Article Text

Abstract
Background Co-stimulatory signals regulate the expansion, persistence, and function of chimeric antigen receptor (CAR) T cells. Most studies have focused on the co-stimulatory domains CD28 or 4-1BB. CAR T cell persistence is enhanced by 4-1BB co-stimulation leading to nuclear factor kappa B (NF-κB) signaling, while resistance to exhaustion is enhanced by mutations of the CD28 co-stimulatory domain.
Methods We hypothesized that a third-generation CAR containing 4-1BB and CD28 with only PYAP signaling motif (mut06) would provide beneficial aspects of both. We designed CD19-specific CAR T cells with either 4-1BB or mut06 together with the combination of both and evaluated their immune-phenotype, cytokine secretion, real-time cytotoxic ability and polyfunctionality against CD19-expressing cells. We analyzed lymphocyte-specific protein tyrosine kinase (LCK) recruitment by the different constructs by immunoblotting. We further determined their ability to control growth of Raji cells in NOD scid gamma (NSG) mice. We also engineered bi-specific CARs against CD20/CD19 combining 4-1BB and mut06 and performed repeated in vitro antigenic stimulation experiments to evaluate their expansion, memory phenotype and phenotypic (PD1+CD39+) and functional exhaustion. Bi-specific CAR T cells were transferred into Raji or Nalm6-bearing mice to study their ability to eradicate CD20/CD19-expressing tumors.
Results Co-stimulatory domains combining 4-1BB and mut06 confers CAR T cells with an increased central memory phenotype, expansion, and LCK recruitment to the CAR. This enhanced function was dependent on the positioning of the two co-stimulatory domains. A bi-specific CAR targeting CD20/CD19, incorporating 4-1BB and mut06 co-stimulation, showed enhanced antigen-dependent in vitro expansion with lower exhaustion-associated markers. Bi-specific CAR T cells exhibited improved in vivo antitumor activity with increased persistence and decreased exhaustion.
Conclusion These results demonstrate that co-stimulation combining 4-1BB with an optimized form of CD28 is a valid approach to optimize CAR T cell function. Cells with both mono-specific and bi-specific versions of this design showed enhanced in vitro and in vivo features such as expansion, persistence and resistance to exhaustion. Our observations validate the approach and justify clinical studies to test the efficacy and safety of this CAR in patients.
- cell engineering
- receptors
- chimeric antigen
- immunotherapy
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Chimeric antigen receptors (CARs) are a hybrid antigen receptor composed of an extracellular antigen-binding domain, hinge and transmembrane domain, and intracellular activation motifs. These activation domains typically consist of a CD3ζ domain and one (second-generation) or more co-stimulatory domains (third-generation). The most common co-stimulatory domains are derived from CD28 or 4-1BB. CD28-based CARs are characterized by faster cytotoxicity and improved antitumor efficacy than 4-1BB-based CARs.1 2 However, CD28 co-stimulation is associated with increased susceptibility to exhaustion and decreased persistence compared with 4-1BB.3–6 We have shown that this lack of persistence is driven by exhaustion-related gene expression, and mutation of the CD28 co-stimulatory domain can alleviate this.7 CARs containing 4-1BB have enhanced proliferation,8 mitigate T cell exhaustion9 and favor longer-term CAR T cell persistence in patients.2–4 Our lab has shown the enhanced function of 4-1BB CAR T cells is driven by nuclear factor kappa B (NF-κB), TRAF1, and TRAF3.10
To further enhance CAR function, groups have examined combining the two co-stimulatory domains into a single CAR to determine if they can recapitulate the strong cytotoxicity and proliferation of a CD28-based CAR with the enhanced memory and persistence of a 4-1BB-based CAR. Third-generation 4-1BB/CD28 CARs showed success against a xenograft model of hepatocellular carcinoma,11 a mouse model of thyroid cancer12 and a solid xenograft model targeting mesothelin.13 Also, in a clinical trial second-generation (CD28) and third-generation (CD28/4-1BB) anti-CD19 CAR T cells were simultaneously administered to patients. Third-generation cells showed enhanced expansion and longer persistence than second-generation.14 Nevertheless, there is still uncertainty over whether third-generation CARs have greater potency than second-generation with several studies in mouse models15 16 and clinical studies17 showing no significant benefit. A second-generation CD28-based CAR against prostate stem cell antigen was found to have a more potent antitumor effect compared with a third-generation CAR with both CD28 and 4-1BB.18 Using an immunoproteomic approach it was found that second-generation CARs could activate additional sources of CD3ζ signaling, which resulted in enhanced signal intensity and superior antitumor function compared with third-generation CARs.19
These studies suggest that additional co-stimulation is not additive but can be detrimental for CAR T cell function. This is supported by work in our lab showing that specific signaling pathways, such as lymphocyte-specific protein tyrosine kinase (LCK) and NF-κB, are critical for CAR function, while excess amounts of others such as NFAT and NUR77 can be detrimental.7 10 How these features may be combined for optimal function is an important question and results have been contradictory. Some have reported that by increasing LCK recruitment to 4-1BB CARs it enhances tumor killing.20 In contrast, reducing LCK recruitment to a CAR with a mutated CD28 endodomain requires 4-1BB co-stimulation to support efficacy.21 We hypothesize that by combining our mutant CD28 (mut06) with 4-1BB, we could build a CAR with the most beneficial aspects of both, mainly cytotoxicity and persistence. We show that 4-1BB combined with mut06 results in CAR T cells with increased central memory (CM) phenotype, polyfunctionality, expansion and cytotoxicity. To further improve the function of our second-generation and third-generation CARs and validate our observations with a different exodomain we designed bi-specific CARs targeting both CD19 and CD20 in a tandem configuration and confirmed the beneficial effects of combining 4-1BB and mut06 co-stimulation.
Materials and methods
Cells
NIH/3T3 cells retrovirally transduced with human CD19 were used as target cells. NIH/3T3 cells were purchased from ATCC. CHO cells retrovirally transduced with human CD20 were used as target cells. CHO cells were purchased from ATCC. K562 cells retrovirally transduced with both human CD19 and CD20 were used as target cells. Nalm6 and RajiWT cells were transduced to express green fluorescent protein (GFP)-firefly luciferase (FFLuc). Raji CD19 antigen panel cell lines which include three genetically modified cell lines involving the CD19 gene (Raji-CD19KO, Raji-CD19Low and Raji-CD19High) were purchased from Sigma-Aldrich and used as target cells. These three cells lines were also retrovirally transduced to express GFP-nanoluciferase (NanoLuc) for their in vivo detection. Human peripheral blood mononuclear cells (PBMCs) were purchased from AllCells. T cells were enriched from PBMCs using the EasySep human T cell isolation kit according to manufacturer’s instructions (STEMCELL). Human T cell complete medium consists of RPMI1640 medium, 10% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin. All media and supplements were purchased from ThermoFisher Scientific. Cell lines were authenticates as previously described7 and used at low passages (3 to 6). Cells were tested for Mycoplasma using the Universal Mycoplasma Detection Kit (ATCC) and were negative.
Genetic constructs and CAR T cell production
The SFG retroviral backbone was modified to include the FMC63 single-chain variable fragment (ScFv) with CD8α transmembrane and hinge domain followed by one or more co-stimulatory domains and CD3ζ for all constructs. The h1928z, h19BBz, and h1906z CAR constructs have been described.7 Mut06 replaces YMNM and PRRP CD28 subdomains with FMNM and ARRA. The h1906BBz construct has the mut06 co-stimulatory domain proximal to the cell membrane followed by 4-1BB and CD3ζ. The h19BB06z construct has the 4-1BB co-stimulatory domain proximal to the cell membrane followed by mut06 and CD3ζ. Bi-specific constructs were included into the SFG retroviral backbone comprizing a CD8α leader peptide followed by a scFv specific for human CD19 (FMC63) linked to a scFv specific for human CD20 (Leu16) engineered in a tandem configuration followed by a CD8α transmembrane domain and either mut06 or 4-1BB plus mut06 and CD3ζ. All plasmids were synthesized externally (Genewiz) and verified by restriction enzyme digest.
