Article Text

Abstract
Background Adoptive cell therapy (ACT) with tumor-infiltrating lymphocytes (TILs) has shown remarkable results in malignant melanoma (MM), while studies on the potential in other cancer diagnoses are sparse. Further, the prospect of using checkpoint inhibitors (CPIs) to support TIL production and therapy remains to be explored.
Study design TIL-based ACT with CPIs was evaluated in a clinical phase I/II trial. Ipilimumab (3 mg/kg) was administered prior to tumor resection and nivolumab (3 mg/kg, every 2 weeks ×4) in relation to TIL infusion. Preconditioning chemotherapy was given before TIL infusion and followed by low-dose (2 10e6 international units (UI) ×1 subcutaneous for 14 days) interleukin-2 stimulation.
Results Twenty-five patients covering 10 different cancer diagnoses were treated with in vitro expanded TILs. Expansion of TILs was successful in 97% of recruited patients. Five patients had sizeable tumor regressions of 30%–63%, including two confirmed partial responses in patients with head-and-neck cancer and cholangiocarcinoma. Safety and feasibility were comparable to MM trials of ACT with the addition of expected CPI toxicity. In an exploratory analysis, tumor mutational burden and expression of the alpha-integrin CD103 (p=0.025) were associated with increased disease control. In vitro tumor reactivity was seen in both patients with an objective response and was associated with regressions in tumor size (p=0.028).
Conclusion High success rates of TIL expansion were demonstrated across multiple solid cancers. TIL ACTs were found feasible, independent of previous therapy. Tumor regressions after ACT combined with CPIs were demonstrated in several cancer types supported by in vitro antitumor reactivity of the TILs.
Trial registration numbers NCT03296137, and EudraCT No. 2017-002323-25.
- immunotherapy
- adoptive
- lymphocytes
- tumor-infiltrating
Data availability statement
Data are available upon reasonable request. Flow cytometry data and genetic data from the tumor material is avaliable to share upon reasonable request within the national legal framework of data sharing and GDPR.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune therapy has advanced from malignant melanoma (MM) into many cancer diagnoses. Adoptive cell therapy (ACT) with in vitro expanded tumor-infiltrating lymphocytes (TILs) is an immune therapy pioneered within MM that has shown remarkable results.1 2 ACT is currently experiencing a renewed interest in its potential use in other solid cancers due to the successful application of immune therapies such as anti-Programmed cell death protein 1 (PD-1) and anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) checkpoint inhibitors (CPIs), also pioneered in melanoma, in several solid cancers.3–5 The interest in TIL-based ACT is further fueled by case reports of tumor regression after TIL therapy in patients without melanoma6–10 and recent breakthroughs with genetically modified chimeric antigen receptor-based ACT in several hematological cancers.11
The feasibility of in vitro TIL expansion has been repeatedly demonstrated in many solid cancers, including bladder, breast, ovarian, renal cell, head-and-neck, non-small cell lung cancers, prostate cancer, sarcoma, and pancreatic carcinomas.12–19 Less is known about the clinical efficacy and safety, as very few ACT clinical trials in patients without MM have been published. Response rates outside MM have generally been lower, and durable complete responses (CRs) have so far been observed only in two patients with cervix cancer and one patient with breast cancer—the latter in combination with anti-PD-1 blockade.6–9 20 Many non-MM clinical trials are currently in progress, including colorectal, pancreatic, breast, head-and-neck, and lung cancers, but most are based on expanded TILs selected for tumor-antigen reactivity (NCT01174121, NCT03215810, NCT03610490, NCT03801083, and NCT03645928).
The TIL-based ACT regime traditionally consists of three steps: lymphodepleting preconditioning, infusion of in vitro expanded cells, and adjuvant stimulation with interleukin (IL)-2.21 Chemotherapy preconditioning and IL-2 therapy both induce transient but substantial toxicities that in almost all cases require hospitalization and prophylactic therapy.22–24 Furthermore, TIL-based ACT requires tumor tissue available for safe resection and adequate in vitro expansion within a relatively short time period, so the patient is still fit for intensive therapy with lymphodepletion and IL-2. These limitations are well described in MM but not in other solid cancers.
