Article Text

Abstract
Background T cell checkpoint immunotherapies have shown promising results in the clinic, but most patients remain non-responsive. CD47-signal regulatory protein alpha (SIRPα) myeloid checkpoint blockade has shown early clinical activity in hematologic malignancies. However, CD47 expression on peripheral blood limits αCD47 antibody selectivity and thus efficacy in solid tumors.
Methods To improve the antibody selectivity and therapeutic window, we developed a novel affinity-tuned bispecific antibody targeting CD47 and programmed death-ligand 1 (PD-L1) to antagonize both innate and adaptive immune checkpoint pathways. This PD-L1-targeted CD47 bispecific antibody was designed with potent affinity for PD-L1 and moderate affinity for CD47 to achieve preferential binding on tumor and myeloid cells expressing PD-L1 in the tumor microenvironment (TME).
Results The antibody design reduced binding on red blood cells and enhanced selectivity to the TME, improving the therapeutic window compared with αCD47 and its combination with αPD-L1 in syngeneic tumor models. Mechanistically, both myeloid and T cells were activated and contributed to antitumor activity of αCD47/PD-L1 bispecific antibody. Distinct from αCD47 and αPD-L1 monotherapies or combination therapies, single-cell RNA sequencing (scRNA-seq) and gene expression analysis revealed that the bispecific treatment resulted in unique innate activation, including pattern recognition receptor-mediated induction of type I interferon pathways and antigen presentation in dendritic cells and macrophage populations. Furthermore, treatment increased the Tcf7+ stem-like progenitor CD8 T cell population in the TME and promoted its differentiation to an effector-like state. Consistent with mouse data, the compounds were well tolerated and demonstrated robust myeloid and T cell activation in non-human primates (NHPs). Notably, RNA-seq analysis in NHPs provided evidence that the innate activation was mainly contributed by CD47-SIRPα but not PD-L1-PD-1 blockade from the bispecific antibody.
Conclusion These findings provide novel mechanistic insights into how myeloid and T cells can be uniquely modulated by the dual innate and adaptive checkpoint antibody and demonstrate its potential in clinical development (NCT04881045) to improve patient outcomes over current PD-(L)1 and CD47-targeted therapies.
- immunotherapy
- antibody affinity
- immunity
- innate
- tumor microenvironment
Data availability statement
All data are available in the main text or the online supplemental materials. The accession number for the mouse and NHP RNA-seq data is GSE177053. Requests for materials should be directed to and will be fulfilled by S-HC, JC-R, or SS-A and the completion of a material transfer agreement with Pfizer.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The PD-1 immune checkpoint is well established in its ability to attenuate T cell-mediated immune responses in patients whose tumors express its ligand, programmed death-ligand 1 (PD-L1). While therapeutic interventions blocking PD-1 or PD-L1 can release antitumor T-cell immunity, the immune response remains suppressed in many tumors.1 In patients harboring these types of tumors, increased antitumor activity cannot be established from PD-1/PD-L1 inhibition alone.2 CD47, a cell surface molecule in the immunoglobulin superfamily, is another established target overexpressed on various malignant cells and has been described colloquially as the regulator of a ‘don’t-eat-me’ signal.3 4 CD47 binds to several proteins, including signal regulatory protein alpha (SIRPα), signal regulatory protein gamma (SIRPγ), integrins, and thrombospondin-1.5 When bound to its receptor, SIRPα, which is present on various myeloid cells, CD47 acts as an innate inhibitory checkpoint by interrupting the phagocytosis of tumor cells and downstream activation of innate responses.4 By suppressing innate immune activation and presentation of tumor antigens and priming of T cell responses, CD47 may enable tumor cells to escape both innate and adaptive immune surveillance.6 MYC oncogene, deregulated in >50% of human cancers, has been found to upregulate both CD47 and PD-L1 expression on the surface of tumor cells by binding to their promoters.7 CD47 overexpression has also been shown to act as a resistance mechanism to PD-1/PD-L1 therapies in preclinical models.8 9 Thus, CD47 has become an attractive target, with various approaches to block CD47/SIRPα interactions in clinical development.10
Treatment with monospecific αCD47 antibodies has been particularly successful in hematological malignancies, such as non-Hodgkin’s lymphoma (NHL).11 Patients with NHL of various cell types treated with αCD47 achieved an objective response rate of 50%–70%, with complete response rates of 36%–40% and markedly increased overall survival.11 Unfortunately, limited efficacy has thus far been observed in patients with solid tumors12 even when αCD47 was combined with αPD-1/PD-L1 agents.9 One explanation for the lack of efficacy in solid tumors is the inability to effectively target the tumor, as CD47 is widely expressed on circulating erythroid, myeloid, and other cells of hemopoietic origin. The high therapeutic doses that would be required have shown the potential to cause significant anemia through increased phagocytosis of red blood cells (RBCs). If the binding of αCD47 to RBCs could be reduced, it might be possible to ameliorate anemia and provide a new method to reverse the innate immunosuppression of myeloid cells by tumors.
We generated an affinity-tuned bispecific antibody (BisAb) targeting CD47 and PD-L1 to antagonize both innate and adaptive immune checkpoint pathways. This novel BisAb was designed with potent affinity for PD-L1 and reduced affinity for CD47 to achieve preferential binding on tumor and myeloid cells expressing PD-L1 in the tumor microenvironment (TME), thus increasing tumor exposure for potent antitumor efficacy and reducing the risk of anemia. The BisAb using an IgG with full effector function further enhanced tumor-killing activity in vitro and in vivo. Moreover, the unique binding selectivity of αCD47/PD-L1 resulted in a distinct macrophage and dendritic cell (DC) activation and an increase in stem-like progenitor and effector CD8+ T cells in the TME, compared with the combination treatment of monospecific αCD47 and αPD-L1 antibodies. Thus, this study provides insight into how myeloid cells and T cells can be uniquely modulated by the dual innate and adaptive checkpoint antibody and demonstrate its potential in clinical development (NCT04881045).
Materials and methods
In vivo mouse models
CT26 cells in serum-free phosphate-buffered saline (PBS) were inoculated in the right flank at 2.5×105 cells per implant and allowed to grow for 14 days. MC38 or B16F10 cells in serum-free PBS were inoculated in the right flank at 5×105 cells per implant and allowed to grow for 7–10 days (MC38) or 9–13 days (B16F10). The mice were then randomized by tumor volume and treated with the described antibodies. Length and width of tumors were recorded using digital caliper measurements. Tumor volume was calculated using the formula tumor volume = width2 × 0.5(Length). Percent of initial body weight was calculated using the formula % of initial body weight=body weight day post-treatment initiation (g)/body weight D0 post-treatment initiation (g) × 100. Percent survival post-treatment was determined using the Kaplan-Meier survival curve in GraphPad Prism. Mice were euthanized if tumor size exceeded 2000 mm3 or had a body weight loss greater than 20% of initial body weight.
Unless otherwise stated, the following treatment antibodies were used for syngeneic tumor experiments: mouse IgG2a isotype control antibody from BioXcell (C1.18.4), and Pfizer in-house generated anti-mouse CD47 antibody (αmCD47), anti-mouse PD-L1 antibody (αmPD-L1), mouse BisAb (mBisAb) or mBisAb mIgG2a Fc null. All antibodies were diluted in PBS and administered intraperitoneally (i.p.). For CT26 antitumor efficacy, survival and bodyweight loss studies, female Balb/c tumor bearing mice were randomized at day 14 post-inoculation. For single agent treatment groups, treatment antibodies were administered at 5 mg/kg. For the combination treatment group, 5 mg/kg of αmCD47 and 5 mg/kg of αmPD-L1 were administered. Treatment antibodies were administered every 3–4 days for a total for three doses (days 0, 3 and 7 post-treatment initiation). For B16F10 antitumor efficacy, female C57/Bl6 tumor bearing mice were randomized at day 9 post inoculation. Treatment antibodies were administered at 20 mg/kg three times a week for a total for nine doses (3Qw ×3 weeks). For MC38 dose-dependent antitumor efficacy in C57/BL6 tumor-bearing mice were randomized at day 7 post-inoculation. Treatment antibodies were administered every 3–4 days for a total of six doses (days 7, 9, 12, 15, 22 and 26 post-tumor inoculation). mBisAb was administered at 10 mg/kg, 20 mg/kg, and 40 mg/kg. Tumors were measured as above twice a week. For MC38 antitumor efficacy with immune cell depletion, female C57/BL6 tumor bearing mice were randomized at day 6 post-inoculation. Vehicle control (PBS) and mBisAb mIgG2a was administered at 40 mg/kg every 3–4 days for a total of six doses (Days 7, 11, 14, 18, 21, 25 post-tumor inoculation). Depletion antibodies were diluted in PBS and administered i.p at 200 ug/mouse on day 6 post-tumor inoculation and every 2–3 days thereafter until the termination of the study for a total of nine doses. The following depletion antibodies were from BioXcell: anti-CD8 (clone 2.43) antibody, anti-CD4 (GK1.5) antibody, anti-NK1.1 (clone PK136) antibody or anti-CSF1R (clone AFS93) antibody. For MC38 antitumor efficacy in Batf3-/- mice, female Batf3-/- tumor bearing mice were randomized at day 10 post-inoculation. Mouse IgG2a isotype control and mBisAb mIgG2a were administered at 10 mg/kg every 3–4 days for a total of four doses (days 10, 13, 17, 20 post-tumor inoculation).
For in vivo binding selectivity, male human FcγR B16F10 tumor bearing mice were randomized at day 13 post-inoculation. Treatment antibodies were generated in-house at Pfizer. The following treatment antibodies were diluted in PBS and administered at 10 mg/kg at days 13 and 16 post-inoculation for a total of two doses: human IgG isotype control, αmCD47 hIgG1, αmPD-L1 hIgG1 and mBisAb hIgG1. Blood and tumor tissue were collected on day 18 post inoculation to assess treatment antibody binding selectivity assessment by flow cytometry.
To assess impact of murine RBCs on treatment, treatment antibodies were administered to male Balb/c mice at 5 mg/kg twice (days 0 and 3). The peripheral blood was collected 2 days post second dose (day 5) and red blood counts were assessed.
Non-human primate models
Cynomolgus monkeys were administrated either vehicle or test-article by slow bolus intravenous injection on study day 1 and day 8. Animals were monitored twice daily for clinical observations and body weights collected on a weekly basis. Scheduled collections of peripheral blood for clinical pathology evaluation included hematology and clinical chemistry assessments. For gene expression analysis, peripheral blood was collected in RNAprotect tubes (Qiagen) according to manufacturer’s instructions for RNA sequencing (RNA-seq).
Statistical analyses
Statistical analysis was applied to biologically independent mice or technical replicates for each experiment. For antitumor efficacy studies, the statistical significance of tumor volumes between experimental groups was calculated using a two-way analysis of variance (ANOVA) with multiple comparison using GraphPad Prism V.8 software. The ordinary one-way ANOVA or Student’s unpaired T cells were used for statistical calculations using GraphPad Prism V.8 software for other statistical analysis as specified in figure legends. All error bars were reported as mean±SEM. The level of significance is indicated as *p<0.05; **p<0.01; ***p<0.001.
More detailed information is available in online supplemental material section.