All SFG constructs were calcium phosphate transfected into H29 cells. Retroviral supernatants of transfected H29 were harvested and used to transduce RD114 cells. Retroviral supernatant of producer cells was harvested, 0.45 µm filtered, and used to transduce T cells as described.22 23 Viability was measured by trypan blue staining and enumerated on an automated cell counter (Bio-Rad). Transduction efficiency was estimated by flow cytometry as a percentage of protein L+live cells. For downstream experiments, CAR T cell doses were normalized based on CAR gene transfer but not sorted to exclude CAR-negative T cells. As a result, the total T cell dose was varied. For all experiments using bi-specific constructs cells were normalized to the lowest CAR expression by accordingly adding varying quantities of untransduced T (UT) cells, reaching the same number of CAR +cells and total T cells per group.
Mice and systemic tumor models
Female and/or male mice at 8–12 weeks of age were used. NOD scid gamma (NSG) mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were purchased from the Jackson Laboratory and bred in our facility. Mice were intravenously injected with 5×105 Raji-GFP/FFluc or Nalm6-GFP/FFluc cells at week −1 and 2×106 CAR T cells were intravenously injected at week 0. For bi-specific CAR T cells mice received 1×106 CAR T cells. Tumors were measured using an IVIS Lumina III In Vivo Imaging System (PerkinElmer) for bioluminescence imaging (BLI) weekly. Mice were monitored for illness daily and sacrificed when there was evidence of leukemia progression, such as decreased activity, hunched posture, or ruffled coat. At certain time points, blood was collected from the submandibular vein into tubes containing K3 EDTA (Sarstedt). At mouse sacrifice blood, spleen, and/or femurs were collected. For blood samples red blood cells were lysed using ammonium-chloride-potassium buffer (ThermoFisher) and stained for flow cytometry as described below. Bone marrow was isolated from femurs by cutting both ends of the femur and flushing it with a syringe containing phosphate buffered saline (PBS) (Gibco). These cells were passed through a 70 µm cell strainer. Red blood cells were lysed as described above and stained for flow cytometry (as described below).
Flow cytometry
The following antibodies with clones listed were obtained from BD Biosciences: anti-hCD3 (HIT3a). The following antibodies were from BioLegend: anti-hCD3 (HIT3a), anti-hCD19 (HIB19), anti-hPD1 (NAT105), anti-hCD45RA (HI100), anti-hCCR7 (G043H7), anti-hCD4 (OKT4), anti-hTIM3 (F38-2E2), anti-CD39 (A1), anti-hCD20 (2H7), anti-hCD45 (HI30), anti-hCD8 (RPA-T8), Streptavidin conjugated with PE/Cyanine7. Streptavidin conjugated with Alexa Fluor 488, Biotinylated Recombinant Protein L, Fixable Viability Dye eFluor 450 were purchased from ThermoFisher Scientific.
Cells were first washed twice with PBS and stained with a fixable viability dye (eBioscience) at room temperature for 30 min. Surface staining was performed at 4°C with Fc block and antibody mix in magnetic-activated cell sorting buffer with 0.5% bovine serum albumin (BSA) (Miltenyi Biotec). For some experiments, Countbright beads (ThermoFisher Scientific) were used for cell quantification following manufacturer’s intructions. All samples were analyzed with a 5-laser LSRII (BD Biosciences), and data were analyzed using FlowJo software (Tree Star).
Cytokine immunoassay
1×106 or 5×104 CAR T cells were washed and co-cultured with different ratios of target cells for 24 hours. Supernatants were collected and analyzed using an ELLA® Assay kit (Multianalyte: interferon gamma (IFN-γ), interleukin 2 (IL-2), IL-6 and tumor necrosis factor alpha (TNF-α)) according to the manufacturer’s instructions.
Cytotoxicity assay
Cytotoxicity assays were run on an xCelligence real-time cell analysis (RTCA) instrument (ACEA Biosciences) according to the manufacturer’s instructions. Briefly, 1×104 adherent target cells (3T3-hCD19 or CHO-hCD20) were plated per well on an E-Plate 96. For non-adherent cells (Raji and K562) tethering antibodies were used to coat xCelligence E-Plate 96 wells according to the manufacturer’s instructions. Briefly, xCelligence E-Plate 96 were coated with anti-CD40 (Raji) or anti-CD71 (K562) antibodies for 3 hours at room temperature then washed and 3×104 target cells were added per well. The next day CAR T cells were re-suspended in fresh complete medium without IL-2 and added onto target cells at various effector to target (E:T) ratios, and growth was monitored.
Isolight
After experimental design and prior to plating cells into chips, CAR T cells were stimulated for 4 hours or overnight with target cells at a 5:1 E:T ratio. For certain experiments total CAR T cells were plated and analyzed in an IsoLight instrument and for others magnetically isolated CD4+ and CD8+ cells were plated independently. Polyfunctionality: co-secretion of 2+ cytokines per cell; Polyfunctional Strength Index (PSI): percentage of polyfunctional cells in the sample multiplied by the intensities of secreted cytokines.
Western blot
A total of 1×107 CAR T cells per CAR were stimulated with 3T3-hCD19 target cells for 24 hours or left unstimulated. Cells were lysed with radioimmunoprecipitation assay buffer and total protein was quantified by ND1000 (Nanodrop); 20 µg of lysate was loaded onto the gel. Proteins were detected using anti-phospho-lymphocyte-specific protein tyrosine kinase (pLCK) (1:1000 5% BSA; phospho Tyr505; Cell SignaliNg), anti-lymphocyte-specific protein tyrosine kinase (LCK) (1:2000 5% BSA; BD), and anti-glyceraldehyde 3-phosphate dehydrogenase (1:5000 5% BSA; Cell SignaLing). Goat antirabbit IgG (H+L) and goat antimouse IgG (H+L) (Cell Signaling) were used as secondary antibodies. The secondary antibodies were diluted in 5% BSA at a 1:10 000 dilution. Primary antibodies were incubated at 4°C overnight, and secondary antibodies were incubated at room temperature for 2 hours. ChemiDocMP Imaging System (Bio-Rad) was used to detect enhanced chemiluminescence (Pierce ECL Western Blotting Substrate) western blotting signals.
Statistics
All statistical analyses were conducted using Prism V.8 software (GraphPad). No statistical methods were used to predetermine sample size. Survival was compared using a log-rank test. Values of p≤0.05 were considered significant. The statistical tests used for each experiment are described in each figure legend.
