Article Text

Abstract
Immunotherapeutic drugs including immune checkpoint blockade antibodies have been approved to treat patients in many types of cancers. However, some patients have little or no reaction to the immunotherapy drugs. The mechanisms underlying resistance to tumor immunotherapy are complicated and involve multiple aspects, including tumor-intrinsic factors, formation of immunosuppressive microenvironment, and alteration of tumor and stromal cell metabolism in the tumor microenvironment. T cell is critical and participates in every aspect of antitumor response, and T cell dysfunction is a severe barrier for effective immunotherapy for cancer. Emerging evidence indicates that extracellular vesicles (EVs) secreted by tumor is one of the major factors that can induce T cell dysfunction. Tumor-derived EVs are widely distributed in serum, tissues, and the tumor microenvironment of patients with cancer, which serve as important communication vehicles for cancer cells. In addition, tumor-derived EVs can carry a variety of immune suppressive signals driving T cell dysfunction for tumor immunity. In this review, we explore the potential mechanisms employed by tumor-derived EVs to control T cell development and effector function within the tumor microenvironment. Especially, we focus on current understanding of how tumor-derived EVs molecularly and metabolically reprogram T cell fates and functions for tumor immunity. In addition, we discuss potential translations of targeting tumor-derived EVs to reconstitute suppressive tumor microenvironment or to develop antigen-based vaccines and drug delivery systems for cancer immunotherapy.
- immune evation
- immune tolerance
- immunotherapy
- tumor microenvironment
- adaptive immunity
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Communications between cancerous and healthy cells play important roles in affecting the progress of oncologic diseases. Cells can communicate with each other via a variety of mechanisms, such as using the soluble factors, directly contacting through adhesion molecules, transferring surface molecules and cytoplasmic components through nanotubules,1 and the usage of extracellular vesicles (EVs).2 According to the guidelines from the International Society for Extracellular Vesicles, EVs refer to a variety of cell-released, circular membrane enclosures that meet the minimal criteria in terms of their methods of isolation and characterization.3 EVs vary in size and have different modes of biogenesis. Among them, exosomes are of endosomal origin with very small size, ranging from 30 to 150 nm in diameter.4 Both in vitro studies using cancer cell lines and in vivo analyses of plasma, urine, and pleural effusions of patients with cancer have revealed the enhanced release of exosomes by tumor cells,5 6 and tumor-derived exosomes are involved in many key aspects of cancer biology such as tumorigenesis,7 drug resistance,8 and metastasis.9 It is important to point out that due to the lack of consensus in the scientific community on both the optimal method for the purification of exosomes and the specific markers for distinguishing different types of EVs, the term exosome used in the literature may or may not accurately describe the specific EVs being studied. In fact, the 2018 minimal information for studies of EVs (MISEV) guidelines have suggested to use the term ‘EVs’ instead of subtype terms.
A major obstacle for immunotherapy is tumor cell escape from immune surveillance, through inhibition of T cell function via either direct contact between tumor cells and T cells or suppressive soluble factors secreted by tumor cells. Tumor cells can use programmed death ligand-1 (PD-L1) and CD80 to directly interact with inhibitory molecules programmed death-1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) expressed in T cells, which leads to an inhibition of T cell activation and function.10 Increasing studies suggest that accumulation of metabolic products and metabolites such as lactate produced from tumor cells can also suppress the function and survival of T cells within the tumor microenvironment (TME).11 12
Emerging evidence indicates that tumor-derived EVs also play a pivotal role in facilitating tumor to escape immune surveillance.13 Tumor-derived EVs can modulate functions of different immune components in the TME to achieve immune suppression. For example, they can drive differentiation of monocytes towards myeloid-derived suppressor cells (MDSCs),14 suppress effector T cell function, generate anergic-like state in natural killer T cells (NKT),15 promote M2-like macrophage polarization,16 17 and stimulate regulatory T cell (Treg) expansion.18 Both MDSCs and Treg cells are well known for their abilities to suppress T cell activation, while polarization of the M2-like macrophage can promote tumor progression.19 In contrast to immunosuppression, tumor-derived EVs also exhibit immune stimulatory properties under some circumstances. For example, tumor-derived EVs can transfer major histocompatibility complexes (MHCs) to dendritic cells (DCs), which results in the activation of T cells, leading to the inhibition of tumor progression.20 In addition, the immunostimulatory function of tumor-derived EVs was mainly achieved either when the tumor cells were exposed to stress conditions or the components of the EVs have been modified. For example, heat-stressed tumor cells have been shown to secret EVs that help inducing the production of interleukin (IL)-6 by DCs and macrophages, leading to a switch of Treg to Th17 cells in the TME.21 Silencing transforming growth factor beta (TGF-β) in leukemia cell-derived EVs promotes maturation and function of DCs and enhances the antigen-specific cytotoxic T lymphocyte responses.22 Modified tumor-secreted EVs with miR-424 knocked down was also reported to increase T cell–mediated antitumor immune response and enhance the immune checkpoint blockade immunotherapy in colorectal cancer tumor models.23 Therefore, the interplay between tumor cells and T cells as mediated by tumor-derived EVs is an emerging and exciting area. However, the molecular mechanisms responsible for the effects on T cell function and differentiation mediated by tumor-derived EVs within the TME have not been well summarized. In this review, we explore the different roles that tumor-derived EVs play in affecting T cell function and the underlying molecular mechanisms. We also discuss how immunostimulatory function of EVs can be utilized to boost the immune therapy efficacy and develop novel strategies for cancer immunotherapy.
