immobilized antibodies to CD3 and CD28, and activation was monitored using the nuclear translocation of NFκB, upregulation of the activation markers CD25 and CD69, intracellular expression of the cytokines IFN-γ and IL-2, or proliferation as described previously7 8 (online supplemental methods). For ExoBlock-treated groups, the exosomes were preincubated with 10 µM ExoBlock for 1 hour at 37°C.
Supplemental material
Isolation of exosomes from mouse sera
Serum was collected from the mice and stored at −80°C. Exosomes were isolated by size column exclusion using a qEVsingle/35+ nm column (Izon Science, Cat# qEVsingle/35 nm) that isolated particles in the range of 35–350 nm according to manufacturers’ instructions. Briefly, 250 µL of serum was loaded on to a qEVsingle/35+ nm column and chased with sterile 1× PBS. Elution fractions of 200 µL were collected and analyzed for particle size and concentration by NTA and protein concentration by Bicinchoninic Acid (BCA) protein assay (Thermo Fisher Scientific, Cat# 23227), respectively. Based on these parameters, the three elution fractions containing the highest concentration of particles and protein levels below 500 µg/mL were combined to an enriched exosome fraction, and small aliquots were frozen at −80°C until further analysis.
Exoglow-NTA and PSVue staining of exosomes
Exosomes were stained with PSVue-480 (Molecular Targeting Technologies, Cat # P-1003) and ExoGlow-NTA (System Biosciences, Cat# EXONTA110A-1) according to manufacturers’ instructions. PSVue binds PS and ExoGlow stains vesicles with intact membranes but not membrane fragments and non-membrane-associated (lipo)-protein aggregates. Briefly, exosomes or an equivalent volume of PSVue staining buffer only control (without exosomes) were stained first with PSVue followed by staining with ExoGlow. Following staining, each sample was divided in two 50 µL aliquots. One was diluted 1:5 in PSVue staining buffer for acquisition on the ImageStream. The second aliquot received an equal volume of 10% Triton-X100, and this was diluted 1:5 in PSVue staining buffer for acquisition acquired on the ImageStream (see online supplemental methods). The Triton-X100-treated samples served as a control to determine the concentration of non-exosome particles insensitive to detergent lysis (eg, protein aggregates or stain aggregates present in the preparations of exosomes and/or the staining reagents.
In vivo studies
The sample size for in vivo experiments was calculated based on the ability to demonstrate with 85% power, a twofold difference in tumor burden, allowing for a 30% SD in tumor size and spread assuming a normal distribution of the samples. The X-mouse model18 and the OTX model19 were established as described previously (see online supplemental methods). For the X-mouse model, the tumor-bearing mice were treated with ExoBlock (32 mg/kg) or nivolumab (10 mg/kg) at various time points as described, and tumor burdens were estimated as described previously18 (online supplemental methods). For the OTX model, tumor-bearing mice were treated with 107 mg/kg of ExoBlock at various time points as described. This dose (107 mg/kg) was also used in the toxicity studies. Untreated tumor-bearing mice served as controls. Five animals were housed per cage, and experiments were terminated on the noted time points based on initial tumor growth kinetics studies, or earlier if signs of duress were observed.22
Analysis of changes in gene expression in the OTX model
On day 25, the mice were euthanized; greater omentum was harvested and snap-frozen for cryopreservation. The cryopreserved samples were thawed; 1 mL of Trizol (Life Technologies) was added; and the tumor-bearing omental tissue was mechanically disrupted using a SCILOGEX D-160 homogenizer. RNA isolation was done as per the manufacturer’s instructions. RNA of 1 µg was used for cDNA synthesis using the iScript cDNA synthesis kit (BioRad) as described by the manufacturer. cDNA of 100 ng was used to set up real-time PCR in a volume of 20 µL to determine the expression of human EpCAM, CD4, CD8, IFN-γ and Foxp3 using the iQ SYBR Green Supermix (BioRad) according to the manufacturer’s instructions using a CFX96 Connect Real Time PCR Detection System (BioRad). The primers used are listed in online supplemental table 1. The thermal cycling conditions included an initial denaturation step at 95°C for 10 min, followed by 40 cycles at 95°C for 30 s, 58°C for 30 s, and plate read. Melting curve analysis of every qPCR was conducted after each cycle, and samples with cycle numbers more than 35 were excluded. Relative gene expression was determined by 2−ΔΔCT method by normalizing to (human) β-actin for the respective sample.
Supplemental material
Statistics
All statistics were calculated using Excel V.2016 (Microsoft) and Prism (Graph Pad). Paired or unpaired Student’s t-test was applied to determine whether the differences between groups could be considered significant. A p value higher than 0.05 was not significant, whereas *p<0.05, **p<0.01, and ***p<0.001 were considered significant.
Results
Design and synthesis of ExoBlock, a novel drug that binds to PS with high avidity
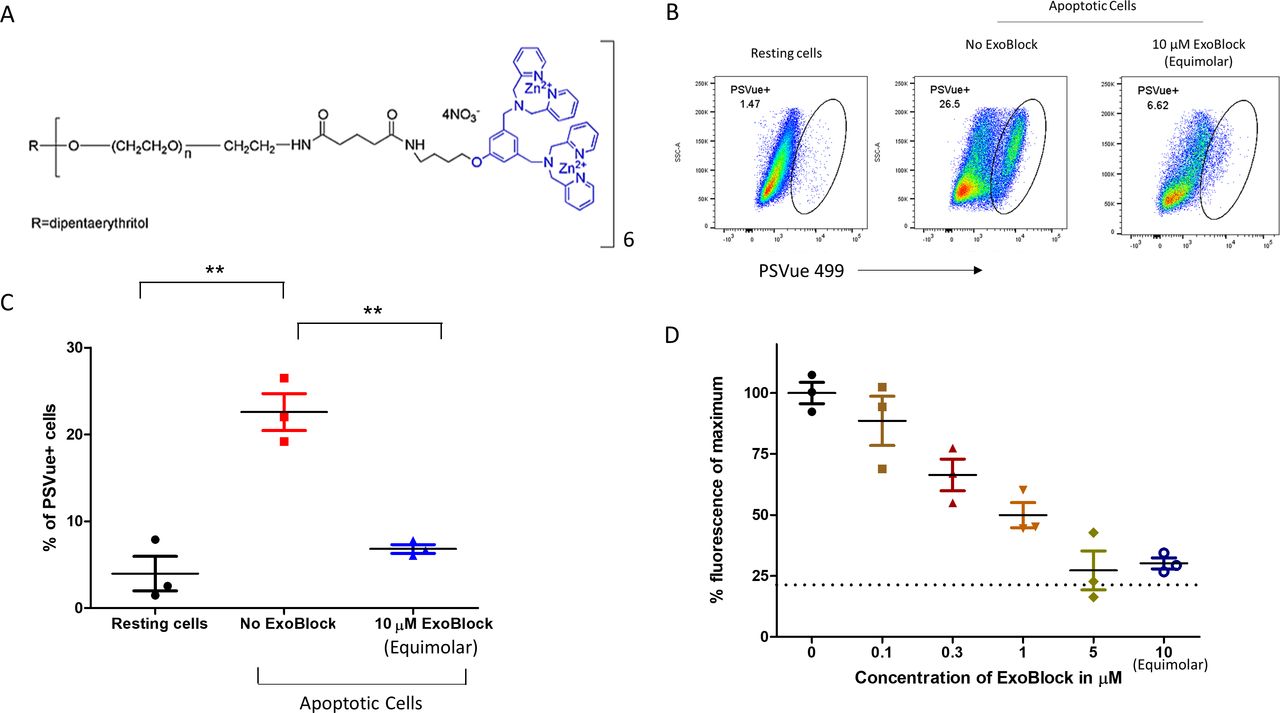
The role of PS in immunosuppression6 23 makes it an attractive immunotherapeutic target. Based on nanoparticle tracking analysis (NTA) data, we have estimated that ovarian ascites fluids contain over 1×1010 PS+ exosomes per millilitre. Synthetic zinc(ii)-dipicolylamine (ZnDPA) co-ordination complexes that mimic annexin V, such as the commercially available PSVue, have been used to bind and visualize PS in different environments including cardiovascular diseases, bacterial infections, and cancers.24 Some of the advantages of these molecules over annexin V include greater stability, faster binding kinetics, and intense labeling due to smaller size. ZnDPA complexes are also highly specific for PS, showing minimal binding to other phospholipids.25 We have synthesized a new molecule, a multimerized version of ZnDPA named ExoBlock ((ZnDPA)6-DP-15K) (patent pending). ExoBlock (figure 1A) was synthesized at the 0.5 g scale via eight synthetic steps with an overall synthetic yield of ~18% (online supplemental figure S1). The final product was purified by a dialysis process and then eventually lyophilized to produce ExoBlock.
