Article Text

Abstract
Recipients of chimeric antigen receptor-modified T (CAR-T) cell therapies for B cell malignancies have profound and prolonged immunodeficiencies and are at risk for serious infections, including respiratory virus infections. Vaccination may be important for infection prevention, but there are limited data on vaccine immunogenicity in this population. We conducted a prospective observational study of the humoral immunogenicity of commercially available 2019–2020 inactivated influenza vaccines in adults immediately prior to or while in durable remission after CD19-, CD20-, or B cell maturation antigen-targeted CAR-T-cell therapy, as well as controls. We tested for antibodies to all four vaccine strains using neutralization and hemagglutination inhibition (HAI) assays. Antibody responses were defined as at least fourfold titer increases from baseline. Seroprotection was defined as a HAI titer ≥40. Enrolled CAR-T-cell recipients were vaccinated 14–29 days prior to (n=5) or 13–57 months following therapy (n=13), and the majority had hypogammaglobulinemia and cellular immunodeficiencies prevaccination. Eight non-immunocompromised adults served as controls. Antibody responses to ≥1 vaccine strain occurred in 2 (40%) individuals before CAR-T-cell therapy and in 4 (31%) individuals vaccinated after CAR-T-cell therapy. An additional 1 (20%) and 6 (46%) individuals had at least twofold increases, respectively. One individual vaccinated prior to CAR-T-cell therapy maintained a response for >3 months following therapy. Across all tested vaccine strains, seroprotection was less frequent in CAR-T-cell recipients than in controls. There was evidence of immunogenicity even among individuals with low immunoglobulin, CD19+ B cell, and CD4+ T-cell counts. These data support consideration for vaccination before and after CAR-T-cell therapy for influenza and other relevant pathogens such as SARS-CoV-2, irrespective of hypogammaglobulinemia or B cell aplasia. However, relatively impaired humoral vaccine immunogenicity indicates the need for additional infection-prevention strategies. Larger studies are needed to refine our understanding of potential correlates of vaccine immunogenicity, and durability of immune responses, in CAR-T-cell therapy recipients.
- immunogenicity
- vaccine
- immunity
- humoral
- receptors
- chimeric antigen
- hematologic neoplasms
- antibody formation
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunogenicity
- vaccine
- immunity
- humoral
- receptors
- chimeric antigen
- hematologic neoplasms
- antibody formation
Background
Chimeric antigen receptor-modified T (CAR-T) cell therapies are increasingly used to treat B cell-lineage lymphoma, leukemia, and multiple myeloma. CAR-T-cell recipients are immunocompromised from their underlying malignancy and prior antitumor treatments, in addition to CAR-T-cell therapy related factors including lymphodepleting chemotherapy and ‘on-target/off-tumor’ depletion of non-malignant B-lineage cells expressing the CAR-T-cell targets.1 2
Strategies to prevent infections after CAR-T-cell therapy are not well established. Vaccination is a potentially cost-effective and durable approach to preventing infection or severe disease from relevant pathogens, but data regarding vaccine immunogenicity in CAR-T-cell therapy recipients are limited.3–5
Respiratory tract infections, particularly with viruses, are the most common infectious complication after CAR-T-cell therapy, and influenza has been reported as a cause of death.2 6 7 Thus, there is an urgent need to understand the utility of influenza vaccination prior to and after CAR-T cell therapy, and to inform the broader question of vaccine immunogenicity in these patients.
We report the results of a prospective observational study of the humoral immunogenicity of the inactivated influenza vaccine (IIV) among CAR-T-cell therapy recipients vaccinated before or after CAR-T-cell therapy compared with controls.
Methods
Study design and participants
We approached all adults ≥18 years planning to receive a 2019–2020 season IIV before or after CD19-, CD20- or B cell maturation antigen (BCMA)-CAR-T-cell therapy at Fred Hutchinson Cancer Center (Fred Hutch). A control cohort included non-immunocompromised Fred Hutch employees. In the pre-CAR-T cohort, the IIV was administered after leukapheresis and ≥2 weeks prior to CAR-T-cell therapy. Exclusion criteria were immunoglobulin replacement therapy (IGRT) within 2 months prior to enrollment, bridging chemotherapy after vaccination, persistent or relapsed disease after CAR-T-cell therapy, or initiation of new antitumor therapies. All participants provided informed consent in accordance with the Declaration of Helsinki.