Biogenesis of EVs and their cargo molecules
There are two major pathways for the biogenesis of EVs: vesicles are formed by outward budding from the plasma membrane, or/and vesicle formation involves the fusion of endosomal-derived multivesicular bodies (MVB) with plasma membrane and the subsequent release of intralumenal vesicle (ILVs) into the extracellular space. The relatively large-sized EVs such as microvesicles are formed by the first pathway, and exosomes are generated by the second pathway.24 Key players for the biogenesis of EVs include the endosomal sorting complex required for transport (ESCRT) machinery and neutral sphingomyelinase (nSMAse). The ESCRT machinery is responsible for the sorting of ubiquitinated proteins into ILVs.24 In addition, both the ESCRT machinery and the ceramide generated by the nSMAse also promote vesicle formation both at the plasma membrane and inside MVBs.25 Besides vesicles formation machinery, members of the Rab family of small GTPases, which play well-established roles in transferring vesicles between intracellular compartments, have also been implicated in regulating the trafficking of MVB to the plasma membrane for exosome release.26 Among the various Rab family members, Rab-27 is the best characterized. Impairing Rab27a or Rab27b can lead to an altered MVB morphology and docking to the plasma membrane.26 Furthermore, there is evidence that the SNARE proteins such as SNAP-23 and SYX-5 are specifically required for exosome release.27 28 Interestingly, exosome biogenesis can be enhanced by a variety of stress conditions such as hypoxia, irradiation, ethanol, or starvation.29 30 Among these different stressors, hypoxia is of particular interest as it is a hallmark of the TME. Studies have shown that hypoxia can both enhance the production of EVs by tumor cells and alter their components.31 As important mediators for near and long-distance intercellular communications, EVs contain a variety of cargo molecules that include lipids, proteins, nucleic acids and metabolites. All EVs share a common group of molecules that pertain to their structure and function.32 EVs derived from different cell types have distinct components, and a certain set of proteins and RNAs are unique to each EV depending on the origins of parent cells (table 1).
Tumor-derived EVs induce alterations of immune-related molecules in T cells
The nucleic acids in the cargo of EVs, including DNAs and RNAs, have been implicated in regulating immune responses. Among different subtypes of EVs, exosomes and the effects of their components on immune cells are the most widely studied. DNAs presented in the exosomes derived from breast cancer cell E0771 have been shown to drive the activation of cGAS-STING signaling and antitumor response in mice.33 DNAs of EVs from the oncogenic genomic DNA sequences transforming protein p21 (HRAS)-driven cancer cells can enhance the ability of circulating neutrophils to activate tissue factor procoagulant activity and IL-8, which are important factors in promoting tumor inflammation and paraneoplastic events.34 T cells can also use its EV mitochondrial DNA as a mediator to prime DCs and enhance their responses.35 Interestingly, DNAs present in the tumor-derived exosomes often contain a variety of clinically relevant information about tumor-specific mutations representing multiple genes such as epidermal growth factor receptor (EGFR), proto-oncogene B-Raf, RAS, isocitrate dehydrogenase 1, and human epidermal growth factor receptor 2 (HER2), which can be used in diagnosis as a promising ‘liquid biopsy’ material for therapy recommendations.36 Multiple types of RNAs have been identified in EVs, including messenger RNAs (mRNAs), microRNAs (miRNAs), and other non-coding RNAs.37 Notably, the majority of exosomal RNAs are degraded fragments with the length of less than 200 nucleotides, while some full-length RNA molecules were also found. The tumor exosomal miRNAs can regulate the immune responses by influencing gene expression and signaling pathways in recipient immune cells via the transfer of miRNAs. For example, exosomal miR-222–3 p was found to downregulate suppressor of cytokine signaling 3 in monocytes and promote STAT3-mediated M2 polarization of macrophages.38 Moreover, exosomal miRNAs can promote cancer progression by stimulating the secretion of angiogenic cytokines, including vascular endothelial growth factor, matrix metallopeptidase 2, MMP9, basic fibroblast growth factor and TGF-β.39 Studies have also shown that tumor exosomal miRNAs, such as adenocarcinoma (PDAC)-specific miR-181–5 p, miR-30a-3p, and squamous cell carcinoma (SCC)-specific miR-10b-5p may serve as diagnostic biomarkers for patients with early non-small cell lung cancer (NSCLC).40 Besides miRNA, exosomal circular RNA-PDE8A was associated with progression and prognosis in patients with pancreatic ductal adenocarcinoma.41
Lipids are extremely prevalent in EVs. Exosomal lipids often mirror that of intraluminal vesicles with more cholesterol, sphingolipids, phosphatidylinositol-3-phosphate, and bis(monoacylglycerol)phosphate.42 These lipids form lipid rafts on the exosomal membrane and mainly sustain the structure of exosomes.43 There is an asymmetry of lipid types between the outer and inner leaflets of the membrane.44 Specifically, sphingolipids and phosphatidylcholine are more prevalent on the outer leaflet than on the inner leaflet, while all other lipid classes are mainly on the inner leaflet.44 It is important to note that high levels of cholesteryl ester, triacylglycerol, and cardiolipin in exosomal preparations may indicate the co-existence of lipid droplets, lipoproteins, or mitochondria within exosomes.42 Recent studies showed that exosomes are capable of directly transporting lipids such as cholesterol, fatty acids, and eicosanoids, from parent cells to recipient cells, which may cause the inflammation, immune or metabolic changes in certain microenvironments.45 46 Adipocytes release lipid-filled exosomes that become a source of lipid for local macrophages and these vesicles were sufficient to induce in vitro differentiation of bone marrow precursors into adipose tissue macrophage-like cells.45 Healthy donor serum circulating exosomes can take up free fatty acid (FFA) directly from serum via CD36 and then deliver FFA into cardiac cells.47
A plethora of membrane proteins have been found in EVs, including tetraspanins, flotillin, PGRL, stomatin, adhesion proteins such as L1CAM, lysosomal associated membrane proteins like LAMP2, integrins, and surface glycoproteins such as fibronectin.48 The most found proteins are membrane transporters and proteins that mediate membrane fusions such as GTPases, annexins, and flotillin, heat shock proteins like HSP70, tetraspanins such as CD9, CD63, and CD81, multivesicular body biogenesis proteins like Alix and TSG101, and phospholipases.49 Tumor-derived EVs often contain proteins that are important for the progression of tumors, which include immunosuppressive proteins (such as death receptor ligands FasL or TRAIL), check point receptor ligands PD-L1, and ectoenzymes engaged in the adenosine pathway (CD39 and D73).50 51 Furthermore, enzymes that control metabolism are among the most frequently identified proteins in proteomics analysis of EVs.52 The dynamic change of enzymes such as ATPase in tumor exosome appears to help generating an environment that benefits tumor progression.53 In addition, tumor-derived EVs carry a unique set of tumor-associated antigens (TAAs), co-stimulatory molecules and the MHC components that could stimulate immune cells and promote antitumor immune responses.54 Thus, proteins carried in tumor-derived EVs could mediate either an immuno-suppressive response or immuno-stimulatory response depending on the specific contexts.