Supplemental material
Results
Design and characterization of αCD47/PD-L1 BisAb
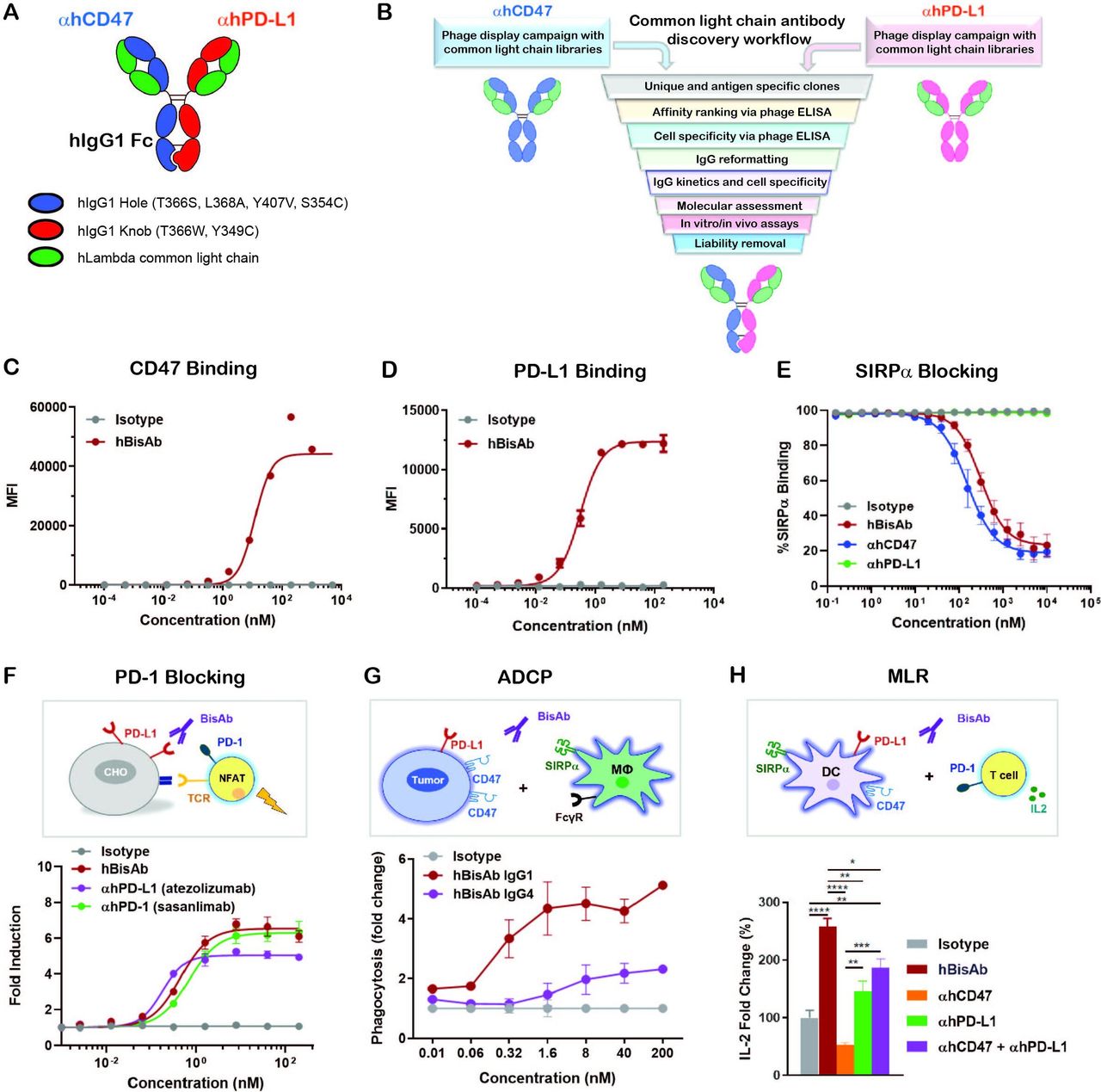
αCD47/PD-L1 is a fully human BisAb (hBisAb) in an IgG1 format using knobs-in-holes technology and a common light chain architecture (figure 1A). Each binding arm was discovered independently using synthetic common light chain phage display libraries13 in two campaigns against extracellular domains of human CD47 and human PD-L1 (figure 1B). After several rounds of optimization, affinity tuning, and liability removal of each binding arm, the final human αCD47/PD-L1 hBisAb with moderate affinity to CD47 and highly potent affinity to PD-L1 was reformatted and purified using standard biochemical procedures. To confirm the dual binding activity of hBisAb to each antigen, the kinetic rate constants were measured by surface plasmon resonance (SPR). The KD of hBisAb to human and cynomolgus monkey CD47 was 59.5 nM and 83.9 nM, respectively, and the KD to human and monkey PD-L1 was 0.227 nM and 0.231 nM, respectively (online supplemental table 1). The EC50 of binding to each target was determined using cell-based binding assays. The binding of the hBisAb to human CD47 ectopically expressed on CHO cells was reduced (CD47 EC50=11.89 nM) (figure 1C) compared with the binding of the hBisAb to human PD-L1 expressed on CHO cells (PD-L1 EC50=0.32 nM) (figure 1D). The increased potency of the PD-L1 EC50 compared with the CD47 EC50 of the hBisAb is aligned with the design of the antibody in which the goal was to develop a BisAb with a moderate affinity to CD47 and highly potent affinity to PD-L1. Despite a reduced affinity for human CD47, hBisAb potently inhibited the interaction of human SIRPα/CD47 as determined by SPR (online supplemental table 1) and in cell-based assays, where hBisAb blocked the binding of SIRPα to CHO-hCD47 cells (figure 1E) with an IC50 of 123.6 nM. Additionally, hBisAb efficiently blocked the interaction of human PD-1/PD-L1 as determined by SPR (online supplemental table 1). The ability of hBisAb to enhance T cell functionality by blocking the PD-1/PD-L1 interaction was also measured by a PD1-PD-L1 T-cell receptor (TCR) nuclear factor of activated T cell (NFAT) reporter assay. The average EC50 induced by hBisAb was 0.451 nM with no statistical difference compared with other human αPD-L1 bivalent antibodies (αhPD-L1) (figure 1F).
Supplemental material
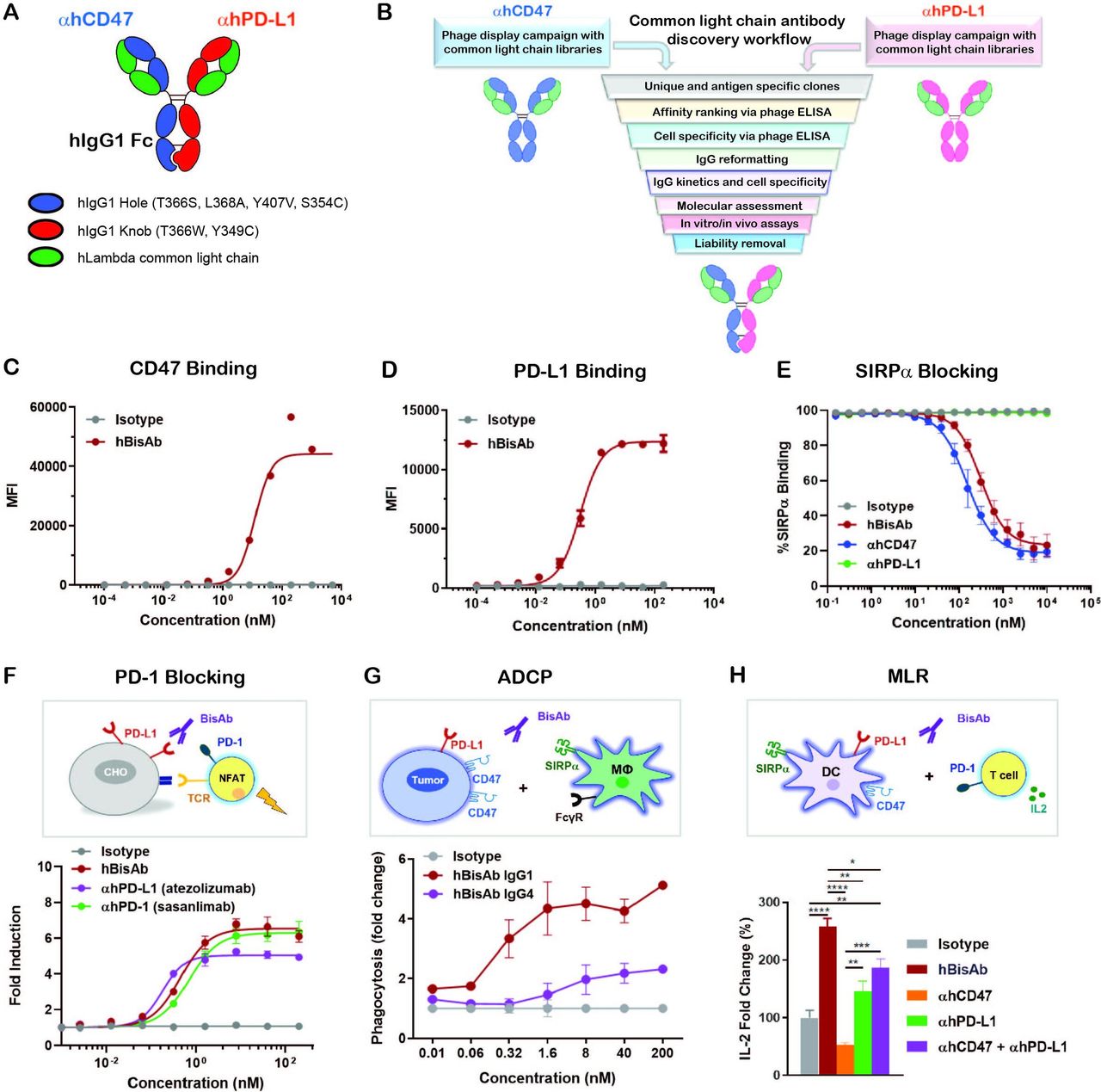
Design and characterization of αCD47/PD-L1 bispecific antibody. (A) The schematic diagram of the human αCD47/PD-L1 bispecific antibody (hBisAb). (B) Workflow of common light chain antibody campaign for the generation of hBisAb. (C, D) Cell based binding of hBisAb on CHO-hCD47 (C), and CHO-hPD-L1 (D), as measured by flow cytometry. (E) In vitro blocking activity of hBisAb on the human CD47/SIRPα interaction as measured by flow cytometry. (F) In vitro blocking activity of hBisAb on the human PD-L1/PD-1 interaction using a PD-L1/PD-1 TCR blocking reporter bioassay. The blocking activity was quantified and normalized as the fold change compared with isotype control. (G) Phagocytosis of NCI-H292 human tumor cells by human monocyte-derived macrophages in the presence of human IgG isotype control, hBisAb in IgG1 or IgG4 format (n=2 donors/group). Phagocytosis of total tumor cells is represented as fold change compared with isotype treatment. (H) MLR assay was conducted to assess concentration of IL-2 at 72 hours in the supernatant by ELISA. A mixture of LPS matured DCs and purified CD4+ T cells were cocultured at a 1:4 ratio in the presence of αhCD47, αhPD-L1, the combination of αhCD47 and αhPD-L1, or hBisAb at 200 nM. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001, one-way ANOVA. ADCP, antibody-dependent cellular phagocytosis; ANOVA, analysis of variance; DC, dendritic cell; IL-2, interleukin 2; MLR, mixed lymphocyte reaction; LPS, lipopolysaccharide; TCR, T cell receptor.
We next performed coculture assays of human CD47+PD-L1+ tumor cells and monocyte-derived macrophages to examine the impact of hBisAb on antibody-dependent cellular phagocytosis (ADCP). We found that hBisAb induced a greater increase in phagocytosis of tumor cells than αhCD47 or αhPD-L1 monotherapy (online supplemental figure 1A), suggesting that engaging both targets on tumor cells increased the efficiency of phagocytic activity. We also compared the activity of hBisAb in the context of IgG1 and IgG4, two IgG isotypes related to phagocytosis. We found that the hBisAb-mediated enhancement of ADCP was significantly more potent with IgG1 than IgG4 (figure 1G). Furthermore, to determine the role of SIRPα/CD47 and PD-1/PD-L1 in DC-mediated T cell activation, we performed a mixed lymphocyte reaction (MLR). In the MLR assay, αhCD47 treatment reduced interleukin 2 (IL-2) secretion while αhPD-L1 single agent treatment and its combination with αhCD47 increased IL-2 secretion (figure 1H). Interestingly, treatment with hBisAb showed a significant increase in IL-2 (figure 1H) and interferon γ (IFNγ) secretion (online supplemental figure 1B) compared with single and combination treatments, demonstrating the superior capacity of hBisAb to enhance DC-mediated T cell activation.