Inactivated influenza vaccines
CAR-T-cell recipients received commercially available trivalent or quadrivalent 2019–2020 Northern Hemisphere IIVs (as detailed in table 1 and online supplemental table S1). Controls received a quadrivalent IIV (Flucelvax, Seqirus).
Supplemental material
Baseline clinical characteristics and immunologic findings of the pre- and post-CAR-T-cell therapy cohorts
Data and blood collection
Data were abstracted from medical records or directly obtained from controls. In the pre-CAR-T cohort, blood samples were obtained before vaccination (baseline), before lymphodepleting chemotherapy, and approximately 30 days and 90 days after CAR-T-cell therapy (online supplemental figure S1). In the post-CAR-T cohort, samples were collected at baseline and once approximately 30–90 days after vaccination. In the control cohort, samples were obtained at baseline and 30 days, 60 days, and 90 days after vaccination. Serum and peripheral blood mononuclear cells (PBMCs) were isolated and stored (online supplemental file 1).

Summary of longitudinal influenza antibody kinetics and geometric mean titers (GMTs). Individual titer results are plotted for each sample collection time point for the pre-CAR-T, post-CAR-T, and control cohort (from left to right in each panel). (A) Neutralization titers to A(H1N1) and (B) HAI titers to A(H1N1), (C) A(H3N2), (D) B(Victoria), and (E) B(Yamagata) are shown. A value of half of the lower limit of detection (LOD) was assigned for values below the LOD (LODs are detailed in the Methods). Data have been jittered to allow viewing of overlapping values. Horizontal bars represent GMT. Symbols on or above the dashed horizontal line represent HAI titers ≥40. Baseline titers to A(H1N1) were significantly lower in the CAR-T cohorts when compared with the control cohort (neutralization assay: pre-CAR-T vs controls, p=0.01; post-CAR-T vs controls, p=0.02. HAI assay: pre-CAR-T vs controls, p=0.02; post-CAR-T vs controls, p=0.004; based on Dunn’s test with the Holm stepwise procedure for multiple comparisons). There were no significant differences in baseline titers between cohorts based on the HAI assay to A(H3N2), B(Victoria), or B(Yamagata) (Kruskal-Wallis, p=0.21, p=0.17 and p=0.36, respectively). CAR-T-cell, chimeric antigen receptor-modified T cell; HAI, hemagglutination inhibition.
Laboratory testing
We performed serum hemagglutination inhibition (HAI) assays for antibodies to all four vaccine strains. Postvaccine results for B(Yamagata) were excluded for individuals without confirmed receipt of a quadrivalent vaccine. The lower and upper limits of detection (LOD) were 10 and 1280, respectively. We also tested serum neutralizing antibody titers for H1 of the A(H1N1) strain.8 The lower and upper LOD ranged from 12.5 to 25 and 2680 to 5369, respectively. We immunophenotyped B cells and T cells from PBMCs and measured total serum IgG, IgM, and IgA. Methods are detailed in the online supplemental file 1.
Outcomes
The primary outcome was an antibody response (seroconversion) to the respective vaccine strains at the first postvaccine time point, defined as an at least fourfold titer increase from baseline (or an HAI titer of ≥40 if the baseline titer was <10).9 We also report the proportion of individuals with an at least twofold titer increase from baseline and HAI antibody titers ≥40, a threshold considered to correlate with seroprotection.10
Analyses
We compared baseline titers between cohorts using Kruskal-Wallis tests, and if significant, Dunn’s test was conducted for pairwise comparisons using the Holm stepwise procedure to account for multiple comparisons. A value of half of the lower LOD was assigned for values below the LOD. We computed the proportion of individuals with an antibody response to ≥1 vaccine strain with Wilson 95% CIs. We explored associations between baseline variables and responses in the post-CAR-T cohort using univariate Firth logistic regression models. Two-tailed p values were calculated and p values<0.05 were considered statistically significant. Analyses were conducted using Stata V.16.0.