Many recent efforts in experimental immune therapy combine non-redundant therapies to improve the resulting clinical efficacy—most prominently illustrated by anti-PD-1 and anti-CTLA-4 combination therapy.25 The possibility to combine CPI with ACT is an appealing therapeutic approach that remains to be explored. Preclinical models demonstrate that CPIs can benefit the phenotype of TILs,26 27 and clinical trials have suggested a positive association between anti-CTLA-4 therapy and in vitro TIL expansion.28–31 Furthermore, several studies have shown overexpression of PD-1 on in vitro expanded TILs, indicating that PD-1 blockade could be a relevant combination strategy to prevent PD-L1-mediated inactivation in vivo.32–35
The addition of CPIs to the ACT schedule may also reduce the need for high-dose IL-2 dose while still supporting optimal in vitro TIL expansion, thus increasing the overall safety and feasibility of ACT. In this clinical trial, we address the potential of TIL-based ACT supported by CPIs in solid cancers in a wide range of solid cancer types.
Methods and materials
Patients
Eligible patients were recruited and treated at the Department of Oncology, Copenhagen University Hospital, Herlev, Denmark. Main inclusion criteria were progressive metastatic cancer disease without available standard therapy options, >1 cm3 tumor lesion available for safe resection, age of 18–70 years, acceptable performance status, and organ function. Exclusion criteria were brain metastases, chronic infections with hepatitis, HIV or treponema pallidum, and ≥10 mg/day corticosteroids (excluding adrenal substitution therapy with hydrocortisone). The trial is registered with clinicaltrial.gov and clinicaltrialsregister.eu.
Study design
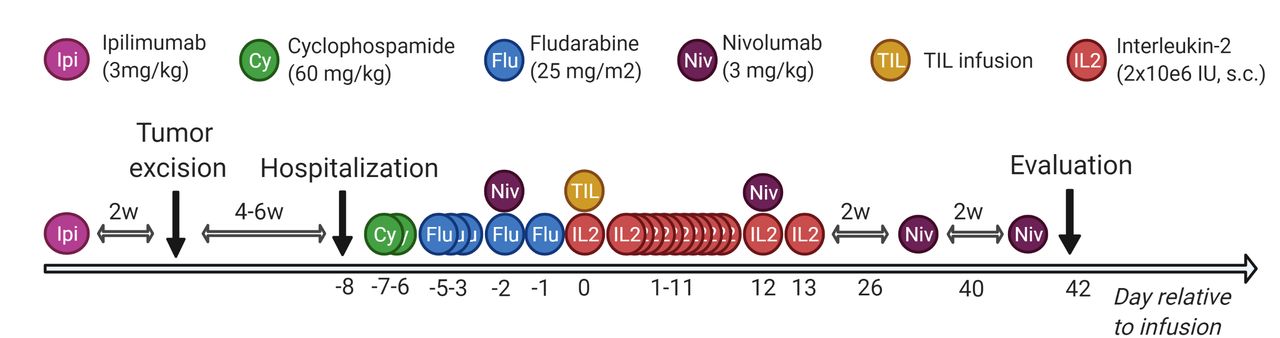
CPIs were added to the course of ACT at specific time points: anti-CTLA antibody ipilimumab (3 mg/kg×1) was administered >2–6 weeks before tumor removal to optimize in vitro expansion. The anti-PD-1 antibody nivolumab (3 mg/kg, q2w×4) was administered 2 days before and in the weeks after TIL infusion to improve in vivo efficacy. At days −7 to −1 relative to the TIL infusion, patients were treated with non-myeloablative conditioning chemotherapy with cyclophosphamide (60 mg/kg×2) and fludarabine phosphate (25 mg/m12×5). At day 0, the entire TIL product was infused and followed by bone marrow stimulation with pegfilgrastim (6 mg×1) and subcutaneous IL-2 injections (2 MIE per day for 14 days). The therapy regimen is illustrated in figure 1. Primary endpoints were tolerability and feasibility of ACT combined with CPI in metastatic solid cancers, secondary endpoints clinical efficacy and immune parameters.