The αCD47/PD-L1 BisAb preferentially binds cells expressing PD-L1 in the TME, reducing binding to PD-L1 negative circulating RBCs
To determine the binding selectivity of the BisAb in vitro, CD47+PD-L1+ human cancer cell line HT-1080 was cocultured with human RBCs, which only express CD47 (figure 2A). The hBisAb demonstrated >1500 fold reduced binding to RBCs (EC50=7.37 nM) compared with tumor cells (EC50=0.0047 nM). In contrast, monospecific αhCD47 antibodies showed similar binding to both tumor and RBCs, with average tumor cell binding EC50 values from 0.006 nM to 0.031 nM and RBC binding EC50 values from 0.027 nM to 1.17 nM (figure 2B). Due to a lack of cross-reactivity of the hBisAb to mouse CD47 or PD-L1, we generated mouse surrogates of αCD47/PD-L1 (mBisAb) with comparable in vitro binding selectivity to tumor cells versus RBCs and blocking activities of CD47/SIRPα and PD-L1/PD-1 as hBisAb (figure 2D and E, online supplemental figure 1C,D). We then used the mBisAb to examine the binding selectivity of BisAb in vivo. αmCD47, αmPD-L1, or mBisAb were administrated in mice inoculated with B16F10 tumor cells expressing CD47 and PD-L1.14 The peripheral blood and tumor were harvested to investigate the antibody distribution among RBCs, tumor cells and tumor-infiltrating leucocytes (TILs). As expected, most RBCs were bound by anti-mouse CD47 bivalent antibodies (αmCD47) with minimal binding on tumor cells or TILs (figure 2E). αmPD-L1 bivalent antibodies tend to bind both murine tumor and immune cells in TME with no binding on RBCs. The mBisAb binding on RBCs was markedly reduced compared with αmCD47 (figure 2E; top panel). In contrast, binding on both tumor cells and TILs was significantly increased relative to αmCD47 (figure 2E; middle and bottom panel), indicating that the PD-L1 arm of the BisAb mediates enhanced selectivity for cells in the TME. Notably, mBisAb exhibited enhanced binding to tumor cells compared with αmPD-L1, demonstrating that the bispecific approach improved targeting of tumor cells over either monotherapy.

The binding selectivity of αCD47/PD-L1 bispecific antibody (A, B) The binding selectivity of hBisAb on tumor cells and RBCs in vitro. The human tumor cell line HT-1080 and RBCs were stained for CD47 and PD-L1 expression by flow cytometry (A). CSFE-labeled HT-1080 tumor cells were cocultured with RBCs in the presence of indicated antibodies and binding selectivity was measured as the percentage of antibody-bound cells per cell type by flow cytometry (B). All experiments were repeated at least twice. (C) Cell-based binding of hBisAb on human tumor cell line HT-1080 and human RBCs in vitro as measured by flow cytometry. (D) Cell-based binding of mBisAb on murine tumor cell line MC38 and murine RBCs in vitro as measured by flow cytometry. (E) The binding selectivity of the αCD47/PD-L1 mouse surrogate mBisAb in vivo. Human FcγR mice bearing subcutaneous B16F10 tumors were dosed IP on days 13 and 16 post-tumor inoculation with αmCD47, αmPD-L1, or mBisAb in human IgG isotype. Blood and tumor tissues were collected at day 18 and therapeutic antibody binding was assessed using an anti-human IgG secondary antibody by flow cytometry (n=3 mice/group).CFSE, carboxyfluorescein succinimidyl ester; hBisAb, human bispecific antibody; IP, intraperitoneally; RBC, red blood cell; TILs, tumor-infiltrating leucocytes.
αCD47/PD-L1 BisAb inhibits tumor growth with better therapeutic window compared with the combination of αCD47 and αPD-L1 and is mediated by CD8+ T cells and DCs in syngeneic models
The antitumor activity of targeting CD47 and PD-L1 in a bispecific format was evaluated in various syngeneic murine tumor models. In the CT26 tumor model with the same antibody doses between groups, mBisAb showed superior tumor growth inhibition than either αmCD47 or αmPD-L1 antibody alone (figure 3A). In addition, an Fc-null version of mBisAb (with inactive mouse IgG2a) exhibited reduced tumor growth inhibition compared with mBisAb with wild-type Fc, suggesting that IgG effector function plays a role in the mBisAb-mediated therapeutic effect (figure 3A). Furthermore, with the same antibody doses between groups the survival rate (75%) of the mBisAb-treated cohort was significantly improved compared with the cohorts treated with αmCD47 (12.5%), αmPD-L1 (12.5%) or their combination (62.5%) (figure 3B), demonstrating that the bispecific approach may provide a more favorable survival benefit. Notably, the administration of mBisAb was tolerated in mice throughout the study with no impact on body weight (figure 3C). Meanwhile, with the same antibody dose, αmCD47 single agent or its combination with αmPD-L1 resulted in transient but significant body weight loss. To further elucidate the potential impact on hematology mediated by CD47-targeting therapeutics, as has been previously described in preclinical models and the clinic,3 12 the blood from each cohort was harvested after the second dose when the transient body weight loss appeared. The number of mouse RBCs was significantly reduced in the cohorts treated with αmCD47 alone and in combination with αmPD-L1 (figure 3D). At the same time, there was no significant difference in the mBisAb-treated group compared with isotype control, indicating that the bispecific targeting approach enhances antitumor efficacy and reduces potential toxicity effects. Single-agent antitumor activity was also demonstrated in B16F10, an immunologically cold tumor model resistant to anti-PD-(L)1 treatment (figure 3E).8 15 Furthermore, the mBisAb resulted in a robust dose-dependent efficacy in the MC38 tumor model (figure 3F).
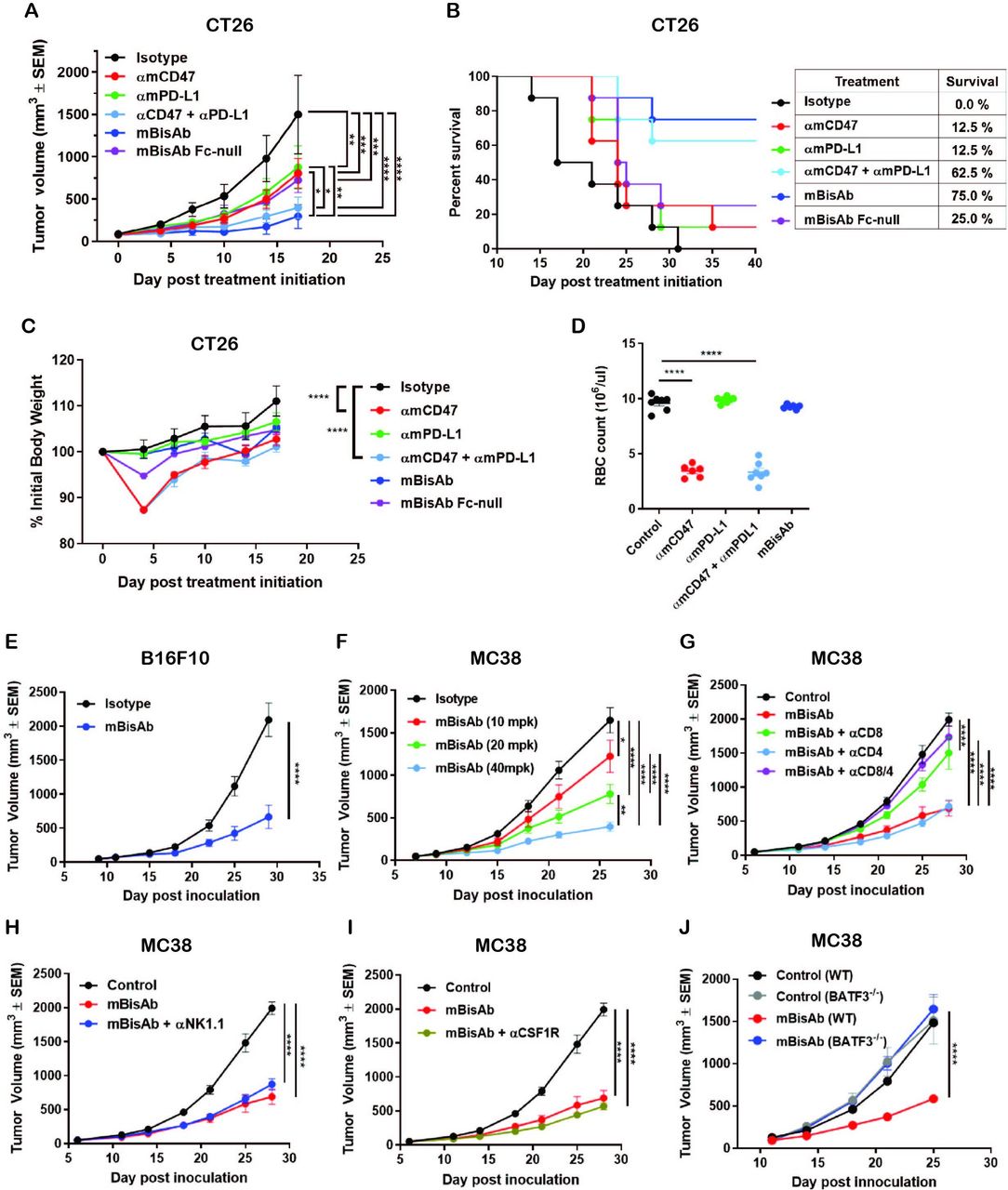
The therapeutic effect of αCD47/PD-L1 mouse surrogates in syngeneic tumor models. (A–C) Antitumor efficacy, survival and body weight post treatment of indicated antibodies in CT26 tumor model. BALB/c mice bearing subcutaneous CT26 tumors were dosed IP with 5 mg/kg of mIgG2a isotype, 5 mg/kg of αmCD47, 5 mg/kg of αmPD-L1, the combination of 5 mg/kg of αmCD47 and 5 mg/kg of αmPD-L1, or 5 mg/kg of mBisAb on days 0, 3, and 7 post-treatment initiation (n=7–8 mice/group). (A) Tumor growth; significance was calculated by two-way ANOVA with Tukey’s multiple comparison test. (B) Kaplan-Meier survival curves post-treatment initiation. (C) Percent of initial body weight reported as a mean±SEM; significance was calculated by two-way ANOVA with Tukey’s multiple comparison test compared with isotype control. (D) BALB/c mice were dosed IP on day 0 and 3 with the same concentration in (A–C). Red blood cells from whole blood on day 5 were counted (n=6–7 mice/group). Significance was calculated by ordinary one-way ANOVA compared with control. (E) Antitumor efficacy in B16F10 tumor model. C57BL/6 mice bearing subcutaneous B16F10 tumors were treated on 9 days post-inoculation with isotype control or mBisAb three times a week for 3 weeks (n=10 mice/group). Significance was calculated by two-way ANOVA with Sidak’s multiple comparison test. (F) Dose-dependent antitumor efficacy in MC38 tumor model. C57BL/6 mice bearing subcutaneous MC38 tumors were treated 7 days postinoculation and dosed IP every 3–4 days for 3 weeks with 10, 20, and 40 mg/kg of mBisAb (n=10 mice/group). Significance was calculated by two-way ANOVA with Tukey’s multiple comparison test. (G–I) Antitumor efficacy following mBisAb treatment and immune cell depletion in MC38 model. MC38-bearing mice were dosed with vehicle (control) or mBisAb every 3–4 days for a total of six doses. Depletion antibodies were administrated IP 1 day prior to treatment with mBisAb and dosed three times a week throughout the study. (n=8–10 mice/group). Significance was calculated by two-way ANOVA with Tukey’s multiple comparison test. (J) C57BL/6 wildtype (WT) and Batf3-/- mice bearing subcutaneous MC38 tumor were dosed IP every 3–4 days for 3 weeks with mBisAb (n=8 mice/group). Significance was calculated by two-way ANOVA with sidak’s multiple comparison test. Asterisks indicate statistical significance (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). ANOVA, analysis of variance; αmPD-L1, anti-mouse PD-L1 antibody; αmCD47, anti-mouse CD47 antibody; mBisAb, αCD47/PD-L1 mouse surrogate; IP, intraperitoneally.