Results
Baseline characteristics
Twenty-six adults received the IIV between October 2019 and March 2020. Baseline characteristics, as well as CAR-T-cell product and vaccine details, are in table 1 and online supplemental tables S1 and S2. The five adults in the pre-CAR-T cohort had relapsed or refractory B cell malignancies and were vaccinated 14–29 days (median, 25) pre-CAR-T-cell therapy. The 13 individuals in the post-CAR-T cohort were 13–57 months (median, 21) from CAR-T-cell therapy and had complete or very good partial remission. The eight controls were 25–62 years old (median, 43). Most individuals in both CAR-T cohorts had hypogammaglobulinemia as well as low CD19+ B cell and CD4+ T-cell counts. The IIV was administered in the prior year to 12 (92%) individuals in the post-CAR-T cohort and all individuals in the control cohort; data were not available for individuals in the pre-CAR-T cohort.
Antibody titers and antibody responses at baseline and at the first postvaccine time point
Baseline influenza antibody titers
Antibody titers to each vaccine strain are in figure 1 and summarized in table 2. Baseline neutralizing antibody titers to A(H1N1) were similar in the pre-CAR-T and post-CAR-T cell cohorts but significantly higher in the control cohort. This was consistent with results from the HAI assay to A(H1N1), which additionally revealed baseline antibody titers above the LOD in only 1 (20%) individual in the pre-CAR-T and 3 (23%) individuals in the post-CAR-T cohorts compared with 7 (88%) in the control cohort. Baseline titers to A(H3N2) were low among all cohorts. Baseline titers to B(Victoria) or B(Yamagata) did not differ significantly between cohorts but tended to be lower in the CAR-T-cell cohorts. Correspondingly, baseline HAI titers ≥40 to A(H1N1), B(Victoria), and B(Yamagata), but not to A(H3N2), were less frequent among CAR-T-cell therapy recipients than controls.
IIV immunogenicity and kinetics of influenza antibody responses
For each cohort, changes in titers by strain are summarized in figure 1 and table 2; titer changes per individual are in figure 2.
{kind=link}
{kind=link}
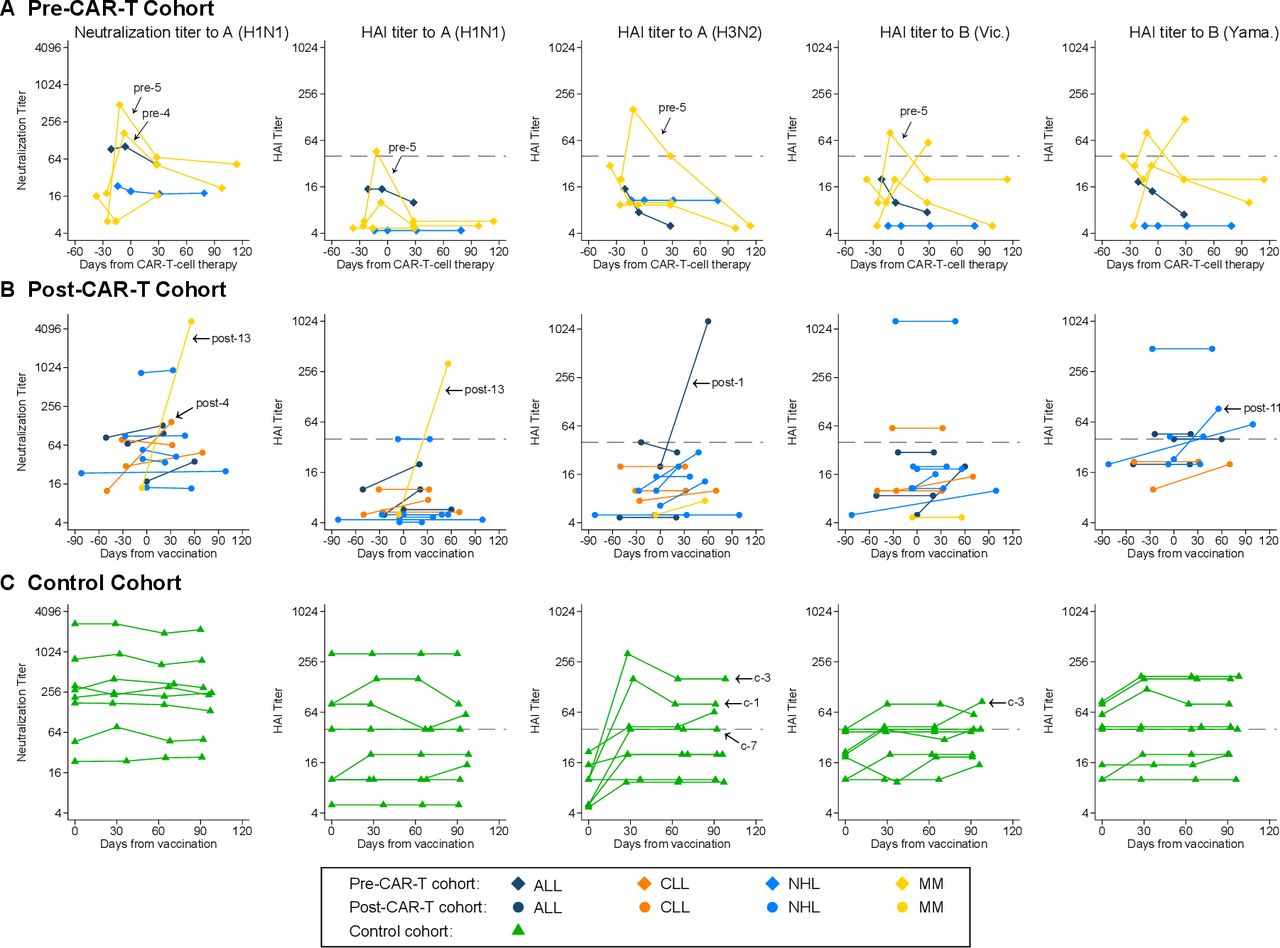