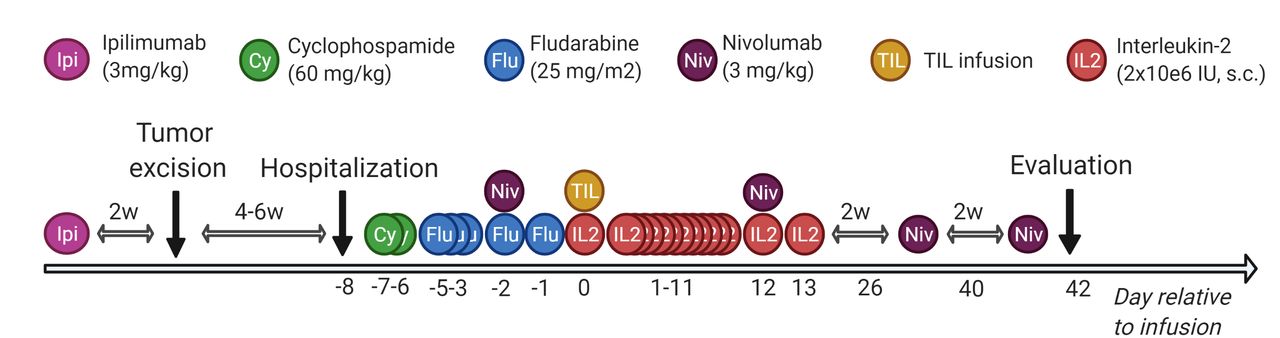
Therapy regimen. Overview of the scheduled adoptive cell therapy in combination with conditioning chemotherapy, low-dose interleukin-2 and checkpoint inhibition. Created with BioRender.com. TIL, tumor-infiltrating lymphocyte.
Assessment of safety, feasibility and clinical efficacy
Patients were continuously monitored for adverse events and graded according to Common Terminology Criteria for Adverse Events (CTCAE) V.4.0. Study drugs and supporting therapy were individually adjusted according to toxicity and management thereof. Feasibility was assessed as successful TIL expansion in vitro, successful TIL infusion and administration of other study drugs. Clinical efficacy was assessed as objective response according to Response evaluation criteria in solid tumors (RECIST) V.1.1. As an exploratory endpoint. In an exploratory analysis, the patients were grouped according to disease control. Disease control was defined as a confirmed partial response (PR) or stable disease (SD) >4.5 months.
Tumor handling and in vitro TIL production
Participants underwent surgical removal of an accessible tumor lesion and the tissue was directly transported in tumor medium containing RPMI-1640 with Glutamax/Hepes (Gibco, Waltham, Massachusetts, USA), 10% Fetal bovine serum (FBS;Gibco), 1% penicillin/streptomycin (Gibco) and 0.5% Fungizone (Bristol-Myers Squibb, New York, New York, USA) to an in-house Good Manufacturing Practice (GMP) facility at the hospital for processing and in vitro TIL expansion. A section of the tumor was sent to the local pathology department for verification of malignancy.
For initial expansion, the tumor was manually dissected into >48 fragments and cultured in 24-well plates (Nunc, Roskilde, Denmark) with 6000 U/mL IL-2 (Clinigen, Staffordshire, Great Britain). When approximately 50×10e6 cells were reached, 20×10e6 cells (young TILs) were set up with irradiated allogenic feeder cells, anti-CD3 antibody (clone: OKT3; Milteny Biotec, Bergisch Gladbach, Germany) and IL-2 (6000 U/mL) in a rapid expansion protocol (REP) for 2 weeks. The production of young and REP TILs has previously been described in detail.9 36
Surplus tumor tissue was frozen in situ or enzymatically digested over 24 hours with collagenase IV (Sigma-Aldrich, St. Louis, Missouri, USA) into a single cell suspension of tumor cells) and TILs (unstimulated TILs). Autologous tumor cell lines were cultured from the transport medium using RPMI-1640 with Glutamax/Hepes (Gibco), 10% FBS (Gibco), 1% penicillin/streptomycin (Gibco) and 0.5% Fungizone (Bristol-Myers Squibb). The tumor cell lines were sent for analysis and diagnosis verification at the Department of Pathology at Herlev Hospital.