To further investigate the mechanism underlying mBisAb-mediated antitumor activity, a series of antibody-based immune cell depletion studies were performed in MC38 tumor-bearing mice. We found that depletion of CD8+, but not CD4+ T cells, abrogated antitumor efficacy of mBisAb (figure 3G). In contrast, depletion of either natural killer (NK) cells or macrophages alone did not impact antitumor efficacy (figure 3H,I). Interestingly, the mBisAb-mediated tumor growth inhibition was also abolished in Batf3-/- mice (figure 3J). Given that BATF3 has been shown to intrinsically regulate the generation of CD8 T cell memory in the context of infection,16 we cannot rule out the influence of BATF3 deficiency on T cells in this model. However, since targeting the CD47 axis depends on a well-established role for DCs in mediating antitumor efficacy,17 18 it is likely that the therapeutic effect of the bispecific involves both CD8+ T cells and DCs.
αCD47/PD-L1 BisAb treatment induces a robust systemic CD8+ T cell response and expansion of the intratumoral CD8+ T cell pool
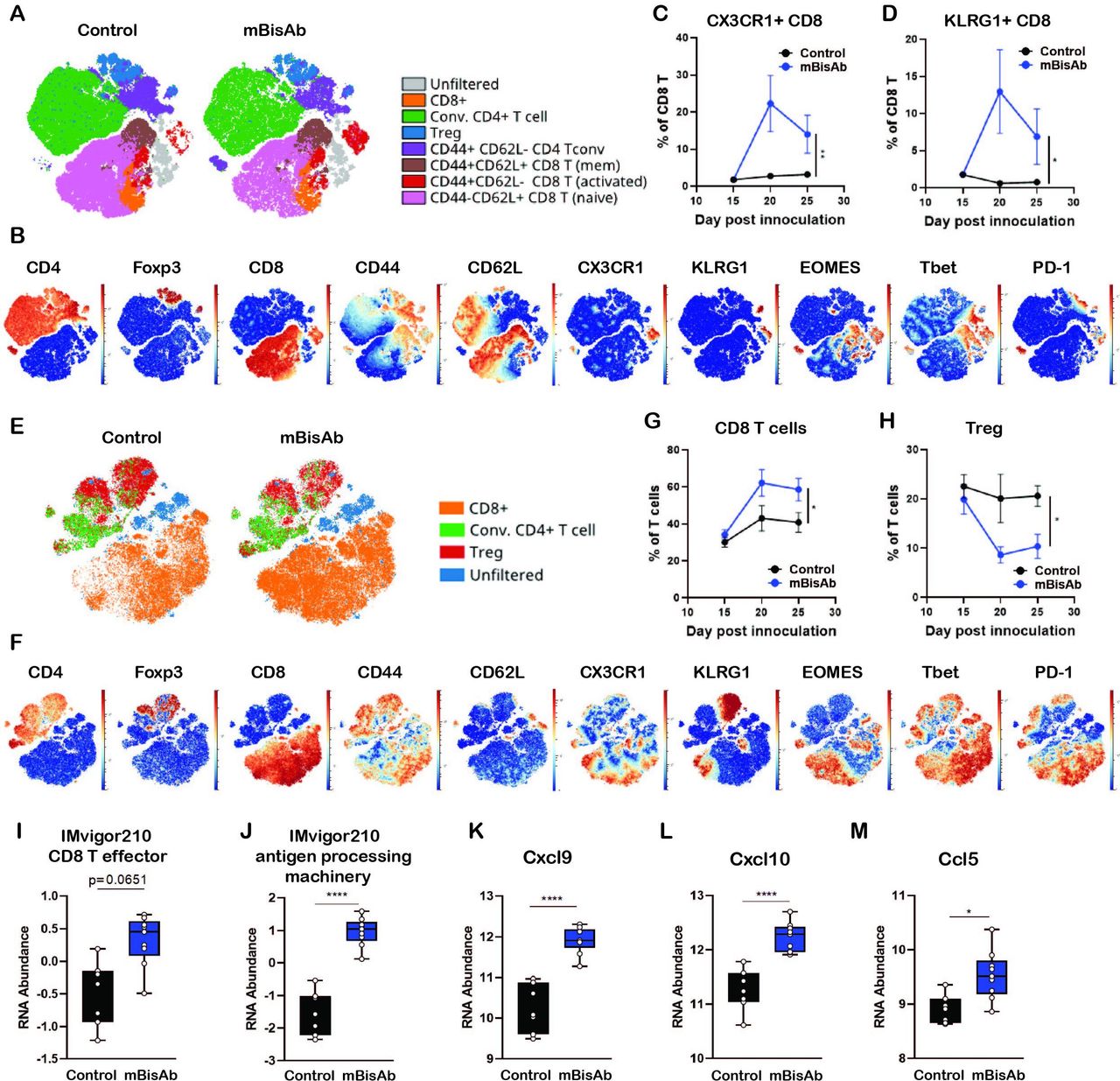
We next investigated the impact of mBisAb treatment on individual immune subsets by performing CyTOF analysis. MC38-bearing mice were treated with isotype (control) or mBisAb every 3–4 days starting at day 10 post-tumor implantation, and tumors and spleens were harvested at day 15, 20, or 25. Intratumoral leucocyte populations were first identified using common lineage-defining markers (online supplemental figure 2A-D),19 and the relative representation of these populations was determined over time (online supplemental figure 2E). We noted an increase in the frequency of CD8 T cells among total CD45+ cells in the tumors of mBisAb-treated mice (online supplemental figure 2E). Within the myeloid compartment, only modest changes in expression of markers typically associated with activation state were observed after mBisAb treatment, except for the apparent transient loss of CX3CR1 and Fc receptor expression (online supplemental figure 2F, compare day 15, 20, and 25). We also observed the same trend for PD-L1 staining in mBisAb-treated tumors and spleens at these time points, which may reflect competition for epitope binding in the presence of mBisAb.
To further understand which T cell compartments are influenced following mBisAb treatment (figure 3G), we generated viSNEs exclusively on T cells (live CD45+CD19−NK1.1−CD3+) from either the spleens (figure 4A–D) or tumors (figure 4E–H) of mBisAb and control-treated mice at day 20 post-tumor implantation, the peak of mBisAb-mediated intratumoral T cell expansion (figure 4G). Compared with control-treated mice, there was a clear enrichment in CD8+ T cells with an activated phenotype (CD44hi, CD62Llo, CX3CR1+, KLRG1+, T-bet+, Eomes+) in the spleens of mBisAb-treated mice (figure 4A,B). We noted a proportional increase in CD8+ T cells expressing KLRG1 and CX3CR1, markers associated with effector T cell responses, among total CD8+ T cells in the spleen of mBisAb-treated mice beginning at day 20 post-implantation, which remained elevated at day 25 (figure 4C,D). We also noted an increase in a population of activated conventional CD4+ T cells (FoxP3−CD44hiCD62Llo) expressing high levels of CX3CR1 (figure 4A,B, online supplemental figure 3A). We further validated these findings using flow cytometry, and found a significant increase in the numbers of activated proliferating (CD44+Ki67+) CD8+ T cells in the spleen on mBisAb treatment, which was not observed on αmCD47 or αmPD-L1 treatment (online supplemental figure 3B). Notably, the frequency of CD103+ cells within cDC1 was also increased in the spleen of mBisAb-treated mice (online supplemental figure 3C). We also found elevated levels of several pro-inflammatory mediators in the serum of mBisAb-treated mice, including IL-1β, IL-12, and IL-18, key cytokines that support CD8+ T cell priming and activation, as well as the key T cell effector cytokine IFNγ (online supplemental table 2). These data suggest that mBisAb promotes increased accumulation of migratory cDC1s in the spleen and a robust systemic T cell response, increasing proinflammatory and effector cytokines systemically. Furthermore, the importance of these increases in migratory cDC1 and effector CD8 T cells induced by mBisAb treatment is highlighted by the loss of antitumor efficacy in the absence of CD8 T cells or Batf3-dependent DCs during mBisAb treatment (figure 3G,J).
Supplemental material
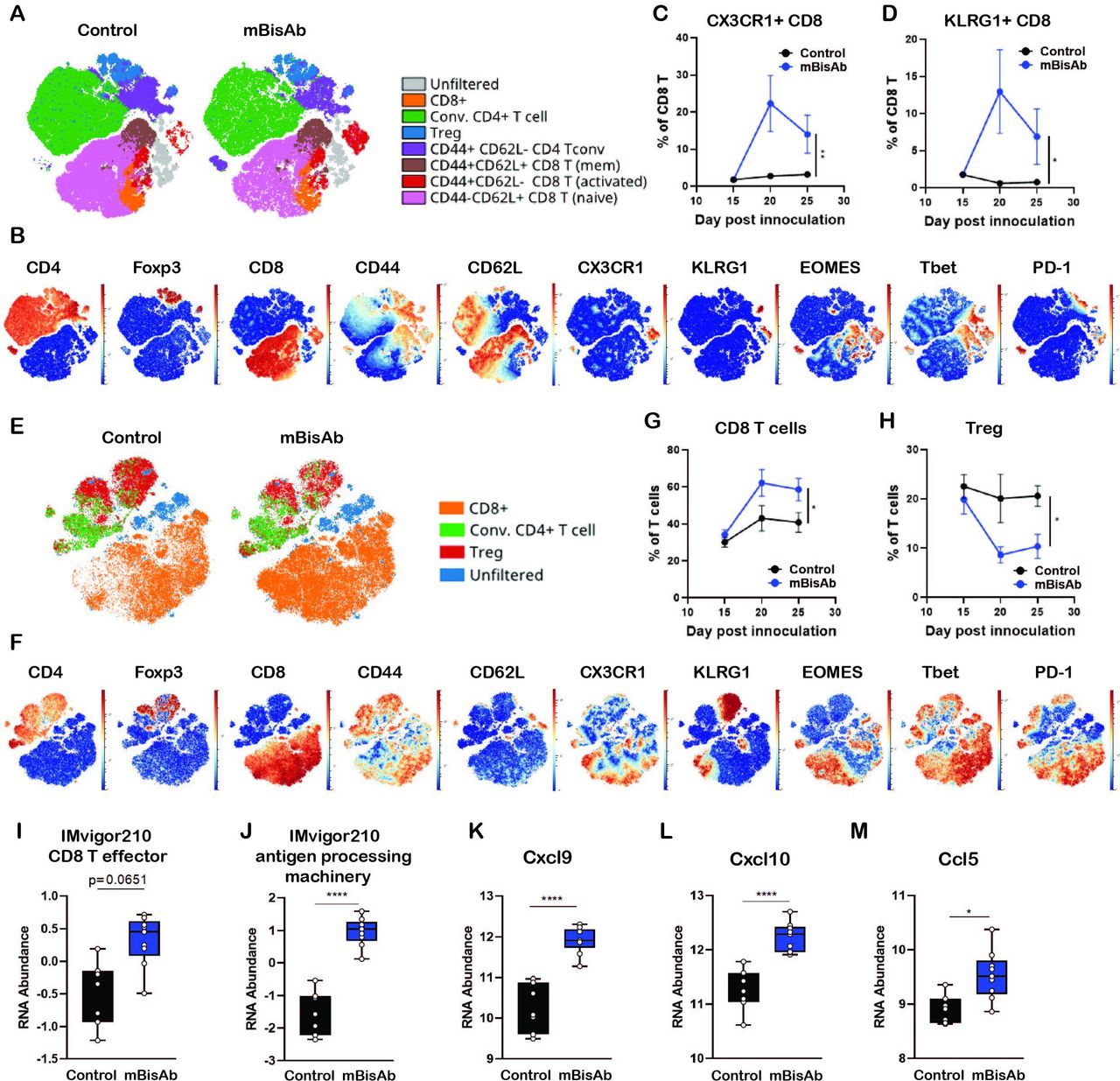
Immunopharmacodynamics of αCD47/PD-L1 mouse surrogate treatment by CyTOF and Nanostring (A–F) MC38-bearing mice were treated with isotype (control) or mBisAb every 3–4 days starting at day 10 post-tumor implantation, and tumors and spleens were harvested at day 15, 20, or 25 and prepared for analysis by CyTOF (n=5 mice/group). (A) ViSNE analysis of total T cells from the spleen of control (left) or mBisAb (right)-treated mice at day 20 post-tumor implantation. Indicated subsets were defined by biaxial gating and position of cells in each subset on the viSNE is represented by a unique color. (B) Expression of indicated markers among total splenic T cells, with each cell in the viSNE colored according to its MMI (mean marker intensity) value of the indicated marker. (C, D) Frequency of CX3CR1+ (C) or KLRG1+ (D) cells among total CD8+ T cells in the spleens of control (black) or mBisAb-treated (blue) mice at the indicated time points. *P<0.05, **p<0.01, Mixed-effects analysis. (E) ViSNE analysis of total T cells from tumors of control or mBisAb-treated mice at day 20 post-tumor implantation. Indicated subsets were defined by biaxial gating and position of cells in each subset on the viSNE is represented by a unique color. (F) Expression of indicated markers among total intratumoral T cells, with each cell in the viSNE colored according to its MMI value of the indicated marker (G, H) Frequency of CD8+ T cells (G) or Tregs (H) among total T cells in control (black) or mBisAb-treated (blue) tumors at the indicated timepoints. *P<0.05, **p<0.01, Mixed-effects analysis. (I–M) MC38-bearing mice were treated with control or mBisAb every 3–4 days starting at day 10 post-tumor implantation. Tumors were harvested at day 20 and prepared for gene expression analysis by Nanostring. Relative RNA abundance of effector CD8+ T cell signature (I) and antigen processing machinery in IMvigor 210 dataset (J). Relative RNA abundance of cxcl9 (K), cxcl10 (L), and ccl5 (M). *P<0.05, ****p<0.0001, Ordinary one-way ANOVA with tukey’s multiple comparison test. ANOVA, analysis of variance; CyTOF, cytometry by time of flight.