Many types of recipient cells use EV internalization as a mechanism to receive the cargos carried by the EVs. Once internalized, EVs can fuse their membranes with those of endosomes in the recipient cells, leading to the horizontal transfer of their content into the target cells.55 The precise mechanisms targeting EV to immune cells and triggering its internalization remain largely unclear, though interactions between EV proteins and membrane receptors in the recipient cells have emerged to play important roles. For example, targeting of EVs to DCs are mediated via the interaction between EV proteins MFG-E8 and the tetraspanins CD9 and CD81 with DC proteins αvβ3 integrin and CD11a/CD54, and blocking the binding sites of CD11a using antibodies can reduce uptake of EVs into DCs.56 Some studies claimed that since T lymphocytes are not phagocytic as macrophages, they receive information from tumor-derived EVs mainly by the interaction between their surface receptors with components in EVs.57 One study has shown that although rat pancreatic adenocarcinoma-derived exosomes are taken up by all leukocyte subpopulations, the amount taken up by CD11b+ and CD11c+ leukocytes greatly exceed that taken up by T cells.58 Similarly, it has been demonstrated that the ability of CD8+ T cells to internalize is not as good as that of other types of immune cells.59 These studies strongly indicate that exosome internalization may not be a critical way for EVs to affect T cell responses. In support of this notion, a recent study has shown that T lymphocytes and its subsets primarily use specific surface receptors to receive information from tumor-derived EVs.57 Specifically, dye-labeled tumor-derived exosomes were incubated with human T cells or NKT cell and B cells respectively, and it was found that T lymphocytes, unlike other mononuclear cells, do not uptake exosomes but rather respond to exosome signals delivered to the cell surface by Ca2+ influx.57 It has shown that naïve CD8+ T cells can uptake EVs shed from antigen-presenting cells via the interactions between T cell antigen receptor, MHC class I/peptide and the intercellular adhesion molecule 1 (CD54) present on the EVs.60 Given the important roles of EVs in regulating immune cell functions, development of more advanced and accurate methods to isolate, characterize, and manipulate EVs to elucidate the mechanisms that govern the specificity between EVs and their recipient immune cells is urgently needed for tumor immunity.
Potential mechanisms responsible for tumor-derived EVs-mediated T cell suppression
Tumor-derived EVs can suppress T cell antitumor immunity through multiple ways, including inhibiting T cell proliferation and responses, promoting Treg expansion, and inducing T cell apoptosis and exhaustion (figure 1). The molecular effects of tumor-derived EVs are achieved via different components and/or molecular processes, including delivery of DNA,34 miRNAs,61–63 metabolites, and amino acid degrading enzymes to T cells,50 64 65 or direct binding with immune suppressive protein ligands on T cells66–68 (table 1). In addition to their direct effects on T cells, tumor-derived EVs can cause T cell suppression by affecting DCs and MDSCs.
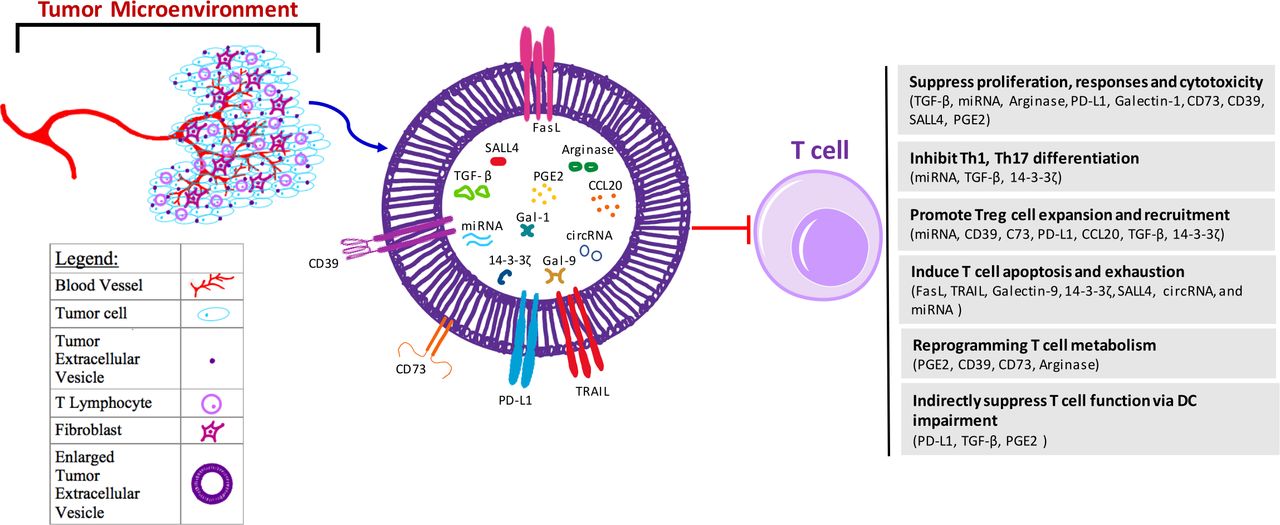
{kind=link}
Mechanisms for tumor-derived EV-mediated suppression on T cells. Tumors are surrounded by different types of stromal cells within a microenvironment that the tumor closely interacts with. Within this microenvironment, there are blood vessels, fibroblasts, and T lymphocytes, as well as environmental secreted factors including exosomes derived from the tumor. These EVs further induce dysfunction of T cells in the suppressive tumor microenvironment mainly through several ways, including (1) inhibition of T cell proliferation, effector immune responses and cytotoxicity through EV components TGF-β, miRNA, arginase, PD-L1, Gal-1, CD73, CD39, SALL4 and PGE2; (2) suppression of T helper cell differentiation through EV components miRNA, TGF-β, and 14-3-3ζ; (3) expansion and recruitment of Treg cells via EV cargoes CD39, C73, PD-L1, CCL20, TGF-β, 14-3-3ζ and miRNA; (4) induction of T cell apoptosis and/or exhaustion through FasL, TRAIL, Gal-9, 14-3-3ζ, SALL4, circRNA and miRNA; (5) reprogramming of T cell metabolism through EV cargos PGE2, CD39, CD73 and arginase to increase cellular adenosine and cAMP levels, or decrease amino acid; and (6) indirect suppression of T cell function via impairing DC maturation, migration, and antigen presentation through components PD-L1, TGF-β and PGE2. DC function impairment also contributes to MDSC and Treg cells expansion, which further lead to T cell suppression. CCL20, C-C motif chemokine ligand 20; circRNAs, circular RNA; DC, dendritic cell; EV, extracellular vesicle; FasL, Fas ligand; Gal, galectin; MDSC, myeloid-derived suppressor cells; miRNAs, microRNAs; PD-L1, programmed death-ligand 1; PGE2, prostaglandin E2; SALL4, spalt like transcription factor 4; TGF-β, transforming growth factor beta; Treg, regulatory T cell.