Supplemental material

ExoBlock competitively inhibits binding of PSVue 499 to apoptotic cells in a dose-dependent manner. (A) Structure of ExoBlock. (B–D) Jurkat cells were treated with 10 µM etoposide for 20 hours to induce apoptosis. The cells were then stained with PSVue with equimolar or titrating molar amounts of ExoBlock. Sytox red was used to eliminate dead cells from the analysis. The experiment was done in triplicate wells. (B) Representative data and (C) quantified data from three wells for equimolar amounts of ExoBlock. (D) Dose dependency of the competitive inhibition highlighting the inverse relationship between ExoBlock dose and detection of PSVue fluorescence. The amount of fluorescence in resting cells is shown as baseline (21.3±5.7) (D). Data shown as mean±SEM. **P<0.01.
With multiple PS binding sites, the hexamer ExoBlock is expected to bind with high avidity to PS. To determine the relative avidity of ExoBlock for PS binding, apoptosis was induced in Jurkat cells, and equimolar or titrating amounts of ExoBlock were coincubated with 10 µM PSVue 499, a commercially available ZnDPA-based PS-binding probe. We found that not only ExoBlock was able to outcompete PSVue at equimolar amounts, but also even amounts as low as 1/30 molar equivalents blocked more than 50% of PSVue binding, confirming its superior avidity for PS (figure 1B–D).
We also performed a preliminary systemic organ toxicity study in mice with ExoBlock. NSG mice were treated with ExoBlock and euthanized 15 days after treatment. Selected organs from treated and control untreated mice were removed, fixed, sectioned, stained, and examined for the evidence for histopathology. No pathological changes were observed at the gross or microscopic level in the lung, spleen, small intestine, kidney, or liver. We conclude that ExoBlock has no observable adverse effects at this dose and schedule in NSG mice.
Human ovarian TMEs are enriched in PS-expressing exosomes
We have previously established that the immunosuppressive activity of exosomes is causally linked with PS expressed on exosomal surfaces. To determine exosomal PS expression, exosomes derived from ovarian tumor ascites fluids were attached to latex beads, and PS was detected using an anti-PS antibody using flow exometry. We observed the abundant expression of PS on these exosomes (figure 2A). To determine whether TMEs are enriched in PS+ exosomes, we quantified ExoGlow+PSVue+ exosomes in plasma from normal donors as well as in ascites fluids from patients with ovarian cancer using imaging flow cytometry (IFC). IFC allows for the detection of fluorescence intensities on individual exosomes as opposed to the flow exometry, which determines the fluorescence intensities of heterogeneous exosome populations immobilized on capture beads. Hence, IFC can visualize heterogeneous expression of PS on exosomes, whereas flow exometry cannot. Ovarian TMEs were found to be highly enriched in PS+ exosomes, and PS+ exosomes comprised more than 90% of the total exosomes in 9 out of 10 ascites fluids tested (figure 2B,C). On the contrary, the proportions of PS+ exosomes were variable but significantly lower in normal donor plasma (figure 2B,C).

TMEs are enriched in PS. (A) PS expression in ovarian tumor ascites fluid-derived exosomes was determined using latex bead flow exometry. Unstained (filled gray histogram), secondary antibody control (open red histogram) and PS-stained exosomes (filled blue histogram). (B,C) The percentage of PS+ vesicles in normal donor plasma as well as in patient-derived ovarian tumor ascites fluids (TME) was determined using imaging flow cytometry. (B) Compiled data (n=10) and (C) representative data for each cohort are shown. Data shown as mean±SEM. Whiskers and outliers were calculated using the Tukey method. **P<0.01. PS, phosphatidylserine; TME, tumor microenvironment.