Among intratumoral T cells, the viSNE analysis revealed a shift toward a higher proportion of CD8+ T cells and a relatively lower proportion of Tregs on mBisAb treatment (figure 4E,F). We further validated these findings by flow cytometry and found that absolute numbers of CD8+ T cells within the tumor were significantly increased by mBisAb treatment compared with control, as well as αmCD47 or αmPD-L1 treatment (online supplemental figure 3D). Although there was a consistent decrease in the proportion of Tregs among T cells in mBisAb-treated tumors, the absolute number of Tregs remained unchanged between mBisAb, αmCD47, αmPD-L1, and control-treated tumors (figure 4H, online supplemental figure 3E). However, this resulted in a significant increase in the CD8+ T cell/Treg ratio in mBisAb-treated but not αmCD47 or αmPD-L1-treated tumors, a measure that has been associated with an effective antitumor response and favorable prognosis (online supplemental figure 3E).20 21 To gain a greater mechanistic understanding of how mBisAb bolstered the intratumoral CD8+ T cell response, we performed comparative gene expression analysis on MC38 tumor tissues of control or mBisAb-treated mice on day 20 post-tumor implantation by Nanostring. We found that mBisAb-treated tumors had a higher representation of gene expression signatures related to CD8+ T cell effector function and antigen processing machinery (figure 4I,J).22 The levels of crucial T cell-recruiting chemokines Cxcl9, Cxcl10, and Ccl5, were also elevated (figure 4K–M, online supplemental table 3) in mBisAb-treated tumors. These data suggest that the mBisAb could augment intratumoral CD8+ T cell responses by enhancing the recruitment of CD8+ T cells into the tumor and enhancing the presentation of tumor antigens.
Supplemental material
αCD47/PD-L1 BisAb treatment reprograms myeloid populations and drives innate activation in the TME
In addition to the enrichment of gene signatures related to T cell activity, we identified many enriched genes in pathways related to innate immune activation in mBisAb-treated tumors. These include type I IFN signaling, pattern recognition receptor (PRR)-mediated induction of IFNα pathways, and FcγR-dependent phagocytosis (figure 5A,B, online supplemental table 3). Using pathway correlation network analysis, we found that several pathways related to innate immune sensing, including toll-like receptor-mediated signaling, were specifically enriched among genes upregulated following mBisAb treatment compared with the combination treatment of αmCD47 and αmPD-L1 (online supplemental figure 4A). Thus, the bispecific targeting approach has distinct impacts on the TME that cannot be recapitulated by blocking CD47 and PD-L1 via separate antibodies.
Supplemental material
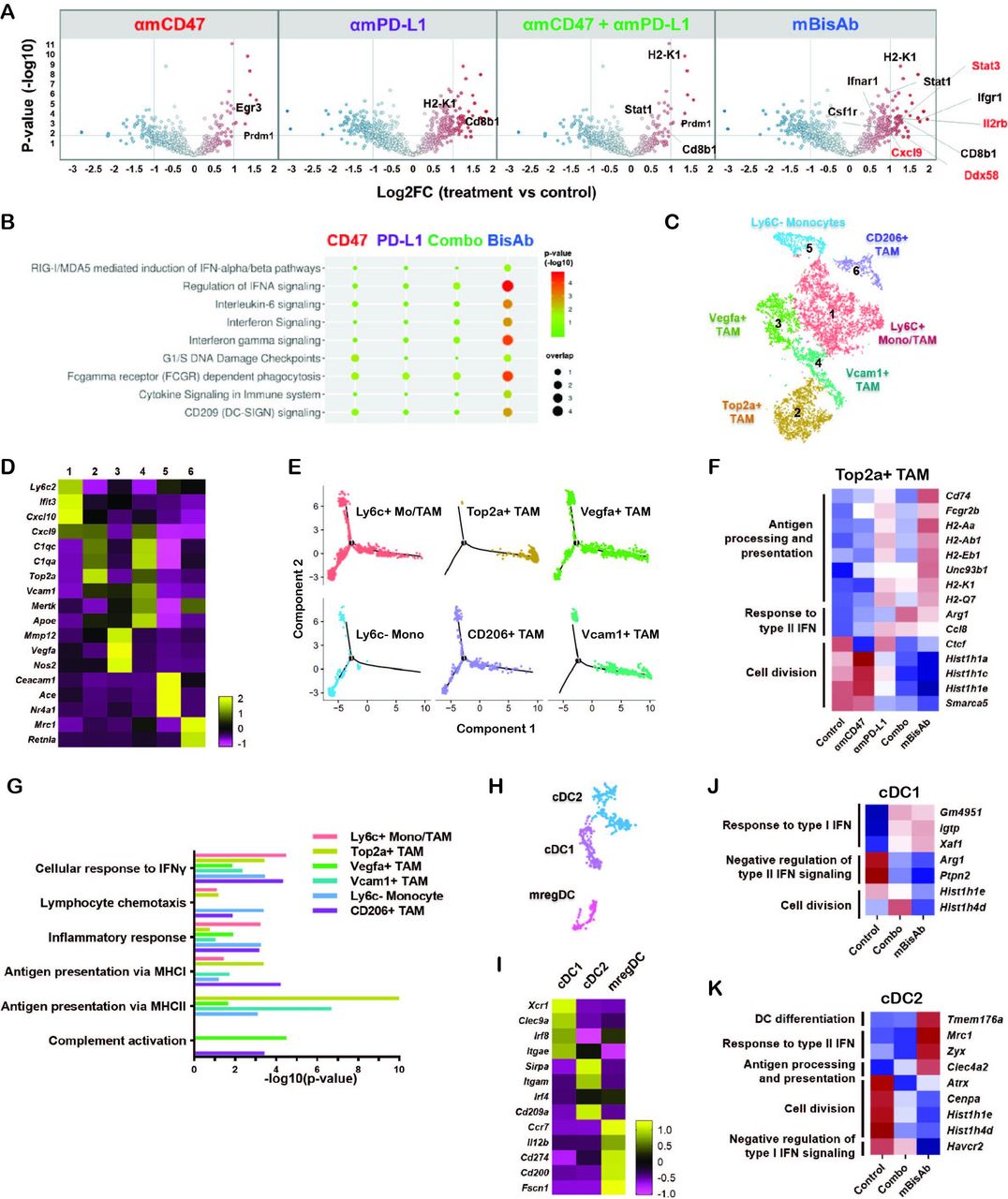
αCD47/PD-L1 mouse surrogate treatment reprograms myeloid populations and drives innate activation in tumor microenvironment (A, B) MC38-bearing mice were treated with the indicated antibodies every 3–4 days starting at day 10 post-tumor implantation. Tumors were harvested at day 20 and prepared for gene expression analysis by Nanostring. Volcano plots show log2 fold change of gene expression in the indicated treatment groups with DEGs compared with the control group (A). REACTOME pathway analysis in the indicated treatment groups compared with the control group (B). (C–K) MC38 tumor-bearing mice were treated with the same condition in (A) and tumor tissues were harvested for gene expression analysis using scRNA-seq. (C) tSNE analysis of intratumoral CD45+ cells from all treatment conditions indicating six distinct clusters of monocyte/macrophage cells. (D) Heatmap displaying the average expressions of the selected DEGs for each cluster of monocyte/macrophage cells relative to other clusters. (E) Single-cell trajectory analysis by Monocle2 of the indicated populations. (F) Relative expression across treatment groups for selected genes that are differentially expressed among Cluster 2 Top2a+ TAM in mBisAb-treated vs control-treated tumors. (G) Bar graph showing selected hits from GO pathway analysis performed on genes upregulated in myeloid clusters from mBisAb-treated tumors relative to the controls. (H) tSNE analysis of intratumoral CD45+ cells from across all treatment conditions, cropped on 3 clusters of DCs. (I) Heatmap displaying the expression of the selected DEGs for each cluster of DC relative to other clusters. (J–K) Relative expression across treatment groups for selected genes that are differentially expressed in mBisAb-treated vs control-treated tumor DC clusters. DC, dendritic cell; DEG, differentially expressed gene; GO, gene ontology; IFN, interferon; MHC, major histocompatibility complex; scRNA-seq, single-cell RNA sequencing; TAM, tumor-associated macrophage; tSNE, t-distributed stochastic neighbor embedding.