Tumor-derived EVs can suppress T cell proliferation. Exosomes derived from breast cancer cell lines have been shown to suppress T cell proliferation, and the effect was mediated via suppressive molecule TGF-β.69 Interestingly, the production of exosomes by breast cancer cell lines can be greatly enhanced by hypoxic condition, which may contribute to therapy resistance of breast cancer.70 Furthermore, recent evidence suggests that microRNAs from tumor-derived exosomes can also alter T cell proliferation. In a study examining the serum of patient with nasopharyngeal carcinoma (NPC), five commonly overexpressed miRNAs were found to alter T cell proliferation and differentiation via downregulation of the MAPK1 signaling pathway.63 In addition, a high miR-24–3 p level was found in the exosomes isolated from patient with NPC sera and those miR-24–3 p-containing exosomes inhibit T cell proliferation via repressing fibroblast growth factor 11 (FGF11).61 Cytokines as central important mediators in the homeostasis of lymphoid cells could promote cell survival, proliferation, and differentiation.71 72 Studies find that mesothelioma cell exosomes strongly impair proliferative responses to IL-2 in CD4+ and CD8+ T cells in vitro, but not in Treg cells.73 Besides these, exosomal PD-L1 secreted by mouse melanoma cells inhibits the proliferation and cytotoxicity of CD8+ T cells by inhibiting IL-2, interferon gamma (IFN-γ), tumor necrosis factor (TNF-α), and granzyme B in T cells in mice.74 Additionally, PD-L1 from tumor-derived EVs suppresses T cell activation in the draining lymph node in TRAMP-C2 mice, and removal of PD-L1 from the EVs inhibits tumor growth.75
Tumor-derived EVs can also induce T cell apoptosis, which has been reported in EVs isolated from melanoma, colorectal cancer, pancreatic cancer, kidney adenocarcinoma tumor cells, as well as sera from patients with oral squamous cell carcinoma and NPC.76–81 Generally, these pro-apoptotic EVs are Fas ligand positive, and they trigger apoptosis of activated T cells at least partially in a Fas ligand-dependent manner. A recent RNA sequencing study demonstrated that many of the genes linked to apoptosis and endoplasmic reticulum (ER) stress are upregulated in T cells by EVs derived from pancreatic cancer cells, meanwhile, those EVs taken up by T lymphocytes can activate p38 MAP kinase signaling, and then induce ER stress‐mediated apoptosis in T cells.79 In addition, EVs from NPC patient plasma contain galectin-9, a ligand of the membrane receptor Tim-3, and can induce apoptosis in mature Th1 lymphocytes and CD4+ T cells.81
Immunosuppressive effects on T cells by tumor-derived EVs can also be indirectly mediated by their effects on dendritic cells. For example, EVs from irradiated prostate cancer impair DC antigen presentation, promoting an adenosine-mediated suppression of CD8+ T cell activation.82 Injection of EVs derived from the Lewis lung carcinoma into mice can block the maturation and migration of DCs to lymph nodes, resulting in immune suppression on T lymphocyte via a PD-L1 dependent mechanism.83 Similar mechanisms of immunosuppression involving DCs and EVs were also observed in melanoma.84 EVs produced by melanoma cells and accessory cells of the TME provoke immune suppression and consequently defective DC functions, which contribute to expansion of Treg cells and MDSCs as well as limitation of the cytotoxicity of T cells.84
These different function arrests caused by tumor-derived EVs are not mutually exclusive in the TME. In fact, they can happen simultaneously. For example, CD45– plasma-derived exosomes isolated from head and neck SCC (HNSCC) patients carry inhibitory factors that induce high levels of CD8+ T cell apoptosis in advanced stage patients, producing high amounts of adenosine and promoting stage-dependent Treg differentiation.85 Likewise, exosomes derived from multiple myeloma similarly mediate T cell suppression through both promoting proliferation of Treg cells and decreasing viability of CD4+ T cells.86
T cell suppression mediated by tumor-derived exosomes is clearly linked to the pathogenesis and disease progression of cancers. Recent study has explored the effects of PD-L1+ tumor-derived exosomes isolated from HNSCCs on immune suppression and disease progression. They identified that PD-L1+ exosomes were positively associated with disease progression in patients with HNSCC and can downregulate CD69 expression of effector T cells.87 Similarly, exosomes derived from highly metastatic murine breast cancer cells have been evaluated for their potential roles in immune suppression and promotion of metastases. Interestingly, these exosomes were taken up by CD45+ bone marrow-derived cells in areas that they accumulated, leading to an accumulation of MDSCs that suppress T cells.88 89 Additionally, tumor-derived EVs can indirectly induce T cell suppression through increasing the level of extracellular adenosine via binding adenosine A2A receptor in T cells.65 The tumor-derived exosomes could inhibit NK cell cytotoxicity in addition to directly suppressing T cell proliferation, leading to a dampened anticancer immune response and promotion of cancer metastases.88
Tumor-derived EVs promote Treg but demote effector T cell differentiation
Upon activation, naïve T cells can differentiate into effector T helper cells (Th), including T-helper 1 (Th1), T-helper 2 (Th2), and T-helper 17 (Th17).90 Naive CD4+ T cells can also differentiate into Treg cells.91 The presence of abnormal quantities of Treg cells in the peripheral blood of patients with cancer is a clear indication of immune suppression.92 In the circulation of patients with ovarian cancer, the number and activity of CD4+CD25highFoxP3+ Treg cells is higher than that in healthy donors, and a higher level of secreted exosomes was also found in the plasma of these patients.93 EVs derived from NPC and patients with melanoma could impede the differentiation of immune-active Th1 and Th17 lymphocytes and induce differentiation of immunosuppressive Treg cells.63 94 This conversion of conventional T cells to CD4+CD25highFoxP3+ Treg cells occurs in a TGF-β1 and IL-10 dependent manner, as the ability of tumor-derived EVs to expand Treg cells is impaired by the neutralizing antibodies specific for TGF-β1 and/or IL-10.93 Furthermore, 14-3-3ζ, a protein that promotes the proliferation of tumor cells, is highly expressed in hepatocellular carcinoma (HCC) tissue, leading to a decrease in the percentages of effector T cells and an increase of Treg cells mediated through exosome.95 Likewise, exosomes derived from NPC tumor cells also contain Treg-attracting chemokine C-C motif ligand 20 (CCL20), which helps to recruit conventional CD4+CD25– T cells, mediate their conversion into inhibitory CD4+CD25high Treg cells, and promote Treg cell expansion.96 In addition to chemokines, the mutant K-RAS gene is involved in the NSCLC exosome-induced switch of naïve CD4+ T cells into a FoxP3+ phenotype in a cytokine-independent manner in a NSCLC xenograft mouse model.97 Notably, on incubation with ovarian cancer-derived EVs, the conversed Treg showed an increased expression of FasL, IL-10, TGF-β1, CTLA-4, granzyme B and perforin, which mediate more potent suppression of responder cell proliferation.93