Results
Co-stimulation with 4-1BB and mut06 results in a favorable memory phenotype
To evaluate how the combination of 4-1BB and mut06 co-stimulation affects CAR function, we designed five human CD19-targeted CARs (figure 1A). All CAR constructs include the FMC63 scFv with a CD8α transmembrane and hinge domain followed by the co-stimulatory domain/s and CD3ζ. h1928z, h19BBz, and h1906z (mut06) have been described previously.7 h1906z replaces the CD28 subdomains of YMNM and PRRP with FMNM and ARRA, leaving only a functional PYAP signaling motif. h1906BBz includes the mut06 domain proximal to the cell membrane followed by 4-1BB and CD3ζ while h19BB06z has 4-1BB proximal to the cell membrane followed by mut06 and CD3ζ. After retroviral transduction and prior to antigen exposure, we found that all CAR T cells had similar proliferation (online supplemental figure S1A) and viability (online supplemental figure S1B) and that CARs with multiple co-stimulatory domains showed higher transduction efficiency, with h19BB06z showing the highest CAR surface expression prior to antigen stimulation (online supplemental figure S1C). We observed that CARs combining 4-1BB and mut06 showed higher CD4/CD8 ratios before CAR engagement (online supplemental figure S1D)
Supplemental material

Multiple co-stimulatory domains enhance chimeric antigen receptor (CAR) T cell memory phenotype. (A) Schematic of CAR constructs. All include 5' long terminal repeat (LTR), CD8 signal peptide (black bar), single-chain variable fragment with a variable heavy chain connected with glycine/serine linker to a variable light chain (VH-G/S-VL), CD8α transmembrane and hinge domain, co-stimulatory, and CD3ζ endodomain, and 3' LTR. (B–D) CAR T cells were stimulated with irradiated 3T3-hCD19 target cells at a 10:1 E:T ratio for 24 hours. Cells were then collected, and phenotype determined by flow cytometry. (B) Surface CAR expression on live CD3+CAR+ cells measured by median fluorescent intensity within protein L+ cells. (C) CD4/CD8 ratio on live CD3+CAR+ T cells. (D) Central memory phenotype (CCR7+CD45RA+) among CD3+CAR+ cells at 24 hours. (E) Central memory phenotype (CCR7+CD45RA+) among CD3+CAR+ cells at weeks 1, 2 and 3 after repeated weekly antigen stimulation with CD19+ target cells. Data are shown from two healthy donors (HD). Data are shown as mean±SD. One-way analysis of variance (ANOVA) was performed with Dunnet’s multiple comparison test against h19BB06z for (B–D). Two-way ANOVA was performed with Dunnet’s multiple comparison test against h19BB06z for (E).**p<0.01, ***p<0.001, ****p<0.0001.
To determine the effect these CARs have on T cell phenotype, we stimulated CAR T cells at a 1:10 E:T ratio for 24 hours with hCD19-expressing target cells (3T3-hCD19). After stimulation, cells were analyzed by flow cytometry and found the highest surface CAR expression on h19BB06z CAR T cells compared with other constructs, same as before CAR engagement (figure 1B). The skew towards CD4+ CAR T cells was also maintained after CAR stimulation in 4-1BB/mut06-contaning CARs (figure 1C).
Previous work has shown that CAR T cells with a less differentiated phenotype such as CM or naïve display better expansion, persistence, and antitumor activity in vivo.24–27 Staining with memory markers after 24 hours of antigen stimulation revealed that CARs combining 4-1BB and mut06 had an increased percentage of CM (CCR7+CD45RA-) CAR T cells compared with other CARs (figure 1D). To further validate that our observations after 24 hours of antigen stimulation reflected cell differentiation at longer time points, we stimulated CAR T cells with CD19-expressing cells weekly for a total of 3 weeks and evaluated their phenotype by flow cytometry each week. We determined that h19BB06z CAR T cells were able to maintain a significantly higher frequency of CM cells while the rest skewed towards a more differentiated phenotype (figure 1E). Together these data show that h19BB06z CAR T cells have a more favorable memory-like phenotype compared with other constructs.
4-1BB and mut06 co-stimulatory domains influence different aspects of h1906BBz and h19BB06z CAR T cell function
To examine the effector function of the different CARs, we quantified cytokine secretion after 24 hours in vitro stimulation with target cells at a 10:1 E:T ratio. Using three healthy donors we found that h1906z had the lowest levels of IFN-γ, IL-2 and TNFα compared with all other constructs, which is consistent with the dampening of CD28 signaling by loss of function mutations.7 When 4-1BB was combined with mut06 we observed an increase in the secretion of IFN-γ, IL-2 and TNF-α, specifically in the configuration where 4-1BB is followed by mut06 (h19BB06z) (figure 2A). IL-6 secretion, associated with cytokine release syndrome in patients,28 was minimal in all constructs although highest in h19BB06z. In line with these observations, we studied CAR T cell polyfunctionality, defined as the ability of each individual cell to secrete two or more cytokines. Several recent reports have demonstrated CAR T cell polyfunctionality is a substantial predictor of in vivo fitness.29–31 To examine this, we used a multiplexed antibody-coated chip that analyzes hundreds of CAR T cells at the single-cell level for frequency and intensity of 28 secreted cytokines. CAR T cells were stimulated for 4 hours with target cells before loading onto chips and analyzed. We found that h1928z, h1906BBz, and h19BB06z had an increased percentage of polyfunctional CAR T cells (figure 2B). The PSI is the percentage of polyfunctional cells multiplied by the intensity of the secreted cytokines. Using this measure, h1928z, h1906BBz, and h19BB06z showed the highest PSI (figure 2B). This demonstrates that combining 4-1BB and mut06 enhances CAR T cell polyfunctionality, which was similar to h1928z.
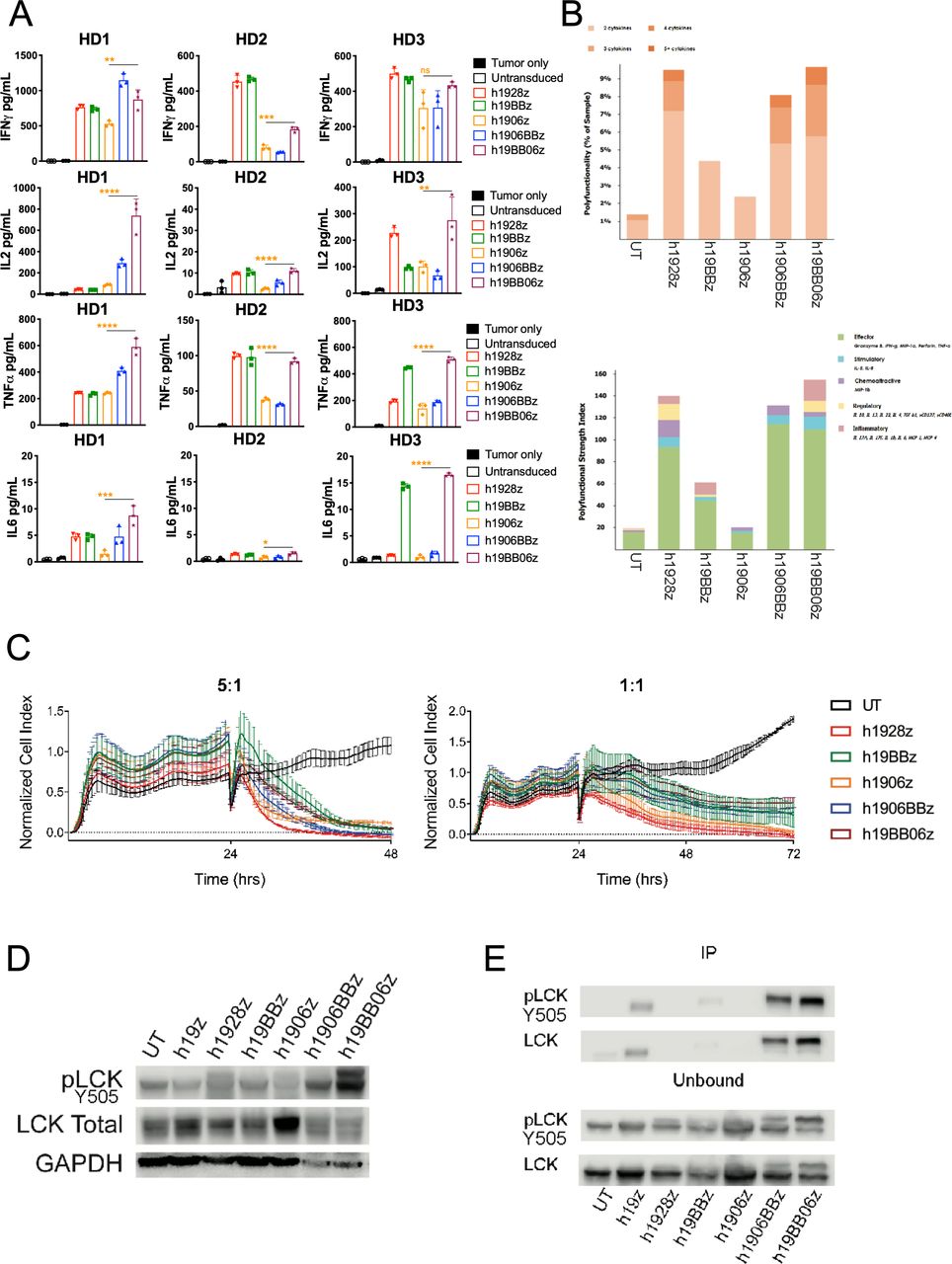
Increased phospho-lymphocyte-specific protein tyrosine kinase (pLCK) enhances in vitro function of h19BB06z chimeric antigen receptor (CAR) T cells. (A) CAR T cells were stimulated with irradiated 3T3-hCD19 cells at a 10:1 E:T ratio. After 24 hours, supernatants were harvested, and cytokines were measured with ELLA. Data are shown from three healthy donors (HDs). (B) A single-cell measure of polyfunctionality (top) and Polyfunctional Strength Index (PSI) (bottom) of CAR T cells stimulated for 4 hours with CD19+ target cells. (C) CAR T cells were co-cultured with irradiated 3T3-hCD19 at indicated E:T ratios. The xCELLigence real-time cell analysis (RTCA) system monitored real-time cytotoxicity. (D–E) CAR T cells were stimulated with irradiated 3T3-hCD19 cells at a 10:1 E:T ratio for 24 hours. Cells were lysed and either total lysate (D) or CAR bound and unbound fractions (E) were western blotted. Data are representative of two HDs (B–E). Data are shown as mean±SD. One-way analysis of variance (ANOVA) was performed with Dunnet’s multiple comparison test against h19BB06z for (A). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
We then examined in vitro killing ability by using a real-time cytotoxicity assay with two different E:T ratios (5:1 and 1:1). We found all CAR constructs had rapid and efficient killing at both ratios (figure 2C). This suggests that in vitro cytotoxicity is not greatly affected by choice or combination of co-stimulatory domain.