ExoBlock rescues T cells from immunosuppression mediated by exosomes isolated from human ovarian TMEs in vitro
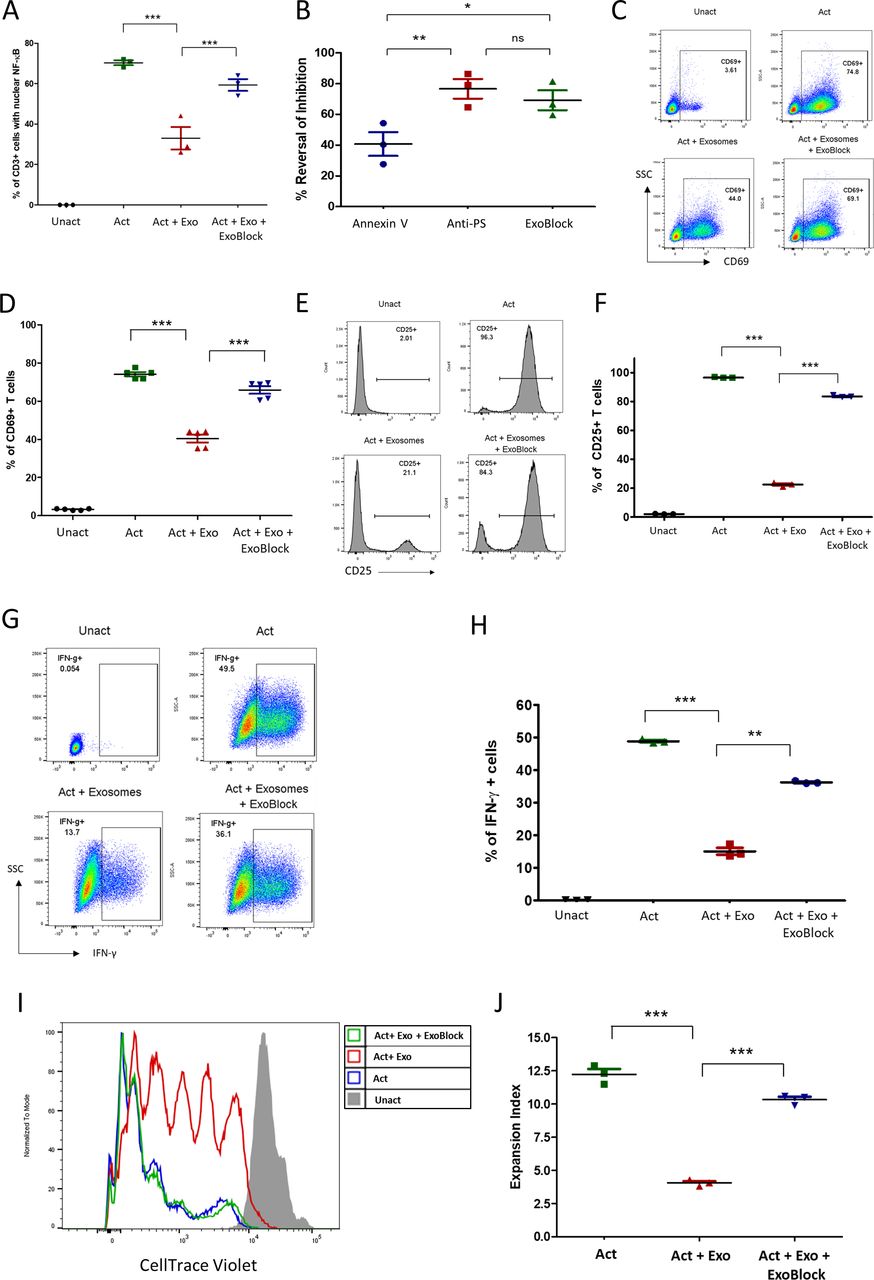
We have previously demonstrated that exosomes isolated from human ovarian tumor ascites fluids arrest CD4+ and CD8+ T-cell signaling through the TCR.6 This signaling arrest is reflected in multiple T-cell activation endpoints, including the nuclear translocation of nuclear factor kappa B (NFκB), upregulation of the activation marker CD69, intracellular expression of the cytokines interferon gamma (IFN-γ) and interleukin (IL)-2, and proliferation.6–8 This inhibitory effect could be significantly reversed by blocking PS using PS-binding molecules such as annexin V or anti-PS antibodies.6 Since ExoBlock has high avidity for PS due to its multivalency, we tested the ability of ExoBlock to reverse the immunosuppressive effect of exosomes derived from ovarian tumor ascites fluids on T-cell function after short-term as well as long-term activation through the TCR. As expected, T-cell activation was significantly inhibited by exosomes regardless of the endpoint, and ExoBlock treatment was able to reverse this effect (figure 3A–J and table 1). This included nuclear translocation of NFκB (figure 3A,B), upregulation of activation markers CD69 (figure 3C,D) and CD25 (figure 3E,F), intracellular expression of the cytokines IL-2 (online supplemental figure S2) and IFN-γ (figure 3G,H), and proliferation (figure 3I,J and online supplemental figure S3). Moreover, a comparison of the in vitro efficacies of different PS-binding molecules revealed that ExoBlock was significantly better than annexin V and was comparable to anti-PS antibody in terms of reversing exosome-mediated inhibition (figure 3B). These results establish that blockade of PS with ExoBlock can rescue T cells from exosome-mediated signaling arrest.
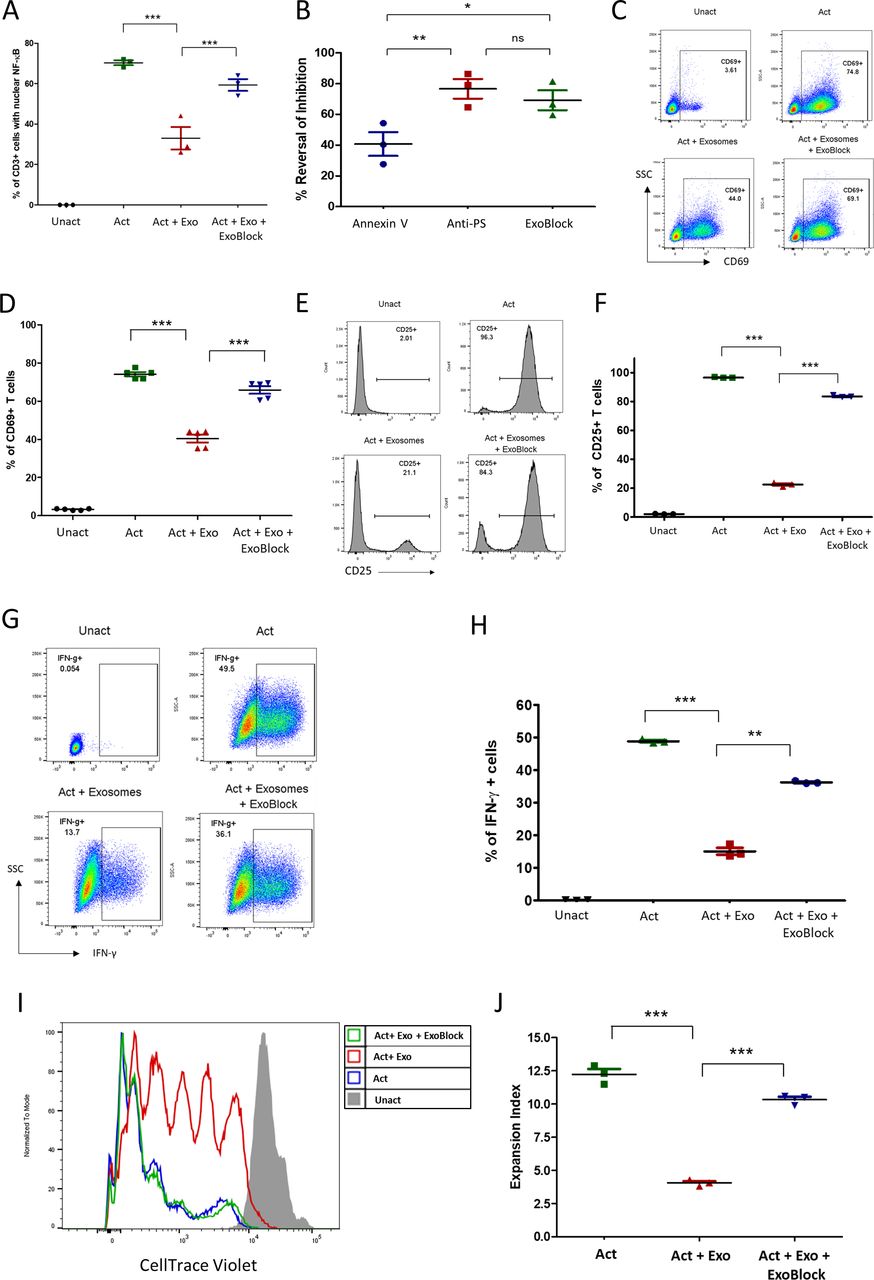
ExoBlock reverses immunosuppression mediated by ovarian tumor-associated exosomes in vitro. (A–D) T cells were either left unactivated (Unact), activated for 2 hour with immobilized antibodies to CD3 and CD28 without exosomes (Act), with exosomes only (Act + Exo), or with exosomes and ExoBlock (Act + Exo + ExoBlock). Activation was monitored by detecting the nuclear translocation of NFκB using confocal microscopy (A,B) or by detecting the upregulation of CD69 on CD3+ T cells using flow cytometry following overnight incubation (representative data shown in C, quantified data shown in D). (B) A comparison of the in vitro efficacies of different PS-binding molecules in terms of reversing inhibition of NFκB translocation. (E–H) T cells were either left Unact, activated for 72hours with immobilized antibodies to CD3 and CD28 without exosomes (Act), Act + Exo, or Act + Exo + ExoBlock. Activation was monitored by detecting the upregulation of CD25 (representative data shown in E, quantified data shown in F) or the expression of intracellular IFN-γ (representative data shown in G, quantified data shown in H). (I–J) T cells labeled with CellTrace violet were either left Unact, activated for 7 days with immobilized antibodies to CD3 and CD28 without exosomes (Act), Act + Exo, or Act + Exo + ExoBlock. Proliferation was assessed by monitoring dye dilution using flow cytometry. (I) Representative dye dilution profiles and (J) expansion indices (depicting fold expansion of the culture) are shown. n=3. Data shown as mean±SEM. *P≤0.05, **P≤0.01, ***P≤0.001. Act + Exo, Act with exosomes only; Act + Exo + ExoBlock, Act with exosomes and ExoBlock;SSC, side scatter; IFN-γ, interferon gamma; ns, not significant; PS, phosphatidylserine; Unact, unactivated.