Immune phenotyping the cell product
The TIL infusion product (REP TILs), young TILs and unstimulated TILs were phenotypically characterized by flow cytometry. REP and Young TILs were thawed 1 day prior to flow cytometry and rested overnight in medium containing RPMI-1640, 1% penicillin/streptomycin (Gibco) and 10% human serum (Sigma-Aldrich), while unstimulated TILs were staining on the day of thawing. After washing, approximately 1×10e6 cells were incubated 5 min at room temperature with near infrared (NIR) live–dead marker (Thermo Fisher, Waltham, Massachusetts, USA) and Fc block, and then incubated in the dark for 20 min at 4°C with the antibody mixes shown in online supplemental table 1. The cells were then washed and acquired on a NovoCyte Quanteon Flow Cytometer (Agilent Technologies, Santa Clara, California, USA).
Supplemental material
In vitro antitumor reactivity
In vitro expanded REP TILs were tested for reactivity against autologous tumor. Autologous tumor cells were either derived from an in vitro cultured tumor cells (TC) or from digested tumor material (TD) as described previously. TILs were thawed and rested the day before reactivity testing as mentioned previously, while TD was thawed on the day of testing. For the reactivity assay, TILs and tumor cells (either TD or TC) were cocultured (3:1 ratio) for 6 hours together with GolgiPlug (BD Biosciences, San Jose, California, USA), GolgiStop (BD Biosciences), and anti-CD107a-BV421 (BD Biosciences, cat 555801). After coculture, the cells were washed twice with phosphate-buffered-saline (PBS) and stained in the dark for 20 min at 4°C with the NIR live–dead marker and the extracellular antibodies listed in online supplemental table 1. Cells were then washed and processed with the Foxp3 Transcription Factor Staining Buffer Set according to the manufacturer’s instructions (eBioscience, San Diego, California, USA), before being stained with the intracellular antibodies listed in online supplemental table 1 in the dark for 30 min at 4°C. After an additional wash with PBS, the cells were acquired on a NovoCyte Quanteon Flow Cytometer (Agilent Technologies). The method is further elaborated elsewhere.37 Antitumor reactivity was defined as simultaneous expression of at least two of the following markers after tumor coculture: tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), CD107a or CD137 (4-1BB). The lower limit for antitumor reactivity in REP TILs was 0.5% of all live CD3+ cells after subtraction of unstimulated background expression of REP TILs.
Tumor mutational burden (TMB)
To assess the TMB of the tumor resection used for in vitro TIL expansion, tumor DNA was sequenced with the pan-cancer genomic assay TruSight Oncology 500 (TSO500; Illumina, San Diego, CA). The tumor material used for TMB analysis was incubated in RNAlater (Life Technologies, Carlsbad CA) during processing, stored overnight and then frozen at −80⁰C until analysis. Tumor DNA was purified using AllPrep DNA/RNA/protein extraction kit (Qiagen, Hilden, Germany). Targeted sequencing was performed using TSO500 HT Library preparation kit according to manufacturer’s instructions (Illumina). Sequencing was done on a NovaSeq6000 (2×100 bp paired-end reads) and sequencing data were processed using the TSO500 assigned software (Local app V.2.2) provided by Illumina.
Data analysis and statistics
Clinical data were collected and stored in a data repository hosted by REDcap (Vanderbilt University, Nashville, Tennessee, USA). Statistical analyses were performed in GraphPad Prism V.9 (GraphPad Software, La Jolla, CA, USA). A log-rank test was used to compare overall survival (OS) between disease control and non-disease control groups. Data correlations were tested for significance with the non-parametric Spearman correlations and grouped data were compared with a non-parametric Mann-Whitney U test. The significance correlations of in vitro reactivity and tumor size regressions were tested with Fisher’s exact test.
Flow cytometry data were analyzed in NovoExpress V.1.4 (Agilent Technologies) and FlowJo V.10.6 (BD Biosciences). Cell subset sizes were compared with a non-parametric Mann-Whitney U test or a Wilcoxon matched pairs test for paired comparisons.