Kinetics of influenza antibody titers by individual. Line plots demonstrating neutralization titers to A(H1N1) and hemagglutination inhibition (HAI) titers to A(H1N1), A(H3N2), B(Victoria), and B(Yamagata) for (A) the pre-CAR-T cohort, (B) the post-CAR-T cohort, and (C) the control cohort. Each line connects results from one individual over time. Individuals with antibody responses at the first postvaccine time point are indicated with an arrow and their study ID. Symbols on or above the dashed horizontal line represent HAI titers ≥40. For the pre-CAR-T cohort, day 0 was set at the day of CAR-T-cell therapy, vaccines were administered between 0 and 8 days after baseline sample collection (median, 0), and time between vaccine and sample collection prior to CAR-T-cell therapy ranged from 13 to 19 days (median, 14). For the post-CAR-T cohort and the control cohort, day 0 was set at the day of vaccination. individuals without confirmed receipt of a quadrivalent vaccine are excluded from the plots showing HAI titers to B(Yamagata). CAR-T, chimeric antigen receptor-modified T cell.
Pre-CAR-T cohort
At the first postvaccine time point, a median of 14 days (range, 13–19) after IIV and before CAR-T-cell therapy, 2 (40%; 95% CI 12% to 77%) individuals demonstrated responses to ≥1 vaccine strain. After CAR-T-cell therapy, titers decreased, but neutralizing titers to A(H1N1) remained above baseline by day 30 post-CAR-T-cell therapy, and one individual maintained an antibody response through day 114. Among the remaining three (60%) individuals, one had at least twofold increases in titers to two strains at the first time point after CAR-T-cell therapy.
Post-CAR-T cohort
The median time between vaccination and postvaccine sample collection was 37 days (range, 20–99). Responses to ≥1 vaccine strain occurred in 4 (31%; 95% CI 13% to 58%) individuals. An additional six (46%) individuals had an at least twofold increase to ≥1 strain each. Three (23%) individuals received IGRT 62, 66, and 95 days prior to the baseline sample, but their baseline titers were similar to those who did not receive IGRT. Two of these individuals received subsequent IGRT prior to the postvaccine sample but none met criteria for a response (online supplemental table S2).
Control cohort
The first postvaccine time point was a median of 29 days from vaccination (range, 27–37). Responses to ≥1 vaccine strain occurred in 3 (38%) individuals, all for the A(H3N2) strain, and were maintained through 90 days after vaccination. Three (38%) additional individuals had an at least twofold increase to ≥1 strain.