ExoBlock reverses exosome-mediated inhibition of different T-cell activation endpoints
ExoBlock rescues T cells from immunosuppression mediated by exosomes isolated from human melanoma microenvironments in vitro
To extend these findings to other human TMEs, we chose DM6-Mut cells—a human melanoma cell line that expresses (melanoma) patient-derived neoantigen peptides. The parent cell line, DM6, is an established melanoma model for immunotherapy with high clinical relevance. Xenografts were established in the greater omentum of globally immunodeficient NSG mice by the intraperitoneal injection of DM6-Mut cells. If left untreated, the tumors progress rapidly and metastasize, and the animals develop ascites fluid in 20–25 days.18 To demonstrate the presence of exosomes in these microenvironments, we isolated and characterized EVs derived from DM6-Mut tumor xenografts. NTA, a technique commonly employed to determine the size of these vesicles, revealed that the DM6-Mut EVs had a modal diameter of 100 nm (online supplemental figure S4A). The presence of exosomal markers was then tested using a commercially available antibody platform called exosome antibody array. We found that these vesicles expressed abundant CD81 and EpCAM, while TSG101, FLOT1, ICAM, annexin V, and ALIX were also detected (online supplemental figure S4B), establishing the identity of these EVs as exosomes. The absence of GM130 indicated that our exosome preparations were not contaminated with cellular debris (online supplemental figure S4B). Exosomal PS expression was detected using latex bead flow exometry as described previously. We found that these exosomes expressed high levels of PS on their surface (figure 4A).
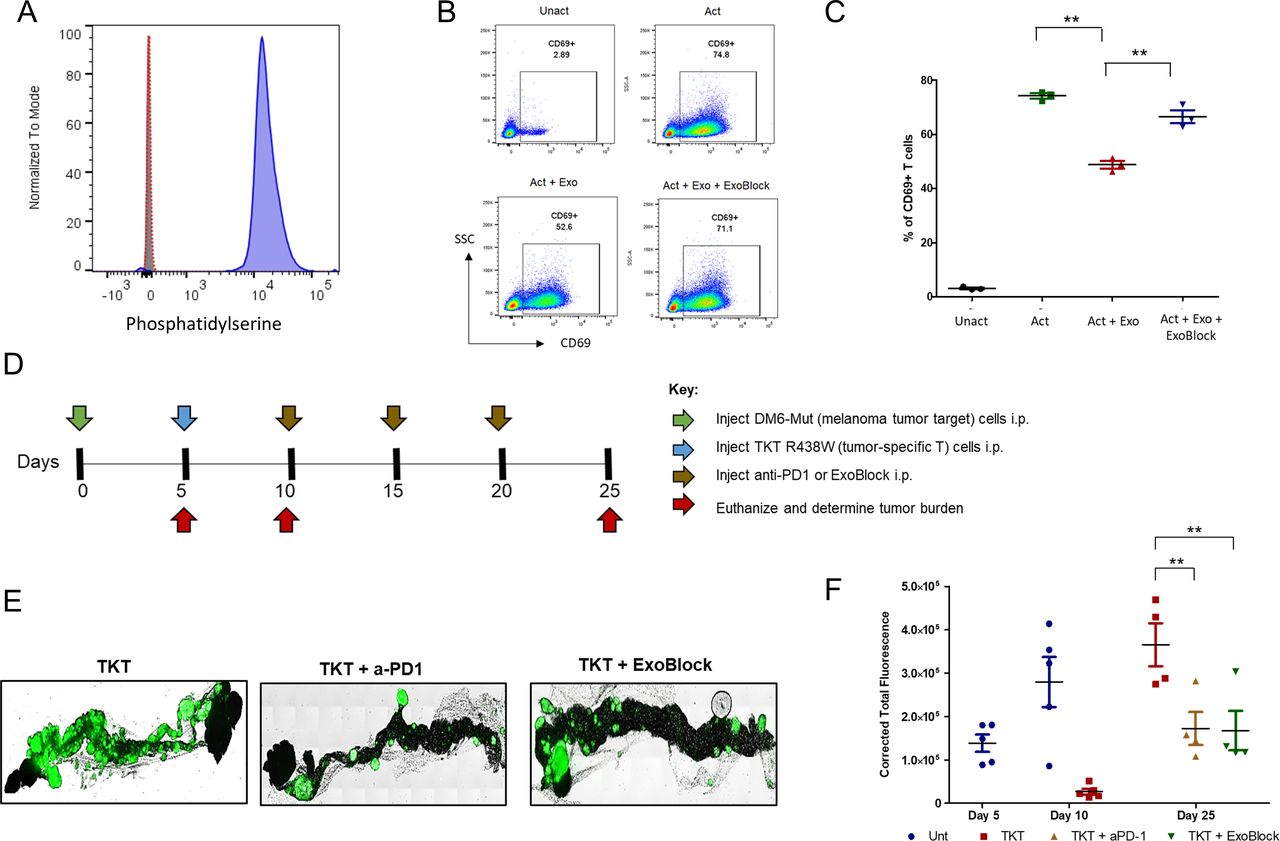
ExoBlock reverses immunosuppression mediated by melanoma-associated exosomes in vitro and suppresses melanoma tumor escape in the X-mouse model. (A) PS expression in melanoma-derived exosomes was determined using latex bead flow exometry. Unstained (filled gray histogram), secondary antibody control (open red histogram) and PS-stained exosomes (filled blue histogram). (B,C) T cells were either left Unact, activated for 2 hours with immobilized antibodies to CD3 and CD28 without exosomes (Act), Act+Exo, or Act+Exo+ExoBlock. Activation was monitored by detecting the upregulation of CD69 on CD3+ T cells using flow cytometry following overnight incubation. (B) Representative data and (C) quantified data from three independent experiments are shown. (D) Experimental scheme indicating the timeline for injection of tumor cells (green arrow), TKT R438W cells (blue arrow), treatment with anti-PD1 or ExoBlock (brown arrows) and estimation of tumor burden (red arrows). (E) Representative images of omental tumor burdens on day 25. (F) Tumor burdens were determined by quantifying the GFP signal using ImageJ software and are represented as corrected total fluorescence. n=4–5 mice per group. There are no data points for untreated cohorts on day 25 as the tumor burdens were too high to determine accurately. Data shown as mean±SEM. **P≤0.01. Act+Exo, activated with exosomes only; Act+Exo+ExoBlock, activated with exosomes and ExoBlock; PS, phosphatidylserine; Unact, unactivated.
We next tested the immunosuppressive ability of these exosomes by determining their effect on T-cell activation, measured by the CD69 upregulation assay described earlier. As predicted, melanoma-derived exosomes significantly inhibited (by 36%) the upregulation of CD69 (figure 4B,C). Treatment with ExoBlock was found to reduce this inhibition to 10%, representing a 71% reversal. We also observed a near-complete reversal of IL-2 expression with ExoBlock (online supplemental figure S4C). We conclude that exosomes present in the DM6-Mut melanoma TME are immunosuppressive, and that treatment with ExoBlock can reverse this exosome-associated immunosuppression.