Results
Patient characteristics
A total of 25 patients across 10 different metastatic cancer types were enrolled and treated with an in vitro expanded TIL product between 2017 and 2019. An additional six patients were included but discontinued prior to TIL infusion. The most common diagnoses were colorectal cancer (CRC, n=7) and choroidea-derived MM (MM choroidea, n=4). Many patients had a long cancer history with several lines of therapy prior to enrolment. The baseline characteristics of all treated patients are listed in table 1.
Baseline patient characteristics
Feasibility and safety
In total, in vitro TIL expansion to therapeutic levels was successful in 30 out of 31 patients (97%). Three patients required a second tumor resection, making the success rate 30 TIL products out of 34 total tumor resections (88%). Six patients were treated with ipilimumab and had surgical removal of tumor tissue but were discontinued due to clinical deterioration (n=5) or failure to produce a TIL product (n=1). All patients received ipilimumab prior to tumor removal with a median interval of 16 days. One patient with MM (patient 11) had previously received ipilimumab as standard therapy 4 months earlier and was not retreated prior to surgical tumor resection for ACT.
Median young TIL expansion time was 23 days (min–max: 15–44 days), and the median number of infused cells was 61×10e9 cells (min–max: 5–114×10e9 cells). All patients received nivolumab on day −2 relative to cell infusion, and 16 patients received all four scheduled doses of nivolumab. Ten of the 25 patients received all 14 doses of IL-2, while 2 patients did not receive any IL-2 after TIL infusion due to fragile clinical condition. In 13 patients, IL-2 was prematurely discontinued due to fragile clinical state (n=6), unacceptable toxicity (n=8) or logistical considerations (n=1). Five of the treated patients also worsened in performance status from trial inclusion to hospital admittance. An overview of TIL expansion and treatment is provided in table 2.
Treatment and response overview
The most severe toxicity was due to the conditioning chemotherapy and IL-2. Grade 3 and 4 adverse events are listed in table 3. Non-ablative myelosuppression with grade 4 neutropenia was seen in all patients, but the duration and the degree of anemia and thrombocytopenia varied greatly. Data on myelosuppression during and after conditioning chemotherapy are further elaborated in a separate publication.38 Two patients suffered from grade 4 dyspnea after TIL infusion and one was transferred to the intensive care unit for temporary assisted ventilation.