MicroRNAs present in tumor-derived EVs can act as a barrier for effective tumor immunity through initiating the differentiation of Th cells. For example, exosomes derived from the sera of patients with NPC have a high level of miR-24–3 p, which can inhibit differentiation of Th1 and Th17 cells via repression of FGF11.61 Furthermore, miR-29a-3p and miR-21–5 p in exosomes derived from epithelial ovarian-associated macrophages synergistically induce the Treg/Th17 cell imbalance through directly targeting STAT3 signaling in CD4+ T cells in tumor-bearing mice.62 Interestingly, heat-stress treatment and the production of heat shock proteins in tumor-derived EVs eliminate the imbalance of Treg/Th17 cells.21 Exosomes derived from heat-stressed colon cancer cells contain high quantities of Hsp70, which strongly stimulates IL-6 secretion from DCs to block Treg cell development but promote differentiation of Th17 cells.21 Blocking TGF-β1 in MC38 colon carcinoma cell-secreted EVs could significantly increase the proportion of Th1 cells and Th1-mediated responses.98
In addition, γδ T cells as a subtype of lymphocyte has been shown to support pancreatic oncogenesis by restraining effector T-cell activation in the TME.99 EVs from oral SCC cells can stimulate γδ T cell expansion and function in a DC-independent manner, while hypoxic condition will attenuate this equilibrium.100
Tumor-derived EVs induce T cell exhaustion
T cell exhaustion is a state of T cell dysfunction with a reduced capacity to secrete cytokines and an increased expression of a panel of inhibitory molecules, including PD1, T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), lymphocyte activation gene 3 protein (LAG3), CTLA-4 and T cell immunoreceptor with Ig and ITIM domains (TIGIT).101 Exhausted T cell responses have been documented following infections by many types of virus, such as HIV, hepatitis B virus and hepatitis C virus, and have also been observed in patients with malignancies.102 The exhausted T cells in the TME lose their effector functions to control tumor growth and display a decreased ability to produce cytokines such as IL-2, IFN-γ, and TNF-α.103 104 Recent research has shown that tumor-infiltrating CD8+ T cells isolated from melanoma-engrafted mice have accumulated dysfunctional mitochondria, which dictate exhaustion in CD8+ T cells.105 Furthermore, studies also focus on the relationship of lipid metabolism and exhaustion. PD-1hiCD8+ TILs from people with non-small-cell lung carcinoma contain greater lipid content than PD-1loCD8+ TILs, thus suggesting that lipid metabolism may contribute to T cell exhaustion.106 In addition, cholesterol can induce CD8+ T cell exhaustion through an ER stress-dependent pathway in the TME.107 Although the phenotypic and functional portraits of T cell exhaustion in the TME are well studied, the role of tumor-derived EVs in inducing T cell exhaustion has not been studied until recently. Studies have shown that miR-146a-5p in HCC-derived exosomes induces T cell exhaustion by activating anti-inflammatory M2-macrophages, while this could be stopped by blocking transcription factor SALL4.108 In addition, it has been documented that a high expression of 14-3-3ζ in HCC-derived exosomes is correlated significantly well with an exhausted phenotype of T cells.95 Exosomes from patient serum of lung adenocarcinoma containing circRNA-002178 could enhance PD-L1 expression via sponging miR-34 in cancer cells to induce CD8+ T cell exhaustion in vitro.67
Tumor-derived EVs reprogram metabolism in T cells
Naïve T cells with a quiescent state predominantly use oxidative phosphorylation to generate ATP to support immune surveillance.109 When activated by antigen and professional antigen presenting cells, T cells must undergo rapid metabolic reprogramming and switch to aerobic glycolysis to exit quiescence and support anabolic metabolism required for proliferation and effector function.109 110 Specifically, activated T cells increase the expression of nutrient transporters, especially glucose transporters 1 and 3 (GLUT1 and 3),110 and use glucose to generate ATP even in the presence of sufficient oxygen. Glycolysis provides fast energy and essential intermediates that are required for cell division and effector function.109 Furthermore, tumor cells also increase the expression level of GLUT1 and uptake more glucose for glycolysis.111 The enhanced utilization of glycolysis by tumor cells leads to an increased consumption of glucose and an accumulation of immunosuppressive metabolites such as lactic acid.112 The sustained consumption of glucose by tumor cells eventually leads to a decrease of glucose levels in the TME and creates an environment with low glucose, low amino acids, and low oxygen. Effector T cells exposed to restricted glucose levels have significantly reduced antitumor activity.113 In addition, elevated mechanistic target of rapamycin kinase activity and PD-1 signaling during the development of T cell exhaustion contributed to metabolic alterations including suppressed respiration, reduced glucose uptake, glycolysis and dysregulated mitochondrial energetics.114 T cells receiving PD-1 signal were unable to engage in glycolysis but displayed an increased fatty acid oxidation (FAO) by increasing carnitine palmitoyl transferase and inducing lipolysis.115 Notably, CTLA-4 could inhibit glycolysis in T cells without augmenting their FAO.115 A recent study has also shown that high cholesterol content in the TME promotes CD8+ T-cell exhaustion in tumor-bearing mice.107 Interestingly, NAD precursor nicotinamide ribose could enhance the antitumor responses of T cells within the TME by producing effector cytokines, reducing the accumulation of depolarized mitochondria, and attenuating mitochondrial ROS (mtROS) levels in CD8+ T cells.105
Increasing evidence suggests that tumor microenvironmental EVs could potentially rewrite metabolism in T cells in the TME116 (figure 1). Tumor-derived EVs help creating an immunosuppressive TME by inducing apoptosis and impairing the function of effector T cells and other immune cells.16 68 74 The effects of the tumor-derived exosomes on impacting the metabolic pathways in the recipient cells contribute significantly to a TME that is suitable for tumor cells to survive. For example, mutant K-RAS colonic cells and hepatic stellate cells release EVs containing GLUT1, which enhances glycolysis in other cells in the TME.117 118 EVs secreted by breast cancer cell line suppress glucose uptake by lung fibroblasts and astrocytes through the downregulation of the expression level and activity of pyruvate kinase isoenzyme (PKM2) and GLUT1.119 Low glucose availability in the TME has been shown to decrease the level of glycolytic metabolites such as phosphoenolpyruvate in T cells, weaken effector function, and render CD8+ T cells less capable of preventing tumor growth.113 120 Furthermore, limiting glucose availability or inhibiting glycolytic enzymes impairs proliferation of effector T cells and their cytokine production.121 122