Having demonstrated the presence of PS+ exosomes in two different TMEs, their ability to suppress T-cell function, and the ability of ExoBlock to reverse this suppression in vitro, we predicted that by targeting PS with ExoBlock, it would be possible to enhance the T cell-dependent antitumor response in vivo. The design and validation of two tumor xenograft models, the X-mouse model,18 and the OTX model19 for evaluating the therapeutic efficacy of ExoBlock in vivo made it possible for us to test our prediction.
In vivo efficacy of ExoBlock demonstrated using the X-mouse model
The in vivo therapeutic efficacy of ExoBlock was first tested in the X-mouse model, a preclinical model designed and validated specifically for the purpose of evaluating immunotherapeutic strategies designed to enhance antitumor T-cell responses.18 This model uses Green Fluorescent Protein (GFP)+ DM6-Mut cells that express patient-derived tumor neoantigens in the context of HLA- A*02:01, making them tumor targets. Xenografts are established in the greater omentum of NSG mice by the intraperitoneal injection of 2.5×106 tumor target cells. As stated previously, these tumors progress rapidly and metastasize if left untreated. The adoptive transfer of 5×105 patient-derived T cells (TKT R438W cells) specific for the peptides expressed on these tumor targets into 5-day-old xenografts results in an immediate suppression of tumor growth. This initial suppression is followed by tumor escape. The X-mouse model allows us to evaluate the efficacy of immune-based therapies that enhance T-cell function by determining their ability to prevent or suppress this tumor escape. This model has been validated by demonstrating the efficacy of two such immune-based therapies—IL-12 and anti-PD-1 (nivolumab) in suppressing tumor escape.18
Having established that DM6-Mut tumors release PS+ immunosuppressive exosomes and that ExoBlock reverses the exosome-mediated T-cell immune suppression, we next used the X-mouse model to test the therapeutic efficacy of ExoBlock in vivo and compared it with checkpoint blockade using the anti-PD-1 antibody nivolumab—a Food and Drug Administration-approved treatment for metastatic melanoma.26 Xenografts were established with DM6-Mut melanoma cells, and tumor-specific T cells (TKT R438W) and ExoBlock or nivolumab was administered by intraperitoneal injection on the indicated days (figure 4D). The dose of ExoBlock was determined based on pharmacokinetic considerations, and the concentrations were used to block the exosome-mediated T-cell suppression in vitro. On day 25, mice were euthanized, and the omental tumor burdens were measured by quantifying the GFP signal as described previously.18 We found that ExoBlock significantly reduced tumor growth (twofold reduction of GFP intensity on day 25), and tumor suppression was comparable to checkpoint blockade using nivolumab (figure 4E,F). The untreated mice developed ascites fluid and extensive metastasis by day 25, necessitating euthanasia, while the ExoBlock-treated mice did not show any of these signs, presumably conferring a survival advantage. We also demonstrate that the efficacy of ExoBlock is dependent on T cells, since ExoBlock had no effect on the growth of DM6-Mut tumors in the absence of TKT R438W cells (online supplemental figure S5). These studies establish the efficacy of ExoBlock in vivo in human melanoma tumors.
In vivo efficacy of ExoBlock demonstrated using the OTX model
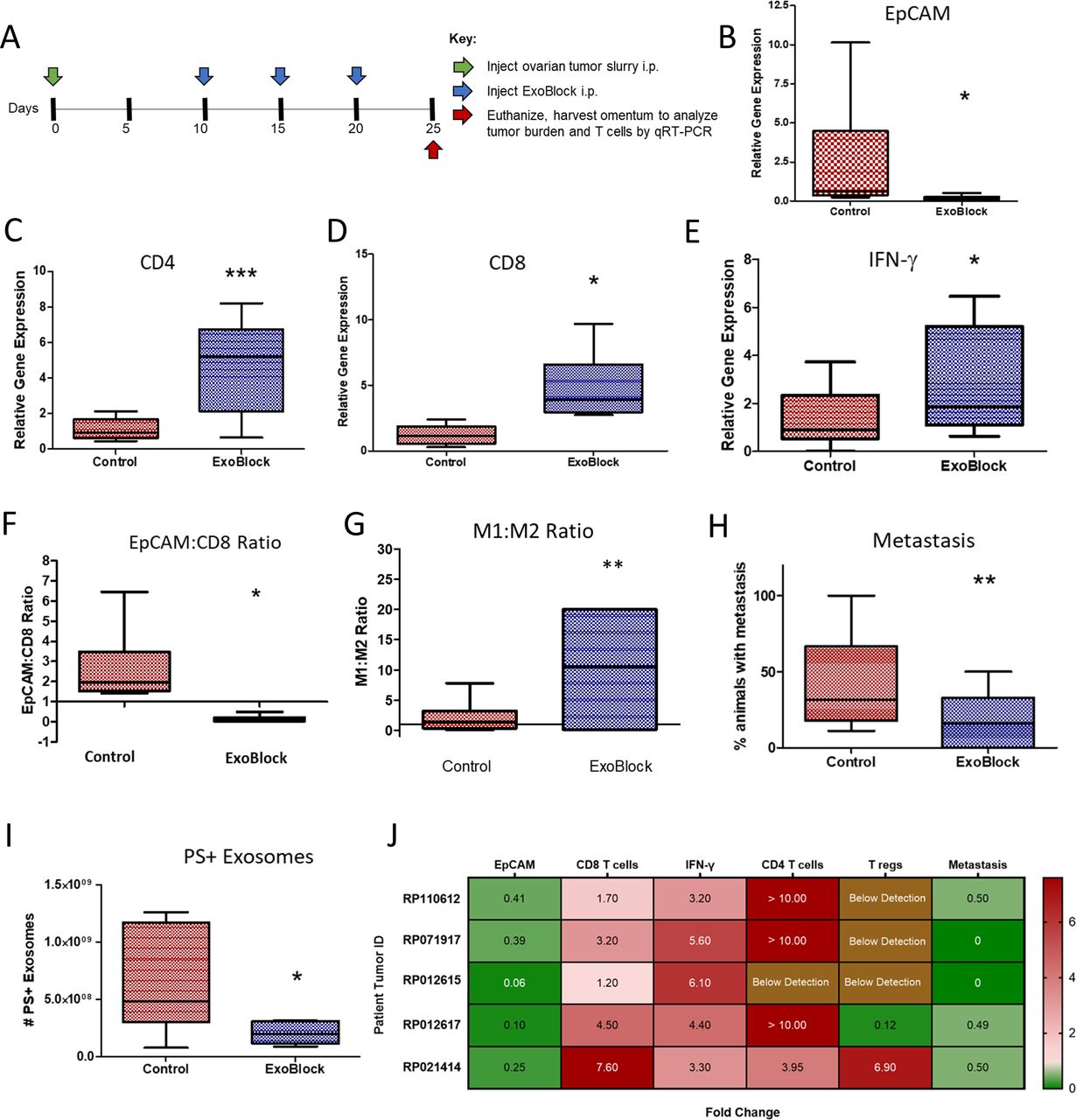
Since adoptively transferred T cells are the only immune cells present in the microenvironment of the X-mouse model,18 we also tested ExoBlock in a more complete TME. This was done using the previously established OTX model,19 which comprises intraperitoneal injection of small aggregates of tumors derived from patients with ovarian cancer into NSG mice, resulting in a rapid and preferential engraftment of a TME including tumor-associated stromal fibroblasts, macrophages, B and T lymphocytes, and tumor cells in the greater omentum of these mice.19 Analysis of tumor burden and T-cell transcripts was performed by real-time quantitiative reverse transcription polymerase chain reaction (qRT-PCR) of the greater omentum following treatment with three intraperitoneal injections of ExoBlock (figure 5A). ExoBlock was not only found to reduce the tumor burden by 92% (figure 5B) but also resulted in a 7.6-fold increase in numbers of CD8 T cells (figure 5C), 4.0-fold increase in CD4 T cells (figure 5D), and 3.3-fold increase in IFN-γ (figure 5E)—a measure of activated T-cell responses in the tumor xenograft, establishing its immune-enhancing effect. As a result, the tumor (EpCAM):CD8 T-cell ratio, which represents the dynamic between tumor growth and immune responses, is reversed on ExoBlock treatment (figure 5F). ExoBlock also affected macrophage polarization by increasing the proportion of the immune-stimulatory M1 macrophages, thus increasing the M1:M2 ratio in the TME (figure 5G, (online supplemental figure S6). Additionally, the incidence of metastasis was found to be significantly reduced in the ExoBlock-treated group (figure 5H), suggesting a possible survival advantage. To determine the effect of ExoBlock treatment on the number of circulating PS+ exosomes, we quantified ExoGlow+PSVue+ exosomes in the circulation of day 25 xenografts using imaging IFC. Sera derived from ExoBlock-treated xenografts displayed a 60% reduction in the number of PS+exosomes compared with sera from the control cohorts (figure 5I and online supplemental figure S7). ExoBlock was tested in multiple xenografts established using ovarian tumors from five different patients and was found to be reproducibly efficacious in reducing tumor burden as well as in enhancing immune responses (figure 5J). The effect of ExoBlock treatment on regulatory T cells, quantified using Foxp3, was found to be variable (figure 5J).