Lymphocyte-specific protein tyrosine kinase (LCK) is a critical molecule for CAR T cell function. Recently it has been shown that recruitment of LCK to 4-1BB CARs can also enhance CAR T cell cytotoxicity.20 We examined the effect of continuous antigen exposure on LCK phosphorylation after 24 hours stimulation and demonstrated that both third-generation CARs had increased pLCK compared with second-generation CARs (figure 2D). Overall, h19BB06z had the highest LCK phosphorylation, suggesting that the co-stimulatory domains' positioning may affect pLCK. We also found that large amounts of pLCK and total LCK were associated with the h19BB06z CAR molecule (figure 2E). These data suggest that the enhanced in vitro function of h1906BBz and h19BB06z results from increased pLCK and that the orientation of the co-stimulatory domains can affect CAR-associated LCK signaling.
Co-stimulatory domain positioning affects in vitro expansion and in vivo tumor-killing
One measure of CAR T cell fitness is its ability to withstand multiple rounds of antigen-stimulation.32 To examine this, we stimulated CAR T cells with target cells at a 5:1 E:T ratio. Re-stimulation of CAR T cells with target cells occurred every 7 days for a total of 4 weeks. We observed a significantly greater expansion of h19BB06z CAR T cells compared with all other groups (figure 3A). To study the function of these cells that have endured multiple antigen challenges, we examined their cytotoxicity and cytokine secretion. To control for the enhanced proliferation of h19BB06z we used the same number of CAR+ T cells for each group in these experiments. We found that h19BB06z had the greatest killing ability compared with all other groups (figure 3B). h19BB06z also showed significantly higher secretion of IL-2 compared with all other groups (figure 3C). Similarly, h19BB06z displayed significantly greater secretion of effector cytokines TNF-α and IFN-γ compared with h1928z and h1906z (figure 3C). h1906BBz CAR T cells displayed lower proliferation, cytotoxicity, and IL-2 production than h19BB06z, again suggesting that co-stimulatory domain positioning can affect CAR function. These data show that after repeated antigen stimulations h19BB06z CAR T cells exhibit the greatest proliferative capacity, cytotoxicity and cytokine secretion, in contrast to single antigen stimulation when the h1928z CAR had the greatest cytokine production and cytotoxicity.
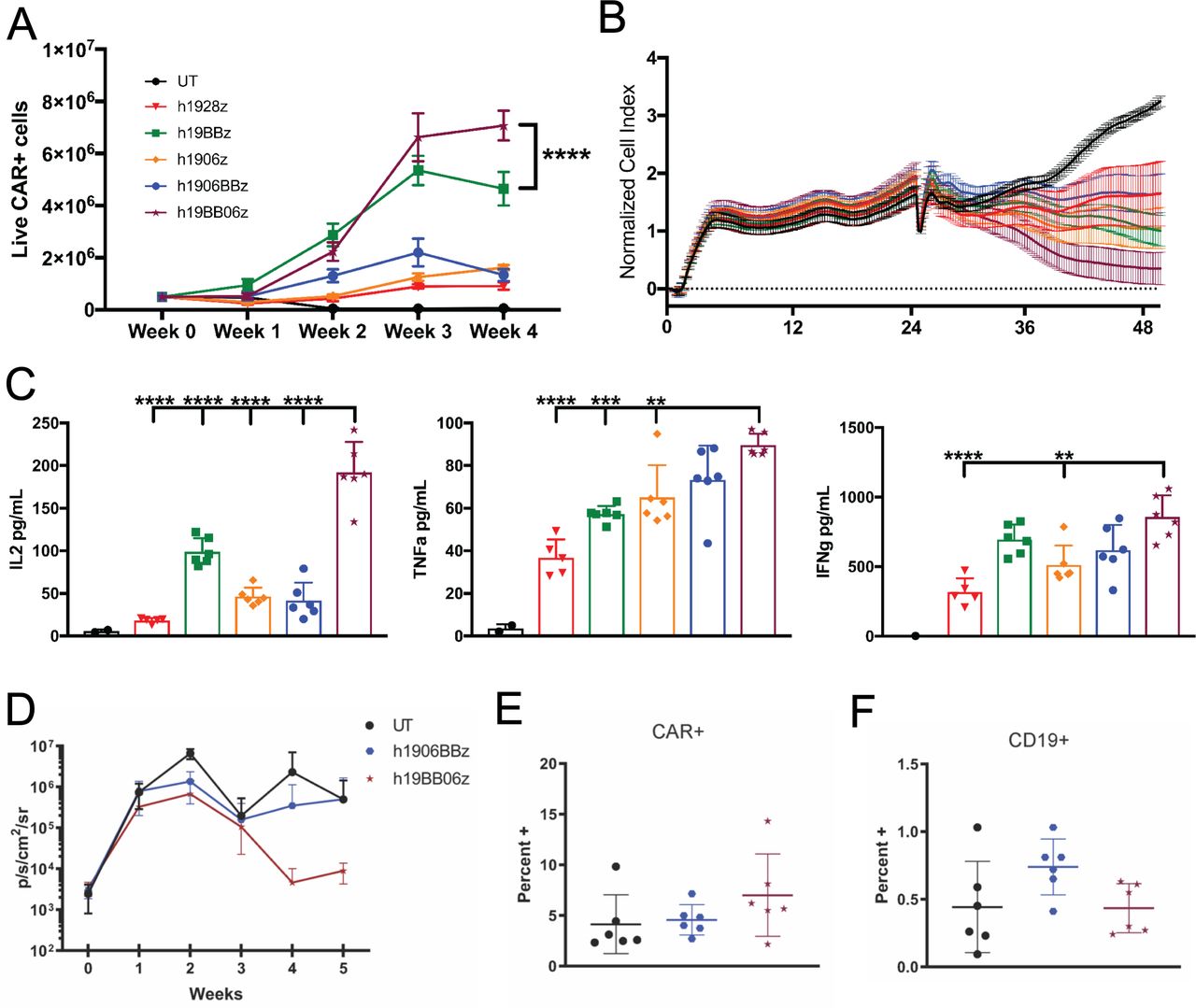
h19BB06z chimeric antigen receptor (CAR) T cells have greater proliferation, cytotoxicity, and IL-2 production after repeated antigen stimulations with improved in vivo antitumor activity. (A) 5×105 CAR T cells were stimulated with 1×105 irradiated 3T3-hCD19 cells. After 1 week, half of the cells were enumerated by flow cytometry and the other half was re-stimulated with 1×105 fresh irradiated 3T3-hCD19 cells. This was repeated for a total of 4 weeks. (B–C) After 4 weeks of re-stimulation the same number of CAR T cells for each group was co-cultured with 3T3-hCD19 cells at a 5:1 E:T ratio and either cytotoxicity (B) or cytokine secretion (C) were measured. n=6. Data are representative of 3 (A) or 2 (B–C) healthy donors. Data are shown as mean±SD. (D) NOD scid gamma mice were intravenously injected with 5×105 Raji-GFP/luc cells at week −1 and 2×106 CAR T cells were intravenously injected at week 0. Mice were then measured for bioluminescence imaging (BLI) weekly. Average BLI for each week is shown. (E) Percentage of Live CAR+ cells at week 5. (F) Percentage of live CD19+ cells at week 5. n=6. Data are representative of two independent experiments. Two-way analysis of variance (ANOVA) was performed with Dunnet’s multiple comparison test against h19BB06z for (A). One-way ANOVA was performed with Dunnet’s multiple comparison test against h19BB06z for (C). **p<0.01, ***p<0.001, ****p<0.0001.