To further elucidate the cellular source of the transcriptional changes in the TME, we next performed single-cell RNA-seq (scRNA-seq) analysis of MC38 tumors following treatment with mBisAb, αmCD47 alone, αmPD-L1 alone, or the combination of αmPD-L1 and αmCD47. The cell-type identity of each cluster was defined by the expression of lineage markers (online supplemental figure 4B,C).19 While most treatment conditions minimally impacted the relative distribution of these populations, we noted an increased frequency of T cells following mBisAb treatment (online supplemental figure 4D). Unsupervised clustering identified six monocyte and macrophage populations (figure 5C), for which differential gene expression analysis revealed characteristic gene signatures that distinguished these clusters (figure 5D, online supplemental figure 5A, table 4). Cluster 5 cells comprised non-classical and classical monocyte populations, while the other clusters likely represent monocyte-derived cells, supported by trajectory analysis (figure 5E). Cluster 1 cells exhibited coexpression of immature monocyte genes Ly6c2 and Ccr2 and the maturation markers Itgax and Cx3cr1.23 Cluster 2 and 4 were identified as tumor-associated macrophage (TAM) populations, characterized by markers for mature macrophages (Vcam1, Mertk, Apoe) and proliferation (Top2a, Mki67). Cluster 3 was distinctly enriched for angiogenic and inflammatory markers (Vegfa, Nos2, Mmp12). Cluster 6 cells exhibited expression of markers typically associated with immunosuppression (Mrc1, Cd200r1). While there were no significant changes in the relative frequency of myeloid clusters following treatment, distinct gene expression changes within these clusters were observed (figure 5F, online supplemental figure 5B–D). Notably, following mBisAb treatment, TAM populations exhibited an increase in the relative expression of genes associated with antigen processing and presentation, concomitant with a decrease in gene expression typically associated with cell cycle and proliferation (figure 5F, online supplemental figure 5B–D, table 4). Gene ontology (GO) pathway analysis comparing mBisAb versus control-treated tumors further illustrated upregulation of genes associated with antigen presentation, response to IFN, and complement activation in multiple myeloid populations (figure 5G, online supplemental figure 5E–J, table 4).
Supplemental material
Unbiased clustering also revealed three subsets of conventional DCs (figure 5H) with gene expression profiles corresponding to cDC1, cDC2, and the recently described mregDC24 populations (figure 5I, online supplemental table 4). Interestingly, mBisAb treatment-induced gene expression changes were similar to those induced by combination treatment of αmCD47 and αmPD-L1 in cDC1 (figure 5J). In contrast, an increase in the relative expression of genes associated with DC differentiation, response to type II IFN, and antigen processing and presentation was observed only in mBisAb-treated cDC2 subsets (figure 5K, online supplemental figure 5K, table 4). Of note, unlike cDC1 cells, cDC2 subsets reportedly coexpress PD-L1 and SIRPα,25 the cognate ligand for CD47, suggesting that this population may be directly impacted by mBisAb therapy. We also noted that compared with other treatments, mBisAb administration induced gene expression changes related mainly to the regulation of translation within the mregDC subset (online supplemental figure 5K).
αCD47/PD-L1 BisAb treatment increases the frequency of stem-like progenitor and effector CD8+ T cell subsets in the tumor and promotes the differentiation of progenitor CD8+ T cells to an effector-like state
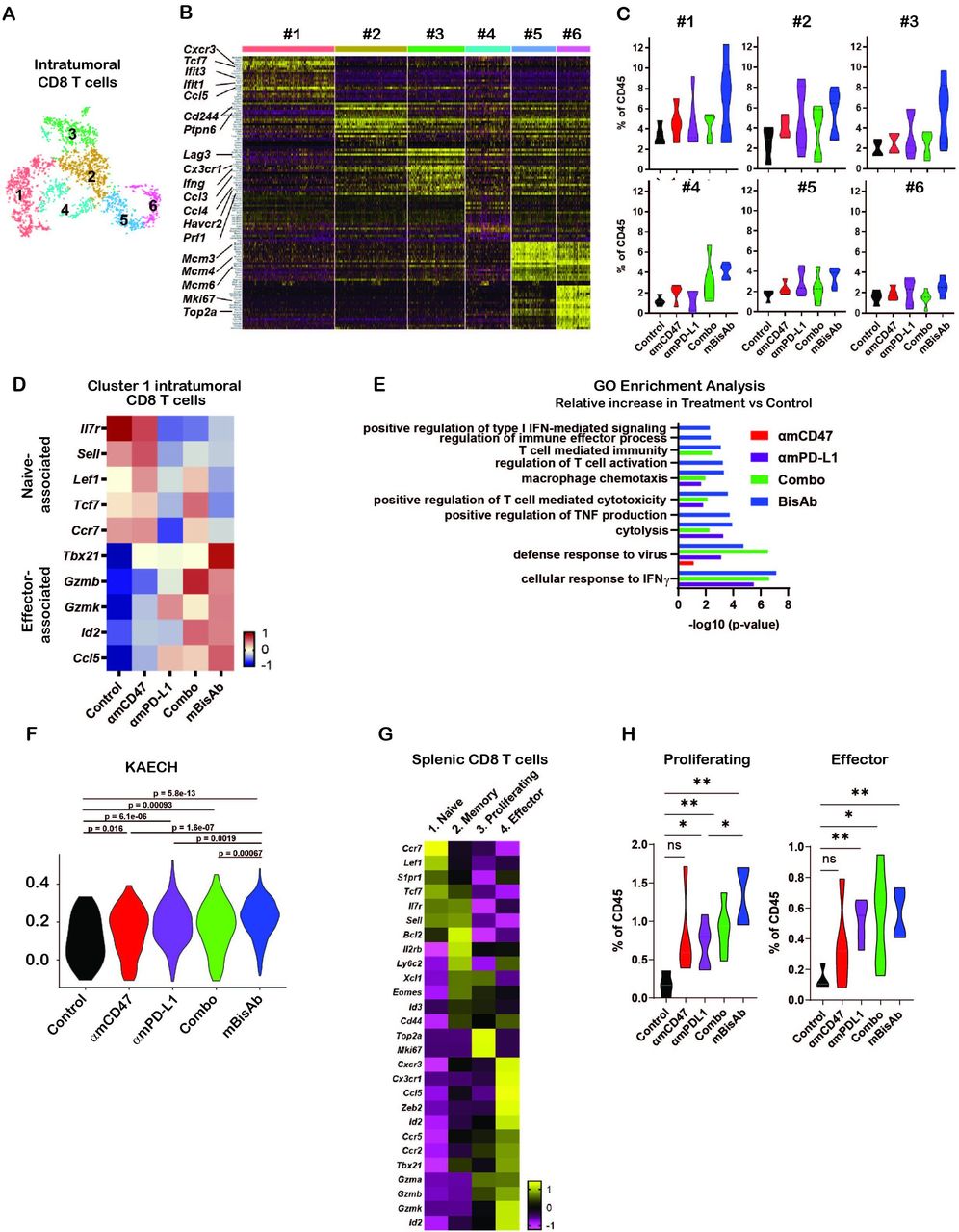
Consistent with our flow cytometry data, we observed significant increases in intratumoral CD8+ T cell populations upon mBisAb treatment by scRNA-seq (online supplemental figure 4D). We next leveraged our scRNA-seq dataset to further understand how mBisAb treatment impacted CD8+ T cells within the TME. We identified six distinct clusters of intratumoral CD8+ T cells (figure 6A). To better understand how these clusters were distinguished from one another, we determined genes that were differentially expressed by each cluster relative to all other intratumoral CD8 T cell clusters (figure 6A,B, online supplemental figure 5). Several genes more highly expressed by CD8+ T cell cluster 1, including the transcription factor Tcf7 and the chemokine receptor Cxcr3, are characteristic of the recently characterized stem-like progenitor CD8+ T cell population. This population has been shown to play a critical role in sustaining an effective antitumor T cell response and is correlated with improved responsiveness to checkpoint blockade therapy.26–28 CD8+ T cell cluster 1 cells also had a lower expression of genes typically associated with T cell exhaustion, including the transcription factor Tox and the inhibitory receptors Pdcd1 (encoding PD-1), Lag3, and Havcr2 (encoding Tim-3), while CD8+ T cell clusters 2 and 3 had relatively high expression of exhaustion-associated genes (figure 6B, online supplemental figure 6A). Notably, CD8+ T cell cluster 3 also had higher expression of genes associated with CD8+ T cell effector functions, including Ifng and Prf1 (encoding perforin), as well as the chemokines Ccl4 and Ccl3, which have been shown to play a role in promoting DC and macrophage recruitment, maturation, and activation.29 30 In addition, CD8+ T cell cluster 3 also had a higher expression of Cx3cr1 (figure 6B, online supplemental figure 6A). This marker has been shown to mark a subset of T cells in chronic viral infection with high effector functions distinct from terminally exhausted cells.31 32 It has also been correlated with a positive response to checkpoint blockade therapy.33 34
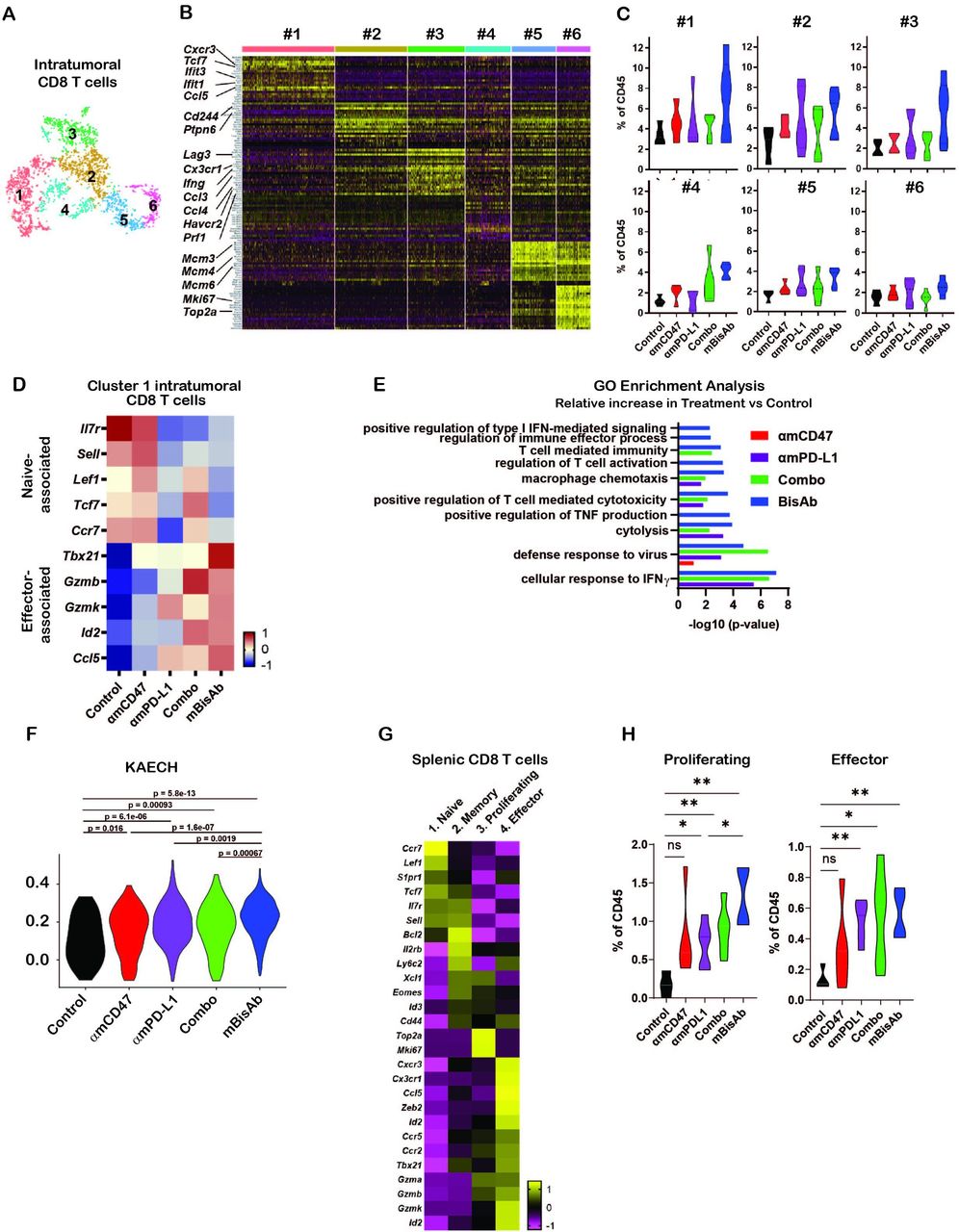
Impact of αCD47/PD-L1 mouse surrogate treatment on intratumoral and splenic T cell compartments (A–K) scRNA-seq samples in figure 5C–K were analysed for CD8+ T cells. (A) tSNE analysis of intratumoral CD45+ cells from across all treatment conditions, cropped on CD8+ T cells, showing six distinct clusters of intratumoral CD8+ T cells. (B) Heatmap displaying the expression of the top 20 DEGs for each cluster of intratumoral CD8+ T cells relative to all other CD8+ T cell clusters. Each column represents an individual cell in the indicated cluster, and each row represents an individual gene. (C) Frequency of each intratumoral CD8+ T cell cluster among total CD45+ cells in each treatment condition. (D) Relative average expression of selected genes that are differentially expressed among cluster 1 CD8+ T cells in mBisAb-treated versus control-treated tumors, shown across all treatment conditions. (E) Selected hits from TopGO pathway analysis performed on genes differentially expressed by cluster 1 CD8+ T cells from mBisAb-treated tumors relative to controls, with -log(10)p value for each pathway among genes differentially expressed by αmCD47, αmPD-L1, or combination-treated tumors relative to controls shown for comparison. No bar indicates that pathway was not significant for the given gene set. (F) Violin plots depicting the relative representation of the KAECH_NAIVE_VS_DAY8_EFF_CD8_TCELL_DN geneset in cluster 1 intratumoral CD8+ T cells across treatment conditions. (G) Heatmap displaying the average expression of selected genes that are differentially expressed among each splenic CD8+ T cell clusters relative to all other CD8+ T cell clusters. (H) Frequency of proliferating and effector splenic CD8+ T cell clusters among total CD45+ cells in spleens from control, αmCD47, αmPD-L1, combination, and mBisAb-treated mice. *P<0.05; **P<0.01, Student’s unpaired t-test. GO, gene ontology; IFN, interferon; ns, not significant; TNF, tumor necrosis factor.