Tumors lead to a microenvironment deficient in many nutrients, which are also required for T cell proliferation and differentiation processes. To offset the deficiency, following their activation, T cells upregulate the uptake of amino acids such as methionine and glutamine as well as enzymes for their metabolism.123 In addition, in some T cell types, cell differentiation and development has been influenced by amino acid deficiency. An amino acid-deprived microenvironment promotes a shift in CD4+ T cells toward Treg differentiation and impairs effector T cell development.123 124 Known mechanisms that contribute to amino acid depletion include sequestration of cysteine and the production of arginase-1 (ARG1), an enzyme that speeds up the breakdown of arginine in the urea cycle. Interestingly, EVs isolated from the plasma and ascites of patients with ovarian cancer were reported to have increased arginase activity, and these ARG1+ cancer EVs are able to inhibit the proliferation of CD4+ and CD8+ T cells, presumably by depleting arginine.64 In addition, the increased arginase content in EVs is correlated with the decreased CD3ζ-chain levels, which is a T cell co-receptor involved in activating both the CD8+ cytotoxic T cells and Th cells.64
One important immunosuppressive factor is adenosine, which exerts T cell immune suppressive function by binding to their receptors A1, A2A, A2B, and A3.125 Adenosine is produced by the action of CD39 and CD73, which catalyze the conversion of ATP to AMP and the hydrolysis of AMP to adenosine, respectively. Studies have shown that tumor-derived EVs can impact adenosine production and signaling by several different ways. CD39/CD73 present on the surface of exosomes can generate extracellular adenosine through the degradation of extracellular ATP.65 Furthermore, CD39/CD73-containing exosomes purified from diverse types of cancers display hydrolytic activity that leads to the production of adenosine from ATP in T cells, thus to inhibit T cell activation.65 126 Furthermore, prostate cancer exosomal prostaglandin E2 induces CD73 expression in DCs thus suppressing T cell function in an adenosine-dependent manner.82 Adenosine generated by tumor-derived EVs can directly regulate intracellular signaling in T cells. For instance, adenosine from mesothelioma exosome has been shown to elevate cAMP level in T cells through the adenosine A2A receptor.65 Interestingly, extracellular adenosine can also promote the development of Treg cells, which in turn produce more extracellular adenosine and thus form a potential positive feedback mechanism that enhances immunosuppression.127 In addition, hydrolytic activity of exosomes leads to the production of other purine nucleosides, including inosine and hypoxanthine in Treg cells.57 126 In support of this notion, exosomes derived from patients with HNSCC’s plasma have shown to contain purine metabolites, including adenosine, inosine, hypoxanthine and xanthine, and immunosuppressive adenosine being the most prominent purine.128 Although many progresses have been made in this field, the effects of tumor-derived EVs on T cell metabolism and their importance in antitumor immunity are still largely unknown. A thorough understanding of the effects of tumor-derived EVs on T cell metabolism and function has the potential to provide new insights for improved immunotherapy for cancer.
Tumor-derived EVs and epigenetic reprogramming in T cells
In order to adapt the shifting environment, T cells dynamically modulate their transcriptional programs, which subsequently influence their differentiation and alter their function and metabolic setup.129 The patterns of heritability of gene expression is mediated by epigenetic mechanisms that involve DNA methylation, histone post-translational modifications and non-coding RNA expression. Abnormal epigenetic patterns correlate with malfunction of effector T cells in tumors.130 131 The epigenetic programming of T cells could be induced by changes of nutrient levels, metabolic status, and external stimuli like growth hormones and cytokines in T cells.132 In addition, tumor-derived EVs may also epigenetically reshape the fate of T cells and contribute to T cell dysfunction in antitumor activity.
Histone acetylation is one the most extensively studied post-translational modifications and is regulated by the opposing actions of histone acetyltransferases and histone deacetylases (HDACs).133 Studies have shown that HDACs are dysregulated in many cancers, making them a therapeutic target for the treatment of cancer.133 Interestingly, histone deacetylase 3 (HDAC3) is required for T cell maturation,134 135 and histone deacetylase 1 and 2 (HDAC1/2) also participate in the proper thymic development of T cells.136 In addition, the HDAC inhibitors vorinostat, romidepsin and panobinostat have been approved for the clinical treatment of cutaneous T cell lymphoma, and belinostat is currently used for treating peripheral T cell lymphoma.137 Besides acetylation, histone methylation also contributes to T cell dysfunction. Melanoma cells have been reported to disrupt methionine metabolism in CD8+ T cells by expressing high levels of the methionine transporter solute carrier family 43 member 2 (SLC43A2).138 Enhanced uptake of methionine by melanoma cells led to a lowering intracellular level of methionine and the methyl donor S-adenosylmethionine in T cells, resulting in loss of dimethylation at lysine 79 of histone H3 (H3K79me2) in T cells and an impaired T cell immunity.138 In recent years, whole-genome methylation profiling has identified a distinct methylome pattern of colorectal cancer-reactive CD8+ T cells, in which tumor-reactive markers CD39 and CD103 are specifically demethylated.139 Latest studies demonstrated that exhausted CD8+ T cells have exhibited distinct epigenetic landscapes, including chromatin accessibility and DNA methylation patterns, from effector CD8+ T cells.130 131
Epigenetic changes play important roles in developing T cell dysfunctional states including exhaustion. Indeed, de novo DNA methylation by the enzyme DNMT3A could promote T cell exhaustion; interestingly, PD-1 blockade treatment can prevent the reprogramming and enhance T cell rejuvenation.140 In addition, interrupting the mitochondrial fitness of CD8+ T cells was able to orchestrate epigenetic reprogramming associated with exhaustion.105 DNA-binding protein TOX and transcription factor NR4a have recently been identified as critical molecules that drive T cell exhaustion by orchestrating exhaustion-linked epigenetic and transcriptomic changes in CD8+ TILs.141 Notably, epigenetic modification was also involved in cytokine release from T cells. Blockade of leukemia inhibitory factor led to release of the epigenetic silencing of C-X-C motif chemokine ligand 9 (CXCL9), which in turn triggers CD8+ T cell tumor infiltration and enhances tumor immunotherapy.142
Studies indicated that many mRNAs and proteins contained in EVs are involved in epigenetic regulation of immune cell functions in the suppressive TME, including DNA methylation, histone modification, and miRNA regulation.143–145 Plasma exosomes containing dual methylated DNAs of CDKN2A and CDKN2B, or CDKN1B transcript contribute to diffuse large B-cell lymphoma pathogenesis.146 As to the aspect on other immune cells, study found that irradiation of glioblastoma cell line cells promotes the tet methylcytosine dioxygenase 2-mediated demethylation of exosomal-miR-378a-3p thus inducing the decrease of granzyme B secretion by NK cells.147 However, molecular mechanisms responsible for tumor EVs-mediated epigenetic regulation on T cells and other immune cells are still under investigation.