To determine the ability of h1906BBz and h19BB06z CAR T cells to control tumor growth in vivo, we injected Raji B lymphocyte tumor cells into NSG mice. We found that h19BB06z CAR T cells were better able to control tumor growth compared with UT and h1906BBz (figure 3D). In the blood of mice receiving h19BB06z there was a slight increase of CAR+ cells (figure 3E) and a corresponding decrease in CD19+ cells (figure 3D) compared with h1906BBz. Together, these data show that h19BB06z CAR T cells have enhanced in vitro proliferation, long-term cytotoxicity, and in vivo tumor-killing compared with h1906BBz. Therefore, we selected the BB06 orientation for further evaluation.
Bi-specific CAR T cells targeting CD19 and CD20 show high surface CAR expression with efficient in vitro cytotoxicity and cytokine production
To further validate our observations and capitalize on dual-antigen recognition we developed constructs encoding for single bi-specific CAR molecules able to recognize both human CD19 and CD20. This multiantigen recognition approach is shown to mitigate disease relapse due to antigen escape.33 34 Antigen-binding domains from the FMC63 (anti-CD19) and Leu16 (anti-CD20) antibodies were linked in a tandem configuration and arranged in different orders, with FMC63 furthest from the cell membrane followed by Leu16 (CD19-CD20) or vice versa (CD20-CD19). Both second- (mut06z) generation and third- (BB06z) generation bi-specific CAR T cells were developed (figure 4A). We observed that all four CAR constructs were significantly expressed on the cell surface at frequencies that ranged from 66% to 91% (figure 4A and online supplemental figure S2A). These four bi-specific constructs also showed different levels of surface CAR expression measured by the median fluorescent intensity within the CAR+ population, with 20–19 Tan BB06z showing the highest expression level (online supplemental figure S2A). Prior to antigen exposure, 20–19 Tan BB06z construct showed a higher frequency of CAR T cells with a CM phenotype (online supplemental figure S2B).
Supplemental material
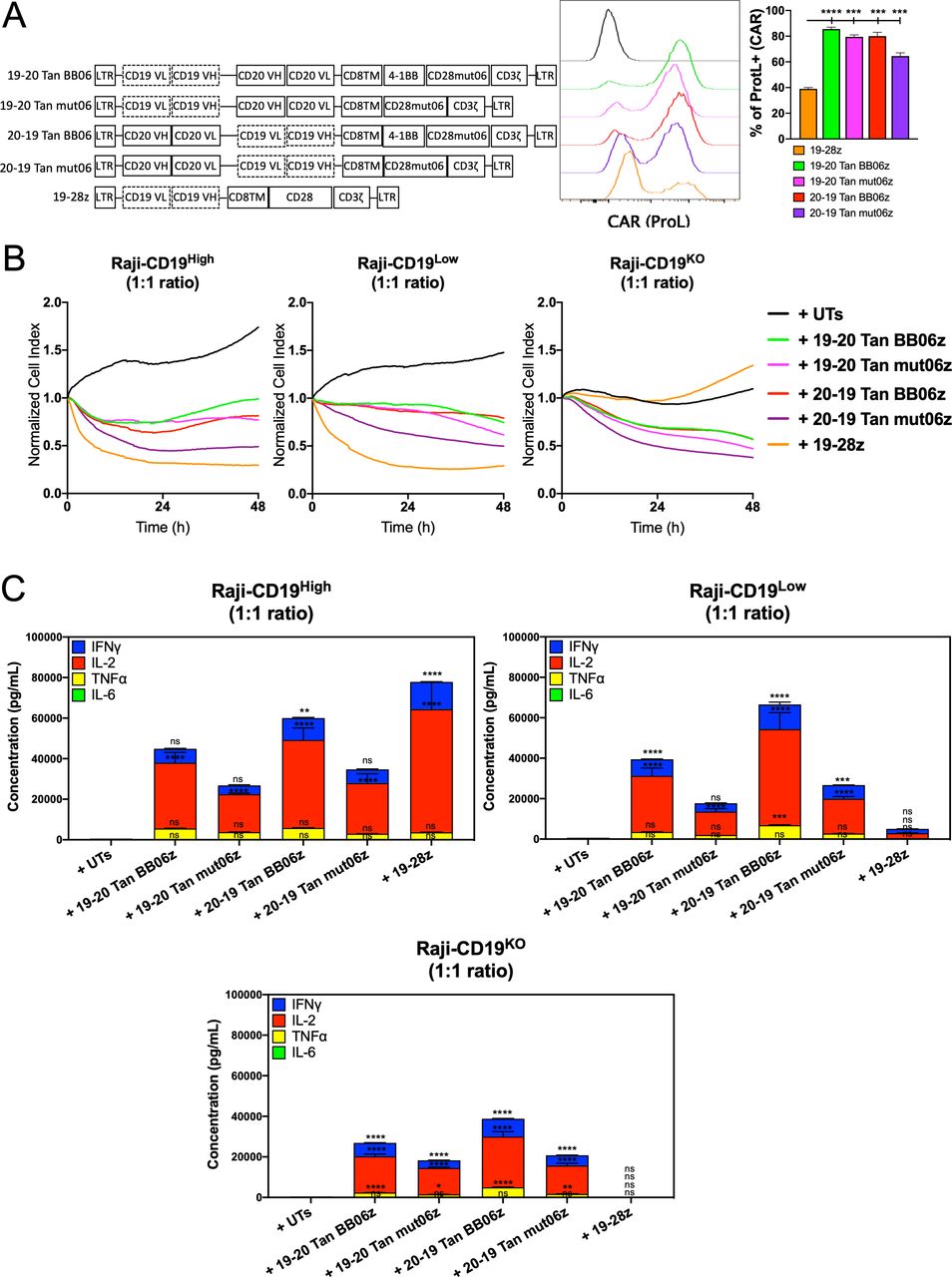
CD19/CD20 bi-specific constructs are highly expressed and confer chimeric antigen receptor (CAR) T cells cytotoxicity and cytokine production. (A) Schematic of bi-specific single CAR constructs targeting CD19 and CD20 in a tandem (Tan) configuration. Histograms showing CAR expression. Quantitative analysis of CAR expression (Protein L staining) at day 7 post-transduction compared with 19-28z cells. (B) Real-time cytotoxicity assay (xCelligence) was performed with the different bi-specific CAR T cells and single 19-28z against Raji-CD19High, Raji-CD19Low and Raji-CD19KO target cells at 1:1 E:T ratio. (C) Concentration of interferon gamma (IFN-γ), interleukin (IL)-2, IL-6 and tumor necrosis factor alpha (TNF-α) measured by ELLA in supernatants from co-cultures of bi-specific CAR T cells and single 19-28z with Raji-CD19-High, Raji-CD19Low and Raji-CD19KO after 24 hours compared with untransduced T (UT) cells. All conditions were normalized to the lowest CAR expression using UT cells, reaching same number of CAR T cells and total T cells per group. Representative results of three independent experiments are shown. Data are shown as mean±SD. NS, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. One-way analysis of variance (ANOVA) was performed with Dunnet’s multiple comparison test against UT.