In contrast, CD8+ T cell cluster 2 had a relatively higher expression of Cd244, a cell surface marker expressed highly on terminally exhausted cells,32 and Ptpn6 (encoding SHP-1) (figure 6B, online supplemental figure 6A). This protein phosphatase inhibits TCR signaling and is a crucial mediator of inhibitory signaling downstream of checkpoint receptors, including PD-1 and B- and T-lymphocyte attenuator.35 Together, these gene expression patterns suggest that while cluster 2 cells may be terminally exhausted, cluster 3 cells retain the potential for high effector function. Notably, mBisAb treatment specifically increased the frequency of cluster 1 stem-like progenitor and cluster 3 effector-like CD8+ T cells to a greater extent than αCD47 and αPD-L1 monotherapies or combination therapies (figure 6C).
We validated the mBisAb-induced increase in the intratumoral stem-like progenitor CD8+ T cell population using flow cytometry (online supplemental figure 6B,C). We found a robust increase in both the frequency of the stem-like progenitor (Tcf1hiLag3lo) population among antigen-experienced (CD44hiCD62Llo) intratumoral CD8+ T cells (online supplemental figure 6B) and in the total numbers of stem-like progenitor CD8+ T cells within the tumors of mBisAb-treated animals (online supplemental figure 6C). To further investigate the impact of mBisAb treatment on this population, we identified genes differentially expressed by cluster 1 CD8+ T cells in the tumors of control versus mBisAb-treated, αmCD47-treated, αmPD-L1-treated, or αmCD47 and αmPD-L1 combination-treated mice. Specific genes downregulated by CD8+ T cell cluster 1 following mBisAb treatment included genes associated with a naïve T cell state, such as the transcription factors Tcf7, Lef1, and Bach2 (figure 6D). In contrast, genes upregulated by CD8+ T cell cluster 1 following mBisAb treatment included genes associated with effector CD8 T cell function and fate such as granzymes (Gzmb, Gzmk), immune cell-recruiting chemokines (Ccl5), and transcription factors Tbx21 and Id2 (figure 6D, online supplemental table 6).
Supplemental material
We next performed GO enrichment analysis on these gene sets (online supplemental table 6) and found that following αmPD-L1, αmPD-L1 and αmCD47-combination, and mBisAb, but not αmCD47, treatment, genes more highly expressed by intratumoral CD8+ T cell cluster 1 were enriched in several pathways related to T cell differentiation and effector function, including cellular response to IFNγ, cytolysis, and T cell-mediated cytotoxicity (figure 6E, online supplemental table 6). However, many of these pathways were more significantly enriched following mBisAb treatment compared with other treatment conditions, and mBisAb treatment also resulted in an enrichment of additional pathways, such as regulation of T cell activation, positive regulation of TNF production, and positive regulation of type I IFN-mediated signaling, which were not enriched in other treatment conditions (figure 6E, online supplemental table 6). We also performed gene set enrichment analysis on genes differentially expressed by cluster 1 CD8+ T cells in the tumors of control vs mBisAb-treated mice. We found enrichment in many pathways associated with differentiation of CD8+ T cells from a naïve to effector state (online supplemental figure 6D, table 6). For example, the gene set most highly represented among genes upregulated by cluster 1 cells following mBisAb treatment was more highly expressed by CD8+ T cells at the peak of acute viral infection than the naïve state (online supplemental figure 6B).36 We compared the collective representation of this gene set within cluster 1 CD8+ T cells across treatment conditions, and we observed the highest representation among cluster 1 CD8+ T cells following mBisAb treatment (figure 6F). Taken together, these findings suggest that mBisAb treatment not only expands the intratumoral stem-like progenitor CD8+ T cell population but also expands a population with high effector potential, perhaps by enhancing the differentiation of the stem-like progenitor population to an effector-like state. Moreover, treatment with mBisAb induced these effects more robustly than αmCD47 or αmPD-L1 monotherapy, as well as their combination.
We also leveraged our scRNA-seq dataset to investigate how mBisAb treatment impacted the splenic CD8+ T cell population. We identified four distinct clusters of CD8+ T cells within the spleen (online supplemental figure 6E). To elucidate the identity of each cluster, we determined the genes that were differentially expressed by each cluster relative to all other splenic CD8+ T cell clusters (figure 6G, online supplemental figure 6E, table 7). Splenic cluster 1 CD8+ T cells exhibited relatively higher expression of genes associated with naïve T cells, including Lef1, Tcf7, Sell, and Il7r. In contrast, cluster 2 expressed intermediate levels of these genes in addition to relatively higher expression of genes associated with a memory-like fate, including Cd44, Ly6c2, Eomes, and Il2rb. Splenic cluster 3 CD8+ T cells exhibited high expression of genes associated with cell cycle, such as Mki67 and Top2a, suggesting that cells in this cluster were actively proliferating. Splenic cluster 4 CD8+ T cells exhibited high expression of genes associated with effector CD8+ T cell function and fate, including key lineage-defining transcription factors (Tbx21, Zeb2, and Id2), effector molecules (Gzma, Gzmb, Gzmk), and chemokine receptors (Ccr5, Cxcr3, Cx3cr1) (figure 6G, online supplemental table 7). We, therefore, defined splenic CD8+ T cell clusters 1, 2, 3, and 4 as naïve, memory, proliferating, and effector, respectively.
Supplemental material
We next investigated how mBisAb treatment impacted the frequencies of these distinct CD8+ T cell populations by comparing the frequency of each cluster among total CD45+ cells in control,αmCD47, αmPD-L1, αmPD-L1 and αmCD47-combination, and mBisAb -treated spleens. We observed that αmPD-L1, αmPD-L1 and αmCD47-combination, and mBisAb, but not αmCD47-treatment, resulted in an increase in the frequency of both proliferating and effector CD8+ T cell clusters relative to control-treated spleens (figure 6H). Notably, mBisAb treatment, but not αmPD-L1 and αmCD47-combination treatment, resulted in a significantly higher increase in proliferating splenic CD8 T cells than PD-L1 treatment (figure 6H). Consistent with our CyTOF and flow cytometry data (figure 4A–D, online supplemental figure 3B), these data reveal that mBisAb treatment induces both a robust systemic, as well as intratumoral, T cell response.
The administration of the αCD47/PD-L1 BisAb modulates both innate and adaptive immunity in cynomolgus monkeys
A non-human primate (NHP) study was conducted to explore the toxicity profile and potential pharmacodynamic markers of the hBisAb. The hBisAb demonstrated comparable binding and activity to cynomolgus monkey CD47 and PD-L1 (online supplemental table 1), making it suitable for in vivo experimentation. NHPs were administrated weekly intravenous doses of vehicle, 10, 30, or 100 mg/kg of hBisAb for 2 weeks. All dose levels were well tolerated during the in-life portion, with no notable clinical signs or changes in food consumption. Time course evaluation of circulating cell populations (standard hematology) following administration of hBisAb revealed minimal non-dose-dependent reductions of circulating RBCs and hemoglobin, and transient modulations of platelets. Absolute cell counts remained in the range of historical controls during the 2-week dosing period, and no changes were considered adverse (figure 7A,B, online supplemental figure 7A). Flow cytometry endpoints supported pharmacological modulation of critical immune effector cells. NHPs following administration of the hBisAb showed significant increases in CD25+CD8+ T cells (figure 7C) and CD69+CD4+ T cells (figure 7D), indicating that hBisAb modulates immune cell populations including T cell activation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Non-human primate study with human αCD47/PD-L1 bispecific antibody (hBisAb) treatment. (A, B) Cynomolgus monkeys received vehicle control and 10, 30, and 100 mg/kg of hBisAb on day 1 and 8 (n=1–3 monkey/group). The peripheral blood was collected at the indicated timepoint for RBC (A) and platelet numbers (B) in response to the treatment. Ordinary one-way ANOVA with Dunnett’s multiple comparisons test. (C, D) Representative flow cytometry plots of CD25+ CD4+ and CD69+ CD8+ T cell populations from NHP peripheral blood on day 8 after receiving vehicle control or 100 mg/kg of hBisAb. (E–H) The NHP PBMCs from peripheral blood were collected with the indicated timepoints after receiving vehicle control or 100 mg/kg of hBisAb on day 1 and 8 for RNA-seq analysis (n=2–3 monkey/group). (E) Selected DEGs associated with innate and adaptive immune pathways in vehicle-treated versus hBisAb-treated samples across all the timepoints during a 2-week period. (F) IPA pathway analysis of the RNA-seq from hBisAb-treated compared with vehicle-treated group on 6 hours post-second dose on day 8. (G, H) LM22 gene signature scores (relative to baseline) of activated DCs and M1 macrophages in vehicle (Veh) vs hBisAb (Ab)-treated group on 0, 6, and 24 hours post-second dose (2D). (I, J) The peripheral blood from cyno received 100 mg/kg of hBisAb and hBisAb2 with reduced CD47 affinity on day 1 and 8 (n=2–3 monkey/group) was collected for RNA-seq analysis. The molecule affinity and enriched IPA pathway scores (I) as well as the selected DEGs (J) expression profiles are listed for the comparison of the two antibodies. 2D, two dimensional; ANOVA, analysis of variance; DC, dendritic cell; DEG, differentially expressed gene; NHP, non-human primate; PBMC, peripheral blood mononuclear cell; RBC, red blood cell.