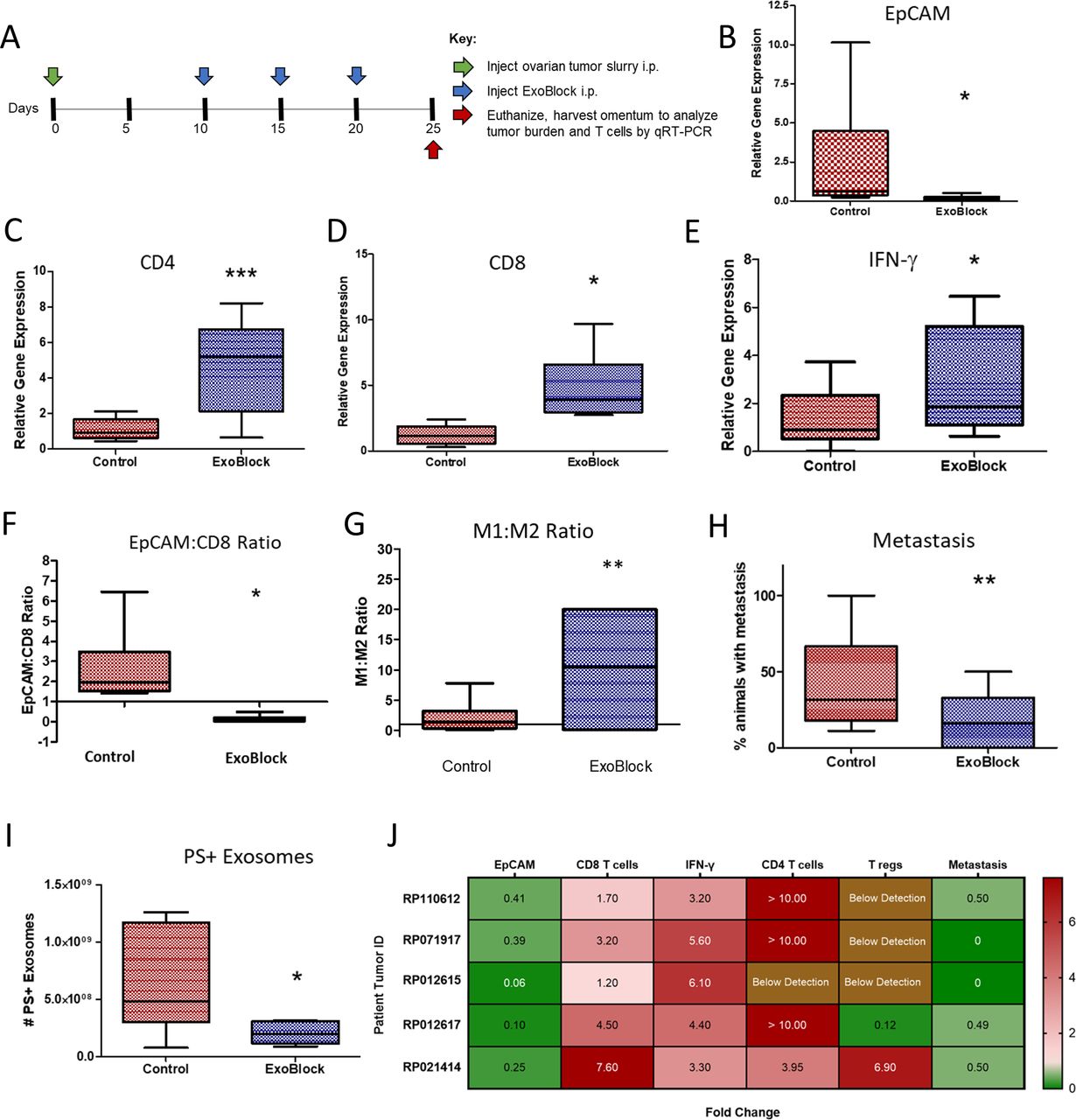
ExoBlock suppresses tumor growth and enhances immune responses in the OTX model. (A) Experimental scheme indicating the timeline for injection of tumor cells (green arrow), ExoBlock (blue arrows), and experimental endpoint (red arrow). On day 25, the animals were euthanized; omenta were harvested; and the expression of different genes were analyzed using qRT-PCR. Relative expression level of EpCAM (for tumor burden) (B), CD8 (C), CD4 (D), and IFN-γ (E) are shown. (F) EpCAM to CD8 ratio and (G) M1:M2 macrophage ratio are shown. (H) Incidence of metastasis and (I) the number of circulating PS+ exosomes per millilitre of serum are shown. Whiskers and outliers were calculated using the Tukey method. n=6 for (G) and n=8 for all the others. Mean fold change in tumor burden, T cells and metastasis in ExoBlock-treated mice normalized to control mice (set as 1) is shown in (J). Xenografts of 5–10 were established for each patient tumor. Data shown as mean±SEM. *P≤0.05, **P≤0.01, ***P≤0.001. IFN-γ, interferon gamma; OTX, omental tumor xenograft; PS, hosphatidylserine.
Flow cytometric analysis of untreated and ExoBlock-treated OTX xenografts (figure 6A) demonstrated a significant increase in the proportions as well as in cell numbers (per xenograft) for total T cells (figure 6B–D), CD4 T cells (figure 6E–G), and CD8 T cells (figure 6H–J), corroborating the results from gene expression analysis using qRT-PCR.