To determine the ability of these four bi-specific CAR T cells to individually recognize both target antigens CD19 or CD20 and trigger effector functions we performed an in vitro evaluation of cytotoxicity and cytokine production. All the subsequent experiments were performed with CAR T cells normalized to the construct expressing the lowest CAR frequency by adding UT cells, reaching the same number of CAR T cells and total T cells per condition. We found that all constructs were able to induce cytolysis against CD19-expressing or CD20-expressing cells with different efficiencies, with 20–19 Tan mut06z showing the fastest response and 19–20 Tan mut06z being the least effective (online supplemental figure S3A). Furthermore, we observed that three bi-specific CAR T cells, 19–20 Tan BB06z, 20–19 Tan BB06z, and 20–19 Tan mut06z, showed significant levels of cytokine production when challenged with either CD19 or CD20 while 19–20 Tan mut06z showed no response (online supplemental figure S3B).
Supplemental material
To investigate bi-specific CAR T cell functionality against a tumor cell that naturally expresses both CD19 and CD20 we used the Raji B cell lymphoma line. To further elucidate the impact of CD19 and CD20 we used a panel of Raji cells expressing varying levels of the CD19 antigen as target cells. These include cells with no CD19 expression (Raji-CD19KO), with low levels (Raji-CD19Low), and high levels (Raji-CD19High) of CD19. We observed that all four constructs showed effective cytotoxicity by RTCA against Raji-CD19KO compared with no effect of the mono-specific h1928z. Furthermore, all four constructs also showed efficient cytotoxicity against Raji-CD19Low and Raji-CD19High with 20–19 Tan mut06 showing the fastest effector function (figure 4B). Additionally, we determined in vitro secretion of IFN-γ, IL-2, TNF-α and IL-6 after 24 hours of co-culture with these three Raji cell lines at a 1:1 ET ratio. We detected secretion of significant levels of IFN-γ, IL-2, TNF-α, and minimal levels of IL-6 upon antigen engagement with the highest levels observed when both antigens were present. Furthermore, BB06z CAR T cells showed the highest levels of cytokines while mut06z CAR T cells the lowest (figure 4C). We observed the same pattern when using K562 cells artificially engineered to express both human CD19 and CD20 (online supplemental figure S4A–B). These results suggest that these four bi-specific CAR T cells can recognize both target antigens and elicit a response that varies depending on both the orientation of the antigen-binding domains and the co-stimulatory motifs.
Supplemental material
CAR T cells combining 4-1BB and mut06 with a CD20/CD19-oriented exodomain show significant in vivo antitumor activity
We next sought to compare the antitumor function of these four bi-specific CAR T cells in an aggressive xenograft model of acute lymphoblastic leukemia. For this we inoculated NSG mice intravenously with Nalm6-FFluc tumor cells and 1×106 CAR T cells were transferred 7 days later. Tumor growth was monitored by quantitative imaging every week. We observed that both CAR constructs with a CD20-CD19 orientation were able to maintain a low tumor burden at early time points (day 11) compared with CD19-CD20 (figure 5A). Furthermore, as the experiment progressed 20–19 BB06z CAR T cells were the only ones that significantly delayed tumor growth compared with UT-treated controls (figure 5B). The kinetics of this antitumor effect were also determined by event-free survival, defined as time in days to an average luminescence equal or greater than 1×106 p/s/cm2/sr on these mice. Here we observed that 20–19 BB06z CAR T cells significantly delayed the progression of the disease while the rest of the groups succumbed sooner (figure 5C). These data suggest that the orientation of the anti-CD20 and anti-CD19 scFvs impact the in vivo antitumor efficiency of bi-specific CAR T cells, as 20–19 BB06z shows better response than 19–20 BB06z. Together, our in vitro and in vivo results focused our subsequent studies on CD20-CD19 mut06z and BB06z CAR T cells.

Bi-specific chimeric antigen receptor (CAR) T cells show different in vivo antitumor efficacy. Nalm6-bearing NSG mice were treated with 1x106 CAR T cells 7 days after initial tumor cell injection. (A) Bioluminescent images of each mouse per condition at specified days after T/CAR T cell injection. (B) Tumor burden (average radiance) of each mouse treated with bi-specific CAR T cells and controls. Two-way analysis of variance (ANOVA) was performed with Dunnet’s multiple comparison test against untransduced T cell (UT). Each line represents an individual mouse. (C) Kaplan-Meier analysis of event-free survival defined as time in days to an average luminescence equal or greater than 1x106 p/s/cm2/sr of mice treated with 1x106 T/CAR T cells compared with UT control (n = 8 mice per group). *p<0.05.
CD20/CD19 bi-specific CAR T cells show enhanced antitumor function compared with standard of care h1928z with increased persistence and lower exhaustion-associated markers
To further investigate the antitumor efficacy of these two bi-specific constructs and evaluate their performance against the standard of care h1928z CAR used in the clinic, we used a Raji B cell tumor model. For this, we inoculated NSG mice intravenously with Raji-FFluc tumor cells and 1×106 CAR T cells were transferred 5 days later. Tumor growth was monitored by quantitative imaging every week. We determined that both bi-specific CAR T cells showed significant antitumor activity compared with UT controls (figure 6A). By analyzing event-free survival we were able to observe that bi-specific and h1928z CAR T cells showed significant control of tumor progression (figure 6A). Furthermore, bi-specific constructs lead to complete tumor regression in some mice while mono-specific CAR T cells controlled tumor growth but with observable tumor burden at the experiment end point (day 42) (online supplemental figure S5A). Moreover, 20–19 Tan BB06z and 20–19 Tan mut06z showed significantly higher number of CAR T cells in peripheral blood compared with h1928z (figure 6B). By performing an ex vivo analysis of these mice, we determined that bi-specific cells showed increased persistence as evidenced by a higher total number of CAR T cells in the bone marrow compared with h1928z (figure 6C). Additionally, bi-specific CAR T cells found in the bone marrow showed a lower frequency of PD1+CD39+ cells, typically associated with an exhausted phenotype. In accordance with this, we found a lower frequency of PD1-CD39- in bi-specific CAR T cells (figure 6D and online supplemental figure S5B). We observed that all three CAR T cells present in the bone marrow of treated mice showed a predominantly CM phenotype. Furthermore, mono-specific h1928z CAR T cells skewed towards CD8+ cells while 20–19 Tan BB06z showed more CD4+ cells and 20–19 Tan mut06z a more balanced ratio (online supplemental figure S5C).