Summary of influenza antibody kinetics
Among both CAR-T cohorts, there was a modest increase in the geometric mean titer (GMT) at the first postvaccine time point (figure 1). The pre-CAR-T cohort had a relatively rapid decrease in GMTs over time to a level below the baseline by the 90-day time point. Some individuals in the post-CAR-T cohort generated antibody titers higher than the controls. The IIV for the 2019–2020 season had relatively low immunogenicity in the controls aside from strain A(H3N2), to which no controls had a prevaccine HAI titer ≥40. Baseline and postvaccine HAI titers ≥40 were more frequent among controls than in either CAR-T cohort (table 2).
Predictors of IIV immunogenicity
To explore possible predictors of IIV immunogenicity, we depict titer fold changes by key clinical and immunologic characteristics (online supplemental figure S2). There was evidence of immunogenicity across most variables, and responses were observed in individuals with very low or no detectable CD19+ B cells and individuals with severe hypogammaglobulinemia. Most responders had IgA and IgM levels below the lower limit of normal. Univariate logistic regression analyses in the post-CAR-T cohort did not identify any associations between an antibody response and variables of age, sex, underlying malignancy, time from CAR-T-cell therapy, IgG level, CD19+ B cell count, or CD4+ T-cell count (data not shown).
Discussion
CAR-T-cell therapy recipients are immunocompromised prior to and for months following therapy, rendering them at high risk for infections.1 6 7 11 Vaccination may be an effective strategy to prevent the acquisition and severity of infections, but there are limited data about vaccine immunogenicity, or predictors of vaccine responses, in this population.3–5 In this prospective study of IIV administered before or after CAR-T-cell therapy, we demonstrated robust antibody responses to ≥1 vaccine strain in 31%–40% of individuals and partial antibody responses in 60%–77%, despite substantial humoral and cellular immunodeficiencies. Our findings support administration of relevant vaccines before CAR-T-cell therapy and for (re)vaccination, as indicated, of individuals in remission, irrespective of serum IgG level and total B cell count. Nevertheless, these data underscore the need for additional preventive measures in CAR-T-cell therapy recipients.
Immunity to influenza prior to vaccination reflects an individual’s history of vaccination and infection.The 2019/2020 H1N1 vaccine strain was similar to the 2018/2019 formulation, and cross-reactive antibodies may explain the high baseline antibody titers to A(H1N1) in controls.12 13 In contrast, a high proportion of individuals pre-CAR-T-cell therapy had undetectable baseline titers to A(H1N1), which may be due to lack of prior vaccination, poor prior responses, or loss of pre-existing immunity. Among individuals in remission after CAR-T-cell therapy, baseline titers to A(H1N1) were also significantly lower than in controls despite a similarly high frequency of prior-year vaccination, suggesting either poor responses and/or rapid waning.14 Baseline antibody titers to the A(H3N2) vaccine strain were low among all cohorts, likely due to a new strain in the 2019/2020 vaccine formulation. Both B strains were unchanged from the previous year, and there was a trend towards lower baseline titers in the CAR-T-cell cohorts. Overall, a higher proportion of controls had detectable influenza-specific antibodies and HAI titers ≥40 at baseline and following vaccination, indicating that CAR-T-cell therapy recipients have higher risk for morbidity from influenza infection.15 16
After receiving the IIV, 31%–40% of individuals in all cohorts had at least fourfold increases in antibody titers for ≥1 vaccine strain, and 60%–77% had at least twofold increases. The limited responses to A(H1N1) and B strains among controls may reflect pre-existing immunity, as individuals with repeated annual IIV have minimal immune responses to the same or similar strains after re-exposure.13 Peak titers generally occurred at the first postvaccine time point. In the pre-CAR-T-cell cohort, we observed relatively rapid antibody decay after CAR-T-cell therapy. Given that the two individuals with antibody responses received plasma cell targeted BCMA-CAR-T-cells, this observation may be related to destruction of newly generated influenza-specific antibody-secreting plasma cells by the CAR-T-cells.17 However, antibody titers generally persisted above baseline for at least 30 days and up to 4 months after CAR-T-cell therapy, which may provide immunity during the highest-risk time frame.7 15 Given the safety of the IIV, these data demonstrate sufficient immunogenicity to consider recommending vaccination.