ExoBlock enhances T-cell numbers in the OTX model. (A) Experimental scheme indicating the timeline for injection of tumor cells (green arrow), ExoBlock (blue arrows), and experimental endpoint (red arrow). On day 25, the animals were euthanized; omenta were harvested; and the percentages as well as total cell numbers of different T-cell subsets in the xenografts were quantified using flow cytometry and cell counts. (B, E,H) Representative data, (C,F,I) compiled percentages; and (D,G,J) cell numbers per xenograft are shown for total T cells (CD3+), CD4 T cells (CD3+CD4+), and CD8 T cells (CD3+CD8+), respectively. Ten xenografts were established for each patient tumor. Data shown as mean±SEM. **P≤0.01, ***P≤0.001.
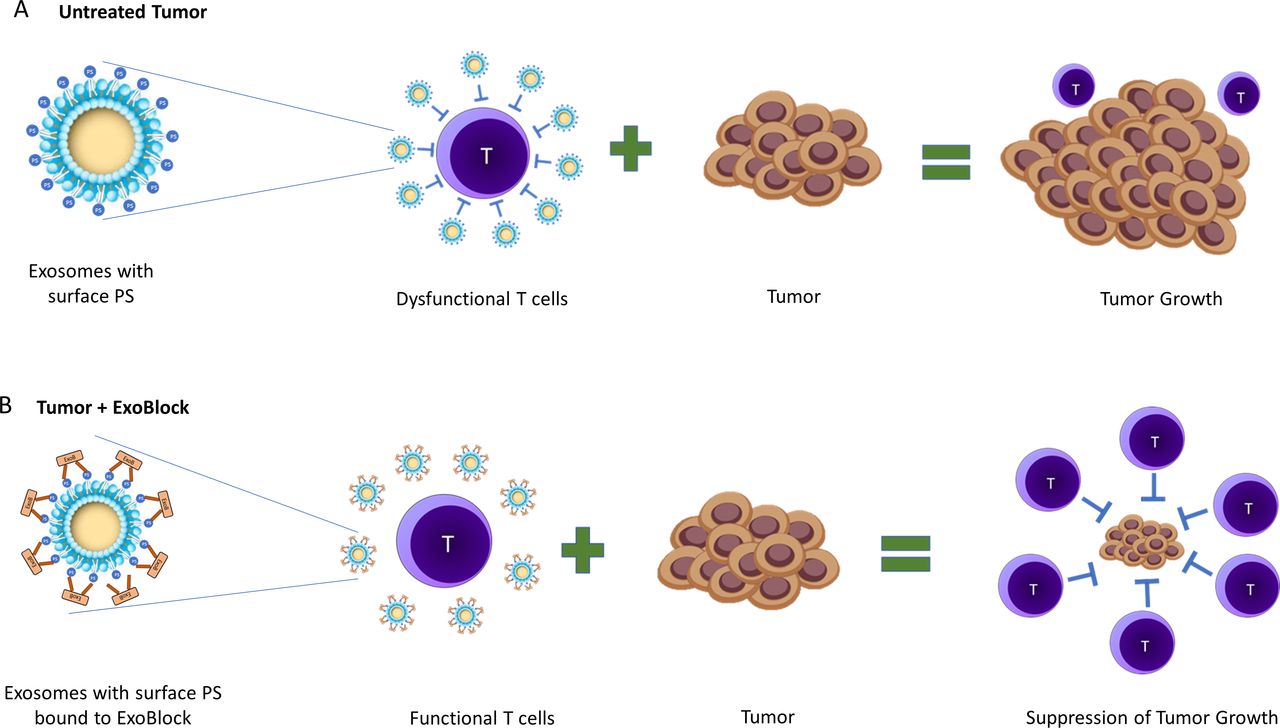
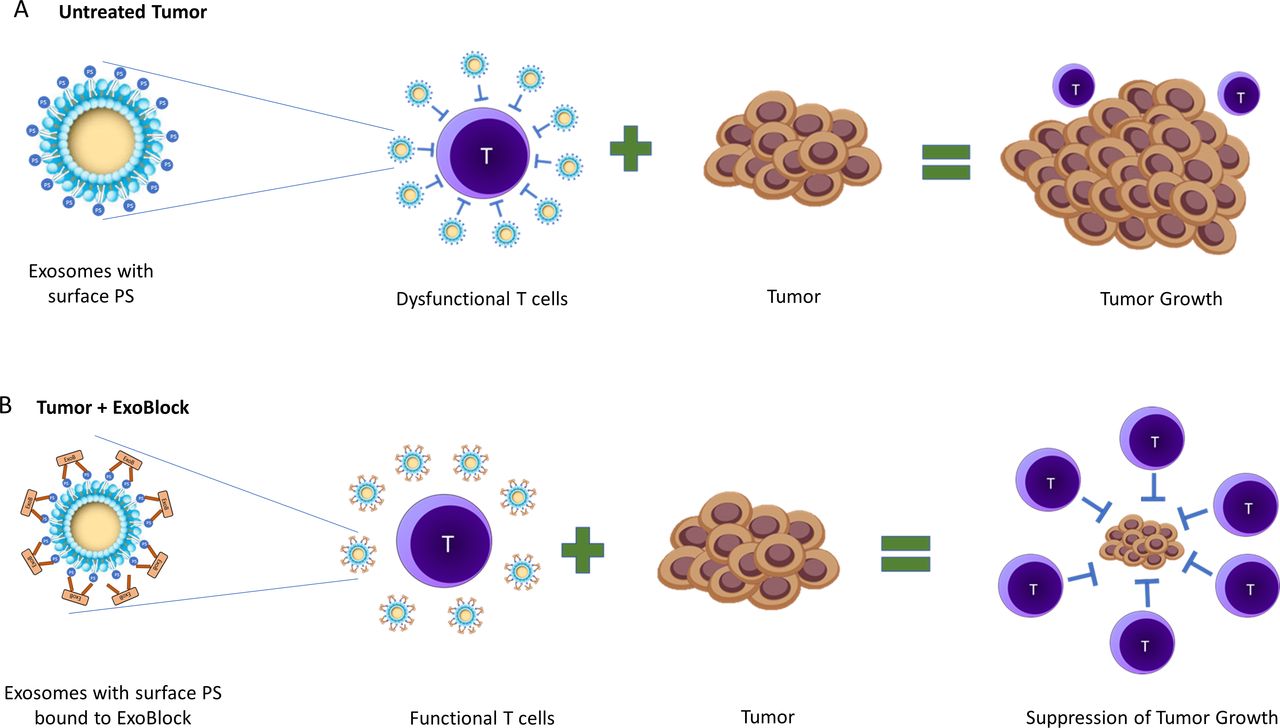
We propose that ExoBlock binds to PS on exosomes, thereby blocking immunosuppression (figure 7). This results in a reactivation or rescue of T cells that are transiently suppressed by the exosomes in TMEs, which in turn suppress tumor growth (figure 7). A reduced tumor burden results in a decrease in PS+ exosomes being produced over time, gradually reversing the immunosuppressive nature of the TME. Together, our results establish the therapeutic potential of ExoBlock as a PS-blocking molecule while also confirming the viability of approaches targeting immunosuppressive exosomes in TMEs.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the neutralization of immunosuppressive PS+ exosomes by ExoBlock. (A) In tumor microenvironments, PS+ exosomes suppress T-cell activation, proliferation and cytotoxic activity leading to tumor growth. (B) Treatment with ExoBlock neutralizes immunosuppressive exosomes, resulting in a restoration of T-cell function and ultimately the suppression of tumor progression. PS, hosphatidylserine.