Supplemental material

CD20-CD19 Tan bi-specific chimeric antigen receptor (CAR) T cells show enhanced in vivo antitumor efficacy and low expression of exhaustion-associated markers. (A) Raji-FFLuc-bearing NSG mice were treated with 1×106 CAR T cells 5 days after initial tumor cell injection. Average tumor burden (average radiance) of mice treated with bi-specific or mono-specific CAR T cells, untransduced T cell (UT) and tumor control. Data are shown as mean±SEM. Two-way analysis of variance (ANOVA) was performed with Dunnet’s multiple comparison test against UT (left). Kaplan-Meier analysis of event-free survival defined as time in days to an average luminescence equal or greater than 1×106 p/s/cm2/sr of mice treated with 1×106 T/CAR T cells compared with UT control (right). *p<0.05. (B) Total number of CAR T cells per μL of blood of tumor-bearing mice treated with the different CAR T cells (each point is 1 mouse). Two-way ANOVA was performed with Dunnet’s multiple comparison test against 19-28z condition. *p<0.05, **p<0.01. (C) Total number of CAR T cells per femur of tumor-bearing mice treated with different CAR T cells (each point is 1 mouse). (D) Expression of PD1 and CD39 among CAR+ cells harvested from the bone marrow of tumor-bearing mice treated with different CAR T cells (n=7 mice per group). Data are shown as mean±SEM. One-way ANOVA was performed with Dunnet’s multiple comparison test against the 19-28z condition. (E) RajiCD19KO-bearing NSG mice were treated with 1×106 CAR T cells 5 days after initial tumor cell injection. Tumor burden (average radiance) of mice treated with bi-specific or mono-specific CAR T cells, UT and tumor control. Each line represents an individual mouse (n=5 mice per group). (F) Kaplan-Meier analysis of survival of mice treated with 1×106 T/CAR T cells compared with tumor only control (n=5 mice per group). P values were determined by a one-sided log-rank Mantel-Cox test. NS, not significant; *p<0.05, **p<0.01. All conditions were normalized to the lowest CAR expression using UT cells, reaching same number of CAR-T cells and total T cells per group. See also online supplemental figure S5.
To further study the in vivo activity of these cells against only the target antigen CD20 we inoculated NSG mice intravenously with RajiCD19KO-Nanoluc tumor cells that lack the expression of CD19; 1×106 CAR T cells were transferred 5 days later and growth was monitored weekly by bioluminescence. We observed that both bi-specific CAR T cells showed significant antitumor effect while mono-specific h1928z showed no effect, comparable to UT cells. Additionally, 20–19 Tan BB06z CAR T cells showed improved tumor control compared with 20–19 Tan mut06z (figure 6E). Both bi-specific CAR T cells were able to significantly prolong the survival of the mice compared with mice receiving UT or h1928z cells (figure 6F). Collectively, these data support the hypothesis that CAR T cell activity can be improved by targeting two independent antigens and that the combination of co-stimulatory domains 4-1BB and a mutated form of CD28 shows an improved function in human CAR T cells.
CD20/CD19 BB06z CAR T cells outperform CD20/CD19 mut06z in vitro
To validate our observations in mono-specific CAR T cells regarding polyfunctionality, we stimulated 20–19 Tan BB06z and 20–19 Tan mut06z CAR T cells with RajiWT cells overnight and isolated CD4+ and CD8+ cells before performing the polyfunctional assay. Analyzing four healthy donors we observed that 20–19 Tan BB06z cells showed higher polyfunctionality and PSI than 20–19 Tan mut06z (online supplemental figure 6).
Supplemental material
To study the response of these CAR T cells to repeated antigenic stimulation we performed an in vitro assay by co-culturing the same initial number of CAR T cells with either Raji-CD19KO, Raji-CD19Low, or Raji-CD19High at a 1:1 E:T ratio. Cells were then counted and analyzed by flow cytometry every 7 days when fresh target cells were added to the co-culture for a total of 4 weeks. We found that 20–19 BB06z CAR T showed significantly superior antigen-dependent expansion than 20–19 mut06z against the three different target cell lines (figure 7A). The total number of T cells also increased significantly more in 20–19 BB06z while UT controls did not expand in the experiment (figure 7B). Additionally, we evaluated the T cell differentiation state along the re-stimulation by investigating CCR7 and CD45RA expression. 20–19 BB06z CAR T showed a significantly higher CM phenotype frequency while 20–19 Tan mut06z skewed towards effector memory and terminally differentiated effector memory cells re-expressing CD45RA (EMRA) after repeated stimulation with RajiCD19High target cells (figure 7C). We observed the same pattern against Raji-CD19KO and Raji-CD19Low (online supplemental figure S7A).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
20-19 Tan BB06z bi-specific chimeric antigen receptor (CAR) T cells show increased expansion, memory phenotype and lower exhaustion-associated markers after repeated antigen stimulation. 5×105 CAR T cells were co-cultured with 5×105 target cells (Raji-CD19KO, Raji-CD19Low and Raji-CD19High). After 1 week half the cells were harvested, enumerated and stained by flow cytometry while the other half were re-stimulated with 5×105 fresh target cells (indicated by arrows). This was repeated for a total of 4 weeks. (A) Number of CAR+ cells at each week. (B) Number of total CD3+ cells each week. Two-way analysis of variance (ANOVA) was performed with Dunnet’s multiple comparison test when comparing to control group (untransduced T cell (UT)) (C) Frequency of central memory (CM—CCR7+CD45RA-), naïve (N—CCR7+CD45RA+), effector memory (EM—CCR7-CD45RA-) and terminally differentiated effector (terminally differentiated effector memory cells re-expressing CD45RA (EMRA)—CCR7-CD45RA+) cells at each week among CAR+ cells. Representative dot-plot for CCR7/CD45RA staining at each week (right). (D) Frequency of PD1+CD39+ cells within total CAR T cells at each week (top) and within CD8+ CAR T cells (bottom). Representative dot-plot for PD1/CD39 staining at each week. (E) After 4 weeks of re-stimulation with Raji-CD19High target cells, the same number of CAR T cells were co-cultured for 24 hours with fresh Raji-CD19High cells at a 1:1 E:T ratio and cytokine secretion was measured. Two-way ANOVA was performed with Sidak’s multiple comparison test when comparing 20–19 Tan mut06z to 20–19 Tan BB06z. Data are shown as mean±SEM. NS, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data are representative of three healthy donors. See also online supplemental figure S7.
We also examined the expression of the exhaustion-associated markers PD1 and CD39 on these cells and observed that 20–19 Tan BB06z CAR T cells were more resistant to acquiring this phenotype (figure 7D). On repeated stimulation with RajiCD19High target cells, 20–19 Tan BB06z CAR T cells showed a significantly lower frequency of PD1+CD39+ cells compared with 20–19 Tan mut06z from week 3 onwards. This lower frequency of PD1+CD39+ 20–19 Tan BB06z cells was observed on total CAR+ cells and CD8+ CAR T cells (figure 7D). The same results were observed against Raji-CD19KO and Raji-CD19Low (online supplemental figure S7B). We also studied the ratio of CD4/CD8 within the CAR+ population and observed that 20–19 Tan BB06z cells showed a ratio close to 1 while 20–19 Tan mut06z cells skewed towards CD8 when challenged with all three Raji cell lines (online supplemental figure S7C). Lastly, after 4 weeks of re-stimulation CAR T cells were counted, washed and re-challenged with RajiCD19High target cells for 24 hours to evaluate cytokine production. To control for the enhanced expansion of 20–19 Tan BB06z we used the same number of CAR+ T cells for each group. We observed that 20–19 Tan BB06z secreted significantly higher levels of IL-2, IFN-γ, and TNF-α than 20–19 Tan mut06z while neither secreted significant levels of IL-6 (figure 7E). The same was observed for CAR T cells that were co-cultured for 4 weeks with Raji-CD19Low and then re-challenged with RajiCD19High (online supplemental figure S7C). These data collectively suggest that 20–19 Tan BB06z CAR T cells can secrete a wider array of cytokines (polyfunctionality) and can expand better after repeated antigenic stimulation than 20–19 Tan mut06z while maintaining a favorable memory-like phenotype. These third-generation CAR T cells can also resist exhaustion and show an enhanced ability to produce cytokines when re-challenged with antigen.