These observations are relatively consistent with a study including 14 CAR-T-cell therapy recipients receiving a two-dose SARS-CoV-2 mRNA vaccine series, in which 5 (36%) developed a positive antibody titer and 6 of 12 tested patients had a T cell response, although absolute titers were relatively low.5 In two other studies including 14 and 3 post-CAR-T-cell therapy recipients receiving SARS-CoV-2 mRNA vaccines, only 11% and 3% had antibody responses, and there were no clear predictors of response.3 4 Comparisons are limited by differences in vaccines, pre-existing immunity, assays, and response definition.
An important observation was the lack of clear correlations between clinical or immunologic characteristics and antibody responses. Key observations included vaccine immunogenicity in individuals with low peripheral CD19+ B cell counts and low serum IgG, IgA and IgM levels. Although some guidelines and clinical heuristics would suggest not vaccinating the majority of individuals in our CAR-T-cohorts, we nonetheless demonstrate clinically relevant immunogenicity of the IIV. This could be due to persistence or recovery of B cells in lymphoid tissue or the bone marrow. Whether responses originated from de novo naïve B cells or boosted memory B cells is unclear. Due to different expression patterns of the CAR-T-cell targets on B-lineage cells, CD19- and CD20-targeting CAR-T-cells may lead to greater depletion of pathogen-specific memory B cells, whereas BCMA-targeting CAR-T-cells may affect the generation of new antibody-producing plasma cells.
This is one of the first reports of vaccine immunogenicity after CAR-T-cell therapy and the first to explore pre-CAR-T-cell therapy vaccine immunogenicity. The prospective study design and inclusion of a control comparator group are additional strengths. Our data support consideration for administration of non-live vaccines before CAR-T-cell therapy for influenza, and by extrapolation, to other relevant pathogens in this clinical context (eg, SARS-CoV-2, pneumococcus). Additionally, vaccinations should be considered in patients in remission after CAR-T-cell therapy. The primary limitation is the small sample size, and observations pertaining to predictors of vaccine response are limited by small numbers and patient heterogeneity. All participants in the pre-CAR-T cohort had a refractory hematologic malignancy, but heterogeneity in the underlying diseases and prior treatments may have influenced responses. These issues were less relevant in the post-CAR-T cohort, as all individuals were in remission for >12 months and received CD19-targeted therapies, except one. Although different CAR-T-cell products with potentially variable kinetics were used, we demonstrated that most individuals shared cellular and humoral deficits. Administration of the IIV was at the discretion of clinical providers, which may have introduced bias, and vaccine types varied. Additionally, timing of sample collection varied based on clinical follow-up; short-lasting responses might have been missed in the post-CAR-T cohort, which had the longest interval between vaccination and postvaccine sample collection. Additional data are needed to determine immunogenicity within the first year after CAR-T-cell therapy. Nevertheless, persistent immunodeficiency is a key concern for the long-term care of CAR-T-cell recipients, adding relevance to our observations. Although HAI titers ≥40 generally correspond to a 50% reduction in the incidence of infection,10 this is not established in immunocompromised individuals. Cellular responses are another critical component of immunity to influenza and other infections;18 T cell responses may demonstrate additional utility of vaccination in this population with impaired B cell immunity.5
In summary, despite the small sample size, these data support consideration for vaccination for influenza and other pathogens before and after CAR-T-cell therapy, irrespective of hypogammaglobulinemia or B cell aplasia. Larger studies are clearly needed to determine optimal timing of vaccination and better define predictors of vaccine immunogenicity and durability. Additional strategies to prevent infections, like vaccination of close contacts and standard precautions, should remain the backbone of infection prevention in these high-risk individuals.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the Fred Hutch Institutional Review Board.
Acknowledgments
The authors thank the study participants, the participants’ local provider for assistance with sample collection and medical records, in addition to Naomi Wilcox (statistician, University of Washington, Seattle, WA), Cassandra Job (BS, Fred Hutchinson Cancer Research Center, Seattle, WA), Jessica Hirianto (undergraduate student, Fred Hutchinson Cancer Research Center, Seattle, WA), Joyce Maalouf (clinical research manager, Fred Hutchinson Cancer Research Center, Seattle, WA), Jessica Morris (data management analyst, Fred Hutchinson Cancer Research Center, Seattle, WA), and Atif Bhatti (BS, Fred Hutchinson Cancer Research Center, Seattle, WA) for assistance with sample processing and data management.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @CarlaWalti
Contributors JAH, CSW, and SAP designed the study; CSW, JK-C, JAH, and TL collected the data; ANL and JDB performed the neutralization assay; KS, CRW, and HYC performed the HAI assay; JB and JJT performed the flow cytometry analyses; CSW, EMK, ANL, KS, JB, and JAH analyzed the data; EMK, CSW, KS, and JAH created the figures; CSW, ANL, KS, EMK, JB, SAP, HYC, JDB, and JAH interpreted the data; CSW, EMK and JAH drafted the initial manuscript. All authors contributed to the writing and revision of the manuscript and approved the final version.