Discussion
The exposure of PS on the surface of apoptotic cells was initially thought of as playing the role of a ‘cleanup crew’, sending an ‘eat-me’ signal to macrophages for the purpose of clearance.23 27 Subsequent studies have established that PS facilitates an immunologically silent clearance and plays an active immunosuppressive role by inducing tolerance to antigens associated with it.23 28 29 The immunosuppressive role of PS has also been demonstrated in exosomes isolated from TMEs, where they have been shown to arrest T-cell activation through the TCR.6 This immune suppression was found to be causally linked to PS on the exosomal surface.6 This makes PS an attractive target to neutralize in order to enhance immune responses in cancer, and possibly also in chronic inflammatory conditions, where a similar role for PS+ exosomes has been described.30 Initial proof-of-concept studies showed that anti-PS antibody could reverse tumor exosome-mediated immunosuppression significantly.6 Strategies to block PS in preclinical studies using anti-PS antibodies and annexin V or to treat lung cancer in clinical trials using a PS specific antibody, bavituximab,23 have met with modest success, perhaps due to the relatively low PS-binding affinity of the molecules used and the relatively high molecular weight of the antibody. ExoBlock is a hexamer that has been engineered to carry six binding sites for PS, which is more than an antibody or annexin V and hence expected to bind PS with a much higher avidity. We have determined that ExoBlock does indeed bind PS with a high avidity and is very effective in blocking immune suppression of multiple T-cell activation endpoints mediated by exosomes derived from two different TMEs in vitro. The fact that ExoBlock was able to reverse 70% of exosome-mediated immune suppression in vitro for multiple activation endpoints indicates that PS expression is critical for exosome-mediated immune suppression. It is however not surprizing that the reversal was not total in these experiments, since various other molecules, including proteins such as PD-L116 and TGF-β,31 lipids such as ganglioside GD3,8 and nucleic acids such as miR-24–3 p32 have been also known to be causally linked to the immunosuppressive nature of these vesicles.
To test the in vivo therapeutic efficacy of ExoBlock on human tumors, we then used two different in-house developed tumor xenograft models. The first, the melanoma-based X-mouse model, was designed and validated for the purpose of preclinically evaluating the efficacy of immune-based therapies that enhance the antitumor responses of patient-derived tumor-specific T cells.18 The X-mouse model has multiple advantages, such as providing a controlled system in which the number of tumor cells used to establish tumor xenografts is known; the timing and number of T cells that are adoptively transferred into tumor‐bearing mice is controlled; and the tumor specificity of the T cells for tumor antigens is well defined.18 Since adoptively transferred T cells are the only immunocompetent cells present in the TME in this model, the suppression of tumor growth can be directly interpreted as being a result of enhanced antitumor T-cell responses.
Since the X-mouse model lacks other immune cells and stroma, which impact therapeutic efficacy, we also tested ExoBlock in a more complete TME represented by the ovarian tumor-based OTX model.19 In all the patient tumors tested, ExoBlock was found to significantly suppress tumor growth and metastasis, with a concomitant increase in CD4 and CD8 T cells, as well as in IFN-γ expression, an indicator of T-cell responses. While there was some variability in the degree of tumor suppression and enhancement of immune responses in different tumors as expected, these data trended in the same direction for all the tumors tested. Since the only T cells in these tumors are the tumor-infiltrating lymphocytes (TILs) present in the original tumor aggregates injected on day 0, these data indicate that the dysfunction of at least a subset of TILs is reversible. This is consistent with previous studies from our lab, given our findings that liposomally delivered IL-12 reactivates hyporesponsive T cells in the TME,33–36 and bodes well for immune-based therapies that are designed to enhance T-cell function. The direct effect of ExoBlock on CD4 and CD8 T cells in the OTX model, along with results from the X-mouse model that demonstrate the requirement for T cells, establishes that the efficacy of ExoBlock is dependent on restoring T-cell function, therefore representing an immune-based therapy.
An enhancement of immune responses is expected to ultimately result in the generation of Tregs, or upregulation of checkpoint molecules such as PD-1 as a natural regulatory mechanism. This raises the possibility of combining ExoBlock with IL-12, which activates T cells while also suppressing checkpoint upregulation,18 and/or checkpoint blockade therapy such as nivolumab to improve the therapeutic efficacy. Future studies will test the efficacies of these combinations in our xenograft models. The effect of ExoBlock on additional immune cell subsets that influence tumor growth such as Th17 cells and myeloid-derived suppressor cells will also be investigated. While there are indications that ExoBlock treatment confers a survival benefit to tumor-bearing mice, we recognize that this needs to be thoroughly tested in one or more tumor models. We plan to extend these studies by investigating the effect of ExoBlock on TME using immunocompetent syngeneic mouse tumor models. These models will also allow us to generate longitudinal tumor growth data, the lack of which is one of the limitations of the current study.
We recognize that PS is present not just on exosomes but also on apoptotic cells and cancer cells as a mechanism of immune evasion. ExoBlock is expected to bind to all PS, therefore representing a multipronged approach to negate PS-induced immunosuppression. Additionally, ExoBlock may also negate other effects PS might be mediating in TMEs. We also recognize that translating this blockade of immunosuppression to clinically efficacious targeting of exosomes in the TME to enhance immune responses will remain a challenge until the biology of exosomes and their role in immune and non-immune cellular interactions is better understood.
To conclude, our results show that targeting immunosuppressive exosomes using a high-avidity PS-binding molecule such as ExoBlock may represent a promising strategy to combat immunosuppression and T-cell dysfunction in human TMEs, providing a rationale for further development and characterization of ExoBlock.
Supplemental material
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
All specimens were collected under an approved IRB protocol (MODCR00004766) at SUNY Buffalo. All in vivo experiments were approved by the SUNY Institutional Animal Care and Use Committee under the protocol MIC17072Y.
Acknowledgments
The authors thank Anthony Miliotto and the Tissue Procurement Facility of Roswell Park for their assistance in providing tumor tissues and ascites fluid. Flow cytometry and confocal microscopy services were provided by the Confocal Microscopy and Flow Cytometry Core Facility at the University at Buffalo. Additional cytometry services were provided by the Flow and Image Cytometry Core facility at the Roswell Park.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MB and GNS contributed equally.
Contributors Since two authors, Maulasri Bhatta and Meghana Bapardekar, have the same initials, MBh will be used for Maulasri Bhatta and MBa will be used for Meghana Bapardekar. Conceptualization: GNS, RBB, and MBh. Methodology: GNS, MBh, HM, AC, MBa, and BDG. Investigation: MBh, GNS, JLL, AC, and MBa. Visualization: GNS, MBh, and RBB. Funding acquisition: RBB, GL, SVB, BMC, LDS, and HM. Resources: KO, RJK, RBB, LDS, GL, BMC, HM, and BDG. Project administration: JLL. Supervision: RBB, SVB-I, RJK, and KYP.
Funding This study was supported by National Institutes of Health grants R43 CA224602 and R43 CA257721-01A1 (to Immune Modulatory Therapies, LLC), CA034196 (LDS), R21 CA205794 (GPL), R01 CA204261 (BMC and GPL), HL-70227 (SVB), R50CA2111108 (HM), Empire State Development funded accelerator grant to support commercialization of technologies at NY state universities (RBB), The University at Buffalo Center for Advanced Technology in Big Data and Health Sciences grant (RBB), and Roswell Park Cancer Center Support grant P30CA016056 (HM).
Competing interests The corresponding author, RB, is an officer and founder of Immune Modulatory Therapies LLC. A patent is currently pending for ExoBlock.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.