Discussion
Previous research shows the design of a CAR can impact its signaling and function.35 Many of these studies focus on how a single co-stimulatory domain, typically 4-1BB or CD28, can determine CAR efficacy. There are conflicting reports on whether combining CD28 and 4-1BB co-stimulation increases efficacy. Several studies in mouse models15 16 and clinical studies36 show no enhancement of tumor killing or patient survival compared with second-generation CARs, in the context of CD19 and B cell malignancies. The concept of finding the perfect balance of signal quantity and quality downstream CAR activation has been extensively examined in recent years. The modification of one amino acid in the CD28 co-stimulatory domain (CD28-YMFM) was able to render CAR T cells resistant to exhaustion and reduced T cell differentiation.37 Another study showed that by strategically mutating immunoreceptor tyrosine-based activation motifs in CD3ζ of a CD28-based CAR the therapeutic effect was enhanced, with CAR T cells showing improved persistence, a less differentiated phenotype and lower exhaustion.38 In this work we show that by designing a CAR that includes 4-1BB and our mutant CD28 (mut06) we could enhance cell function by optimizing signaling and have the most advantageous aspects of both co-stimulatory domains.
It has been previously shown that including the CD28 co-stimulatory domain can increase CAR expression.39 We observed that a co-stimulatory domain composed of 4-1BB followed by mut06 resulted in higher surface CAR expression in both mono-specific and bi-specific CAR T cells. This increase in CAR expression may allow better clinical outcomes by reducing the number of initial T cells to produce a viable product. It was recently reported that tisagenlecleucel (4-1BB-based) showed reduced ability to perform against low-antigen density targets compared with axicabtagene ciloleucel (CD28-based);40 higher levels of CAR expression on 4-1BB/mut06 cells could increase this threshold and improve the efficacy of 4-1BB-based CARs. Elevated CAR expression can also amplify tonic signaling resulting in poor persistence.41–43 However, while CAR T cells combining 4-1BB and mut06 (mono-specific and bi-specific) have the highest CAR expression, they show the greatest expansion after 4 weeks of repeated antigen stimulation. Several studies support the concept that T cells with a less differentiated state and memory features are associated with an improved antitumor effect and persistence in adoptive cell therapy (ACT).38 44 Previous work shows that 4-1BB-based CAR T cells show an increased CM phenotype.45 Here we show that constructs combining 4-1BB with mut06 maintain a memory-like phenotype after repeated antigenic stimulation that is superior to mut06 or 4-1BB alone.
We observed that h1906z CAR T cells secrete lower levels of cytokines compared with h1928z which is consistent with our previously published mouse data showing mut06 reduces cytokine secretion compared with unmutated CD28 co-stimulation.7 Here we also show that mut06 CAR T cells have lower polyfunctionality, which can be increased by combining it with 4-1BB. This ability to produce a wider range of cytokines at a single-cell level is associated with an improved clinical response.31 While cytokine secretion of h19BB06z CAR T cells was increased compared with mut06, their short-term cytotoxicity was comparable to 4-1BB CARs. However, on repeated antigen stimulation, h19BB06z CAR was able to maintain the greatest cytotoxic ability and cytokine secretion. Different strategies have been studied in order to exploit the beneficial aspects of the IL-2 axis on ACT. Some include mutated versions of IL-2 with higher affinity for IL-2Rβ,46 or the use of an engineered IL-2 cytokine conjugated to polyethylene glycol that enhances ACT.47 Here we show that by combining 4-1BB and mut06, both in mono-specific or bi-specific CAR T cells, we can obtain a cell able to secrete high levels of endogenous IL-2 after repeated stimulation that could act paracrinally and/or autocrinally improving the efficacy of these transferred cells.
A recent report demonstrated that 4-1BB CAR T cell cytotoxicity can be enhanced by recruiting LCK to the CAR complex20 suggesting that a third-generation CAR which increased LCK would be more effective. We found that co-stimulation with both mut06 and 4-1BB increased pLCK in CAR T cells. We also found that a portion of this pLCK was associated with the CAR and that this depended on the order of the co-stimulatory domains.
Our in vitro data suggest that h1906BBz and h19BB06z CAR T cells have similar functional abilities after initial antigen exposure. However, after multiple challenges with antigen, h19BB06z CAR T cells showed the ability to maintain a memory-like phenotype and expanded significantly more than the other constructs. These observations support a recent study where 4-1BB activation of the NF-κB pathway enhances the in vitro survival of CAR T cells with lower expression of apoptotic factors compared with CD28-based co-stimulation.5 When we examined these CARs in vivo we found h19BB06z had enhanced tumor killing compared with h1906BBz. All this suggests that the positioning of the co-stimulatory domains is critical for CAR function. Previous work shows that having CD28 proximal to the cell membrane followed by 4-1BB increases B-cell acute lymphoblastic leukemia tumor regression in xenograft models.2 Likewise, our data show that having the 4-1BB co-stimulatory domain proximal to the cell membrane can enhance CAR T cell function. This may be due to differences in signaling cascades triggered by 4-1BB and mut06 where this orientation appears to be more effective.
We validated our observations using a different extracellular domain able to recognize both CD19 and CD20. These bi-specific CAR T cells independently recognized both target antigens and elicited an efficient effector function, which may prove beneficial in scenarios where CD19 is lost or downregulated.33 48 Moreover, both antiCD20/CD19 BB06z and mut06z cells showed efficient in vivo antitumor activity compared with standard anti-CD19-28z CAR T cells. These cells also showed a more favorable memory phenotype associated with increased antitumor potency and persistence44 49 with less exhaustion-associated markers than second-generation 20–19 mut06z cells.
In a recent report, tandem anti-CD19/CD20 CAR T cells with 4-1BB as co-stimulation were evaluated in a phase-1 study for relapsed, refractory B cell malignancies. They demonstrate therapeutic safety and efficacy, however these cells skew towards an effector-like phenotype prior to infusion.50 These observations suggest a possible clinical application of our newly developed third-generation bi-specific CAR T cells which showed an improved memory-like phenotype that may even be more beneficial by enhancing the efficacy of this approach. Together our work demonstrates that the activity of CAR T cells can be enhanced by fine-tuning the signaling domains, specifically, by combining 4-1BB and an optimized form of CD28 co-stimulation. Plans to evaluate the clinical relevance of these findings in patients with cancer are currently underway.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Animal studies were performed under protocols approved by the Institutional Animal Care and Use Committee of the University of South Florida.
Acknowledgments
The authors thank the team at Atara Biotherapeutics for helpful and critical scientific discussions. This work has been supported in part by the Flow Cytometry Core Facility at Moffitt Cancer Center, an NCI-designated Comprehensive Cancer Center (P30-CA076292).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @marcoldavila
ER and JCB contributed equally.
Contributors Conceptualization: MLD, ER, JCB, GL. Methodology: ER, JCB, NT, SBL, KS, KR, HK, EC. Investigation: MLD, ER, JCB, YB, GL. Visualization: ER, JCB, KS. Funding acquisition: MLD. Project administration: MLD, BY. Supervision: MLD. Writing—original draft: JCB, ER, MLD. Writing—review and editing: MLD, FL, YB, JCB, ER. Guarantor: MLD.
Funding This study was supported by research funding from Atara Biotherapeutics to MLD.
Competing interests The mut06 CAR has been patented by Moffitt Cancer Center and licensed to Atara Biotherapeutics. MLD has received licensing fees and research support from Atara Biotherapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.