To further elucidate the immune modulation induced by hBisAb, we also performed gene expression analysis in NHP peripheral blood samples by bulk RNA-seq. Samples were collected at multiple time points, including pre-first/post-first dose (day 1) and pre-second/post-second dose (day 8) of hBisAb and vehicles over 2 weeks (figure 7E). Using principal component analysis (PCA) of the bulk RNA-seq data, the hBisAb and vehicle-treated samples clustered separately (online supplemental figure 7B). Analysis of the top differentially expressed genes (DEGs) between hBisAb and vehicle-treated samples revealed that hBisAb treatment increased the expression of genes associated with type I IFN signaling (STAT1, BATF2, IRF7, RSAD2, IFIT3) and myeloid cell activation (CD80, MX1, TMEM173, DDX58) (figure 7E, online supplemental figure 7D, table 8). Furthermore, many genes related to T cell activation (CD28, ICOS, LAG3, CTLA4) and cell migration (CXCL10, CCR7) were also enriched in the hBisAb cohort (figure 7E, online supplemental figure 7C,D). Indeed, ingenuity pathway analysis (IPA) of the top DEGs identified IFN signaling and PRR-mediated innate immune responses as the most enriched pathways in hBisAb cohorts relative to vehicle controls (figure 7F, online supplemental figure 7E). Using gene correlation network analysis among top upregulated DEGs in hBisAb-treated samples, STAT1 and STAT2 were identified with the most connections as a hub (online supplemental figure 7F). These findings are consistent with preclinical and clinical reports showing that upregulation of STAT1 on immune cells in the tumor is positively correlated with response to αPD-(L)1 therapy in melanoma and urothelial cancer.22 37 38
Supplemental material
To deconvolute which hematopoietic cell types are responsible for the immune-modulating effects of hBisAb treatment, we applied the LM22 gene signature, a specific gene signature that can distinguish between 22 immune cell subsets, to our datasets. Interestingly, among all immune subsets, activated DCs and M1 macrophage phenotypes are the most enriched gene signature in hBisAb-treated samples relative to vehicle (figure 7G,H). These data are consistent with the TME data in mouse models (figure 5) and clinical evidence in patients39 that innate immune modulation, particularly the type I IFN response, renders tumors sensitive to current immune checkpoint inhibitors.
To further elucidate the relative contributions of the CD47 and PD-L1 arms of hBisAb to the enrichment of innate activation pathways observed on treatment, we performed gene expression analysis in samples treated with a version of hBisAb, which has reduced CD47 affinity (hBisAb2) in part of the NHP studies. IPA analysis comparing the two hBisAb with different CD47 affinities revealed a distinct enrichment in pathways related to type I IFN signaling and myeloid cell activation among samples treated with hBisAb relative to hBisAb2 (figure 7I). Consistent with this, hBisAb induced the upregulation of several key innate immune-modulating genes which were not induced by hBisAb2 (figure 7J). Taken together, these findings indicate that the CD47 arm is the primary driver of the robust innate immune activation induced by hBisAb treatment.
Discussion
Mounting evidence has shown that antagonizing the CD47-SIRPα myeloid checkpoint can prevent tumor cell evasion from innate immune surveillance. However, the liability of targeting CD47/SIRPα using a monospecific antibody approach is the hematological antigen sink and toxicity.11 The ubiquitous expression of CD47 on normal cells, especially RBCs, significantly reduces the exposure of therapeutic antibodies on solid tumors overexpressing CD47. In this study, we described a novel affinity-tuned αCD47/PD-L1 BisAb with three key features. First, the design of moderate CD47 and potent PD-L1 affinity for the bispecific improved its selectivity to the TME versus peripheral blood. Second, the bispecific treatment resulted in a more favorable therapeutic window than αCD47, αPD-L1 monotherapy, or their combination in syngeneic tumor models. The administration of αCD47/PD-L1 bispecific antibodies in NHP and murine systems showed minimal anemia but maintained strong innate and adaptive immune responses. Finally, we identified unique myeloid and T cell activation patterns following the bispecific treatment in murine and NHP systems, demonstrating its potent ability to engage both innate and adaptive immune responses for antitumor efficacy.
Our comparative gene expression analysis revealed that distinct from αCD47 and αPD-L1 monotherapies or combination therapies, mBisAb-treated tumor tissues displayed robust upregulation in pathways associated with innate activation, including PRR-mediated induction of IFNα pathways, type I IFN signaling, and FcγR dependent phagocytosis. PRR signaling, including activation of STING by sensing of cytosolic dsDNA, can promote type I IFN and other proinflammatory cytokine production in DCs and TAMs by inducing the nuclear translocation and activity of transcription factors NF-kB and IRF3/7.17 40–44 Indeed, recent research utilizing IFNAR-/- and STING-/- mouse models highlight the necessity of these pathways in mediating CD47 blockade-induced tumor rejection by enhanced innate sensing of tumor cells via cGAS-STING signaling, leading to enhanced cross-priming of CD8 T cells.14 18 Our data indicate that both innate and adaptive responses are necessary for mBisAb efficacy, in concordance with previous work co-targeting the CD47/SIRPα and PD-1/PD-L1 axis in preclinical tumor models.14 45 46 However, how myeloid and T cells are modulated transcriptionally at the single-cell level has not been extensively evaluated in the previous studies. Our scRNA-seq data revealed an upregulation in the expression of genes associated with a response to type I IFN within intratumoral cDC1 cells. cDC2, which coexpress PD-L1 and SIRPα and may be directly impacted by mBisAb therapy, instead displayed an increase in the relative expression of genes associated with antigen processing and presentation. Increased antigen processing and presentation were also observed within mregDCs, a population particularly enriched for PD-L1 expression and associated with an immunosuppressive phenotype,24 suggesting possible reprogramming of this population by mBisAb targeting.
In addition to impacts on DC populations, our scRNA-seq analysis of TAM clusters also revealed a dramatic increase in gene expression associated with antigen processing and presentation and a downregulation of genes associated with cell cycle following mBisAb treatment. Notably, this signature is consistent with exposure to IFNγ47 48 and may in part be explained by the expansion of IFNγ producing CD8+ T cell population following mBisAb treatment. Indeed, TAM repolarization following αPD-(L)1 treatment is mediated mainly by increased IFNγ signaling and required IFNγR expression on TAMs.48 Furthermore, type I IFN sensitizes TAMs to IFNγ through increased STAT1 signaling,49 which gene signature was enriched in mBisAb-treated tumor tissues. Interestingly, the combination of monotherapies similarly induced the relative loss of cell cycle-specific transcripts but did not increase antigen processing and presentation genes, suggesting that mBisAb may be more efficient at targeting or activating these TAM populations. Our data support a model in which BisAb treatment activates innate immunity to stimulate adaptive responses within the TME, which may in turn act to repolarize TAM populations further to support antitumor immunity.
Previous studies have demonstrated enhanced cross-priming and activation of tumor antigen-specific CD8+ T cells by DCs and macrophages following SIRPα-CD47 pathway blockade.17 18 50 However, the impact of SIRPα-CD47 pathway inhibition on the size and distribution of intratumoral CD8+ T cell populations remained unknown. Here, we found that mBisAb treatment induced robust expansion of the intratumoral CD8+ T cell pool in vivo. Notably, populations that have been positively correlated with patient responsiveness to checkpoint blockade therapy, including the Tcf1+ stem-like progenitor26–28 and effector-like CD8+ T cells, were more significantly expanded by mBisAb than αPD-L1 or its combination with αCD47. In addition to the robust expansion of the Tcf1+ stem-like progenitor population, mBisAb also induced its differentiation into an effector-like state. Furthermore, mBisAb treatment resulted in the most significant expansion of an effector-like CD8+ T cell population characterized by high expression of Ifng that was transcriptionally distinct from the terminally exhausted T cell population. Notably, a recent study demonstrated that a signature of activation and differentiation among the intratumoral Tcf1+ stem-like progenitor CD8+ T cell population was a hallmark of patients successfully responding to immune checkpoint blockade (ICB).51 Moreover, signatures related to T cell cytolytic activity and IFNγ signaling, which include genes highly expressed by the effector-like CD8+ T cell population (Ifng, Prf1) that was robustly expanded by mBisAb treatment, have also been correlated with patient responsiveness to ICB therapy.37 39 Taken together, these findings suggest that mBisAb treatment successfully induces an intratumoral CD8+ T cell landscape highly reflective of patients undergoing an effective response to ICB therapy by both expanding the Tcf1+ stem-like progenitor population and promoting its differentiation to an effector-like state.
PD-1/PD-L1 pathway blockade is known to expand the Tcf1+ stem-like progenitor population,26 27 but the exact mechanism by which mBisAb expands this population further remains unclear. However, one likely explanation is that enhanced cross-priming by intratumoral APCs mediated by CD47 blockade17 18 could further bolster the expansion of the Tcf1+ stem-like progenitor population mediated by PD-L1 blockade. Indeed, an increased gene expression signature related to antigen processing and presentation following mBisAb treatment and loss of mBisAb-mediated efficacy in Batf3-/- mice both indicate that DCs may play a vital role in the stem-like progenitor CD8+ T cell expansion. Given the enhanced binding selectivity of the αCD47/PD-L1 BisAb to the TME relative to αCD47, an increase in antibody exposure for sufficient SIRPα-CD47 blockade within the TME could explain the expansion of the Tcf1+ stem-like and effector-like CD8+ T cell populations in response to mBisAb relative to αPD-L1 and its combination with αCD47. A recent study has demonstrated that Tcf1+ stem-like progenitor CD8+ T cells reside primarily within APC-rich niches within the TME.52 Robust activation of APCs by BisAb-mediated tumor-directed SIRPα-CD47 pathway blockade could help create such niches that could support the persistence and/or differentiation of the Tcf1+ stem-like progenitor population further to its expansion by the PD-L1 arm. Taken together, these findings indicate that targeting both innate and adaptive checkpoints using a bispecific approach can synergistically remodel the intratumoral immune landscape to a state that promotes potent antitumor activity. Our findings suggest that this therapeutic approach holds promising potential to improve patient outcomes over current PD-(L)1 and CD47-targeted therapies.
Supplemental material
Data availability statement
All data are available in the main text or the online supplemental materials. The accession number for the mouse and NHP RNA-seq data is GSE177053. Requests for materials should be directed to and will be fulfilled by S-HC, JC-R, or SS-A and the completion of a material transfer agreement with Pfizer.
Ethics statements
Patient consent for publication
Ethics approval
All animal experiments were approved by the Institutional Animal Use and Care Committee of Pfizer and in compliance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Office of Laboratory Animal Welfare.
Acknowledgments
The authors thank Robert Rickert, Premal Patel, and Barbra Sasu for their useful discussions. The authors also thank Cindy Li, Breanna Jung, Pamela Santiago, Sophanna Kong, Parul Gupta, Jatin Narula, Qinghai Peng, Qing Zong, Jianying Wang, Vicki Markiewicz, Guoyun Zhu, Christopher Dillon, Eleanore Hendrickson, Lynn Wang, Nancy Chau, Muamera Zulcic, Rupesh Kanchi Ravi, Xianxian Zheng, Jennifer Kinong, David Schaer, Dirk Zajonc, James Patterson, Chiu Lau-Barre, Matthew Kunicki, Allison Vitsky, Yvonne S. L. Mak, Sindy Liao Chan, Tina Mistry, Colleen L. Brown, Sherman M. Chin, Joanna Ngo, James Apgar, Daniel McDonough, Willie Wang, Tao Sai and Xi Yang for technical assistance or guidance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors S-HC, PKD, JS, SD, JC-R, and SS-A led the design of the molecule and experiments. S-HC, PKD, JS, NK, RL, and JH performed experiments, analysed data, interpret the data, and wrote the manuscript. CW, ZM, TVB, and KCL assisted with design of the molecule, conducting, and interpretation of the experiments. WY, S-HC, NK, and RL assisted with design of the computational methods and/or with interpretation of the analyses. S-HC, JC-R, and SS-A led the design, interpretation, and the editing of the manuscript.
Funding This study was funded by Pfizer.
Competing interests All authors are employees of Pfizer which developed antibodies presented in this manuscript.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.