Funding This work was supported by grants from the Swiss National Science Foundation (P2BSP3 188 162 to CSW), the National Institutes of Health National Cancer Institute (NIH/NCI; U01CA247548 to JAH and P01CA018029 to DJG and CJT), the NIH/NCI Cancer Center Support Grants (P30CA0087-48 and P30CA015704-44), the American Society for Transplantation and Cellular Therapy (to JB), a Washington Vaccine Alliance Pilot Grant (to SAP), and Juno Therapeutics (a Bristol-Myers Squibb company). JDB is an Investigator of the Howard Hughes Medical Institute.
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding institutions.
Competing interests JJT received research funding from Vir Biotechnology for research unrelated to this study. RAG received consulting fees from Novartis, served on ad hoc advisory boards for Janssen and Pfizer and has patents licensed to Juno Therapeutics. DJG has received research funding, has served as an advisor and has received royalties from Juno Therapeutics, a Bristol-Myers Squibb (BMS) company; has served as an advisor and received research funding from Seattle Genetics; has served as an advisor to GlaxoSmithKline, Celgene, Janssen Biotech, Bristol-Myers Squibb, Neoleukin Therapeutics and Legend Biotech; and has received research funding from SpringWorks Therapeutics, Sanofi and Cellectar Biosciences. AJC received research funding from Janssen, Sanofi, BMS, Harpoon, Nektar; and received consulting fees from Janssen, Cellectar, Sanofi, GlaxoSmithKline, and Abbvie. DGM has served as a consultant for A2 Biotherapeutics, Amgen, Bioline Rx, BMS, Celgene a BMS company, Genentech, Gilead, Janssen, Juno Therapeutics a BMS company, Kite Pharma, Legend Biotech, MorphoSys, Novartis, and Pharmacyclics; has received research funding paid directly to the institution, including salary support, from Kite Pharma, Juno Therapeutics/BMS, and Celgene/BMS and has patents with Juno Therapeutics/BMS (pending, not issued, licensed, no royalties, no licenses); and has stock options in A2 Biotherapeutics. CJT received research funding from Juno Therapeutics, Nektar Therapeutics, AstraZeneca, TCR2 Therapeutics; is a member of scientific advisory boards for Precision Biosciences, Eureka Therapeutics, Caribou Biosciences, T-CURX, Myeloid Therapeutics, ArsenalBio, and Century Therapeutics; has served on ad hoc advisory boards for Nektar Therapeutics, Allogene, Asher Biotherapeutics, PACT Pharma, Astra Zeneca; has stock options for Precision Biosciences, Eureka Therapeutics, Caribou Biosciences, Myeloid Therapeutics, ArsenalBio; and has patents licensed or optioned to Juno Therapeutics. SAP received research support from Global Life Technologies, Inc., and participated in research trials with Chimerix, Inc and Merck & Co. He also participated in a clinical trial sponsored by the National Institute of Allergy and Infectious Diseases (U01-AI132004); influenza vaccines for this trial are provided by Sanofi-Aventis. HYC reported consulting with Ellume, Pfizer, The Bill and Melinda Gates Foundation, Glaxo Smith Kline, and Merck. She has received research funding from Gates Ventures, Sanofi Pasteur, and support and reagents from Ellume and Cepheid outside of the submitted work. JDB is on the scientific advisory board of Oncorus and has performed consulting for Moderna. JAH received consulting fees or honoraria from Gilead Sciences, Amplyx, Allovir, Allogene therapeutics, CRISPR therapeutics, CSL Behring, OptumHealth, Octapharma, and Takeda and research funding from Takeda, Allovir, Karius, and Gilead Sciences.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.