Targeting tumor-derived EVs is a novel strategy for tumor immunotherapy and diagnosis
Immune checkpoint blockade therapy has emerged as a promising anticancer strategy for different tumor types. Many anticancer drugs have been developed based on the immune checkpoint molecules.148 Nevertheless, a majority of patients with cancer fail to respond to current checkpoint blockade immunotherapies. The immune suppressive function of tumor-derived EVs drives researchers to rethink the failure of current immune checkpoint blockade therapies. It has been shown that the presence of PD-L1 in tumor-derived exosomes is positively correlated with tumor activity and progression, and blocking exosome signaling in T cells with anti-PD-1 antibody effectively attenuates immune suppression mediated by the PD-L1+ exosomes.74 75 87 Recent studies have demonstrated that directly targeting β-catenin could be an effective strategy to enhance anti-PD-1 based therapy in hepatocellular carcinoma.149 EVs derived from ovarian tumor ascites contain phosphatidylserine (PS) which suppresses T cell activation and the signaling arrest in T cells could be stopped by a blockade of PS.150 EVs from estrogen receptor-binding fragment-associated antigen 9 (EBAG9)-overexpressing prostate cancer cells can facilitate immune escape of tumors by inhibiting T cell cytotoxicity and regulating immune-related gene expression. This specific immune suppression could also be rescued by EBAG9 neutralizing antibody treatment.151 In addition, other immune checkpoint molecules Tim-3 and Galectin-9 have been found to be present in large quantity in the exosomes isolated from the plasma of the patients with non-small cell lung cancer.152 Interestingly, the exosome-based immune checkpoint blockade strategies have now been explored to develop for cancer therapy. Exosomes engineered to contain signal regulatory protein α (SIRPα) variant can block the interaction between SIRPα and CD47, which limits the ability of macrophages, thus makes macrophages to engulf tumor cells again and promotes an intensive T cell infiltration in vivo.153 CD47 is abundantly expressed in many tumor cells, and its interaction with SIRPα on the macrophages confers a ‘don’t eat me’ signal to allow the tumor cells to evade the attack by macrophages. Interestingly, CD47 is also present in many EVs, which likewise protects EVs from phagocytosis by monocytes.154 A new type of exosomes that capsule two distinct types of antibodies could simultaneously activate T cell surface CD3 and cancer cell-associated EGFR thus displaying excellent antitumor activities.155
In addition to enhancement of immune checkpoint blockade therapy, tumor-derived EVs could be utilized as a source of cellular antigens for development of novel tumor therapeutic vaccines.20 156 Many molecules present in tumor-derived EVs can trigger antitumor immunity, and these include TAAs, damage-associated molecular patterns, and MHC-peptide complexes.157–159 Immunization of mice with those tumor EVs containing TAAs and MHC-peptide complexes can enhance antigen presentation, leading to tumor-specific T cell stimulation and enhanced T cell infiltration.157 158 It has been suggested that use of EVs as tumor antigens for the development of cancer vaccines is superior to the use of irradiated tumor cells or tumor cell lysates, which can express more concentrated antigens and lead to a better recovery in T cell proportion and is easier to store.160 161 Furthermore, antigen-loaded exosomes can induce specific T cell responses with a Th1-type shift.162 Exosomes loaded with immune cell ligand α-galactosylceramide (αGC) and antigen ovalbumin (OVA) can not only induce potent NK and γδ T cell innate immune responses, but also lead to synergistically amplified T cell and B cell responses.163 In addition, treatment of tumor-bearing mice with αGC/OVA-loaded exosomes decreased tumor growth, increased antigen-specific CD8+ T cell tumor infiltration and prolonged the survival of tumor-bearing mice.163 In fact, EV-based vaccines have recently been developed for cancer immune therapy. A heterologous human/rat HER2-specific exosome-targeted T cell vaccine has been demonstrated to stimulate CD4+ T cell responses, leading to increased induction of HER2-specific antibody and a remarkable eradication of 90% of HER2-expressed target breast cancer cells.164 Additionally, a method for increasing the yield of EVs by cyclophosphamide treatment has been reported, which may be used to produce antigens for future design of cancer vaccines.161 Exosome engineering approaches, such as decorating tumor peptides on the exosome surface and microfluidic technique, have also been utilized to enhance the ability of harvested exosomes in antigen presentation and T cell activation in future vaccine design.165 166 One attractive approach is engineering EVs to allow their specific uptake by DCs and induce antigen-specific T cell responses. This can be achieved by modifying the EV glycan surface so it can bind CD209, a DC-specific receptor that mediates antigen uptake and induction of CD4+ and CD8+ T cell activation. This approach has been successfully applied to EVs from glioblastoma and melanoma, which represent an enriched cell-free source of TAA to pulse DCs for efficient antigen delivery and initiation of antitumor immune responses.167 168
Tumor-derived EVs also hold great promise in cancer diagnosis, and there are some ongoing clinical trials that explore biomarkers present in these EVs for cancer diagnosis (latest information can be tracked in https://clinicaltrials.gov). EVs derived from tumors that are in pre-metastatic niches often contain pro-metastatic factors, which can be used to determine the metastatic organotropism of tumors. EVs from liquid fluids such as plasma, ascites and urine are potential reservoirs of tumor biomarkers that could be clinically useful, which enable liquid biopsies in cancer diagnosis and prognosis. In recent years, several studies have highlighted different markers contained specifically in EVs derived from different cancer subtypes. For example, EVs from pancreatic cancer contain a very promising clinical prognostic marker, macrophage migration inhibitory factor.169 EVs from the plasma of patients with breast cancer are enriched with miR-21 and miR-1246, which can be used for the diagnosis of breast cancer or for monitoring the effectiveness of treatment.170 In addition, ongoing clinical trials are evaluating the diagnosis marker for EVs from plasma of lung cancer (NCT04529915), and tumor-derived EVs for thyroid cancer (NCT04742608), and early prognosis marker for colorectal cancer (NCT04523389). Tumor-derived EVs may also be used to monitor the response of a patient to cancer treatment. The level of PD-L1 in the circulating EVs from patients with melanoma correlated well with the tumor response to checkpoint blockade therapy.171 A lower level of TGF-β in EVs corresponded to an increased immune activity after chemotherapy.159 A very recent study found a significant correlation between high levels of urokinase-type plasminogen activator receptor (uPAR) in EVs and poor clinical outcomes of immunotherapy in patients with melanoma.172 Thus, the level of exosomal uPAR can be potentially used as a biomarker of resistance to checkpoint inhibitor immunotherapy in melanoma.
Conclusions and perspectives
Tumor-derived EVs have emerged as important mediators between malignant tumors and tumor-infiltrating immune components. It is well established that T cell function is dramatically impaired by the suppressive TME in patients with cancer. In addition, a large proportion of patients does not respond to or does not achieve durable responses to the current immunotherapies because of the dysfunction of effector T cells caused by T cell exhaustion, anergy, and senescence.173 Increasing evidence suggests that tumor-derived EVs are the critical factors to promote immune suppression and maintain a suppressive TME. They can suppress the function of T lymphocytes via a variety of mechanisms that include regulating T cell metabolism, affecting the differentiation of T cell subsets, and delivering immune suppressive molecular signals from the parent tumor cell to distantly situated T cells (table 1). Tumor-derived EVs have been reported to affect T cell exhaustion, however, whether they also induce other states anergy, senescence, and stemness of T cells remains largely unknown. Improved understanding of these molecular regulations will be critical for the development of effective therapeutic strategies for cancer treatment.
Although great progress has been made in understanding the effects of tumor-derived EVs on immune cells, however, one important concern in the field of tumor-derived EVs is the heterogeneous nature of these vesicles, especially those isolated from the patients. Although new assays for distinguishing cancerous from non-cancerous plasma samples have been reported,174 175 the method for separating tumor-derived EVs and non-malignant cell-derived EVs from body fluids are not generally available. As a consequence, the total EV fractions isolated from the plasma or body fluid of patients with cancer are often referred as tumor-derived EVs. In reality, these EVs are a mix of both tumor cell-derived and non-malignant cell-derived EVs, which may not recapitulate the content, phenotypic and functional spectrum of tumor EVs in patients with cancer. These may explain the conundrum that the same tumor EVs appear to carry both suppressive and stimulatory cargoes, as these tumor EVs being investigated could be heterogeneous and stemmed from both malignant and non-malignant cells. This notion was supported by a study on EVs from the plasma of patients with melanoma.175 Using a three-step strategy that involves immune-based capture, researchers were able to separate malignant cell-derived EVs (MTEX) and non-malignant cell-derived EVs (non-MTEX) and found that MTEX has an enrichment in immunosuppressive proteins and a paucity of immunostimulatory proteins. On the other hand, the non-MTEX has a higher ratio of stimulatory/inhibitory proteins, and this ratio varies widely in patients and negatively correlates with disease progressions.
In addition, several other challenging issues are still unknown in the field. First, the effects of individual component present in tumor-derived EVs on immune cells are unclear and worth to be studied in great details. Majority of previous studies have focused on the effects of DNA, miRNAs and immune suppressive proteins present in tumor-derived EVs on T cells, but little is known about the carbohydrates on the surface of EV, such as polysaccharides and oligosaccharides on immune cells.176 In addition, tumor-derived EVs contain some immune suppressive metabolites, such as purines, lactic acid, fatty acids, and eicosanoids,11 45 46 128 as well as increased levels of metabolic proteins and enzymes, such as GLUT1, PKM2, aldolase A and aldehyde dehydrogenase 3.118 177 178 However, the precise roles of these metabolites and molecules from tumor-derived EVs in controlling T cell fate and function are still under investigation. Second, whether and how the tumor-derived EVs metabolically and epigenetically rewrite T cell differentiation and functions in tumor immunity is also not fully understood. Increasing evidence suggests that the metabolic barrier imposed on T cells by the TME is an important factor that triggers T cell suppression.179 Furthermore, different subsets of T cells may utilize different metabolic regulation for their survival and effector function.180 Precisely dissecting the metabolic changes in T cells induced by individual component in the tumor-derived EVs will provide critical insights for a better understanding of the failures of current immunotherapies and development of effective tumor treatments.115 In addition to metabolic regulation in T cells, very limited information is known about how tumor EVs epigenetically regulate the factions in T cells in tumor immunity, which should be put to more efforts in the future studies. Third, novel technologies and combination strategies should be utilized to develop effective EV-based immunotherapies for future tumor treatments. Given the specific characters of EVs, nanomedicine could be as a promising form to efficiently deliver endogenous and exogenous therapeutics.181 In addition, the applications of exosomes for combination therapy in tumors have been reported in the preclinical models. Specifically, chemotherapy drug doxorubicin and cholesterol-modified miRNA21 inhibitor are co-embedded into the lipid bilayer of healthy mice blood exosomes can be delivered to tumor cell for enhanced tumor therapy.182 Furthermore, tumor EVs as a reliable source of tumor antigens may constitute a better immunological strategy for the development of novel tumor immunotherapeutic vaccines.
Ethics statements
Patient consent for publication
Acknowledgments
We apologize since we cannot cite all the relevant references in this research area because of space limitations.
References
Footnotes
Contributors FM, JV and GL wrote the manuscript and prepared the figure and table. YW and GP advised the paper construction and revised the manuscript.
Funding This work was partially supported by grants from the Melanoma Research Alliance (https://doi.org/10.48050/pc.gr.86351 to GP) and the NIH (R15GM141652 to YW; R01CA184379, R01CA242188, R01CA237149, and R21AG067441 to GP).
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.