Toxicity of study drugs and interventions
Clinical efficacy
Five patients had sizeable tumor regressions of 30%–63% (figure 2A) including two patients (16%) with confirmed PRs. The PRs consisted of one patient (patient 8) with cholangiocarcinoma (CC) that demonstrated total regression of a smaller lung metastasis and another patient (patient 19) with head-and-neck squamous cell carcinoma (HNSCC) who experienced total regression of several sizeable lung metastases (figure 3). Both patients eventually suffered from progressive disease. Two patients had unconfirmed PR. The disease control rate was 32% with eight patients experiencing either PR or SD of >4.5 months.
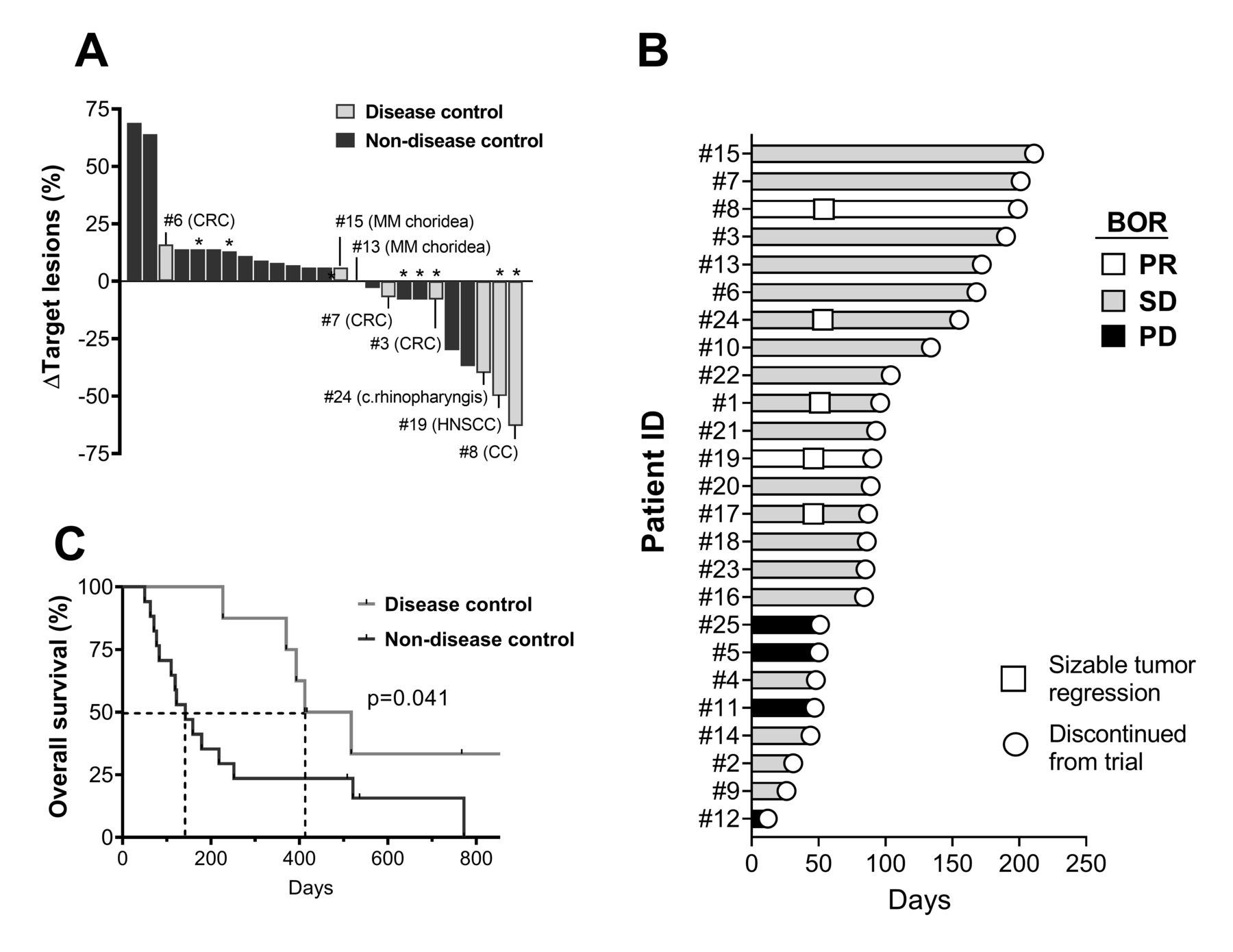
Clinical efficacy and OS. (A) Best change in target lesion sum of all patients. The patients with disease control (PR and/or SD >4.5 months) are marked with light gray and listed with cancer diagnoses. (B) Swimmer’s plot showing the duration of response in the individual patients and indicating start of tumor regressions >30% of target lesions. (C) Kaplan-Meier plot of the OS in the benefit and non-benefit groups. The survival curves are compared with a log-rank test. *The patients with antitumor reactive cells in vitro. BOR, best overall response; CC, cholangiocarcinoma; CRC, colorectal cancer; HNSCC, head-and-neck squamous cell carcinoma; MM, malignant melanoma; OS, overall survival; PD, progressive disease; PR, partial response; SD, stable disease.

Computerized tomography (CT) scan images from patients with an objective response to therapy. Two patients had a partial response according to RECIST V.1.1. Images from baseline and 6 weeks after cell infusion in patient 8 with cholangiocarcinoma and patient 19 with HNSCC are shown. HNSCC, head-and-neck squamous cell carcinoma.
The median progression-free survival was 89 days and median OS was 227 days. The patients in the disease control group had a significantly longer OS (p=0.041) than the non-disease control group (figure 2B), with a median survival of 465 days vs 142 days, respectively. Treatment feasibility regarding in vitro TIL expansion was not significantly altered between the two groups, but the benefit group received significantly more of the scheduled IL-2 and nivolumab doses (table 2).
TIL phenotype of infusion product
The TILs were phenotyped at different stages of expansion using flow cytometry. The REP TILs used for therapy were almost exclusively CD3+ T cells with a median of 98.5% (min–max: 92.1%–99.6%) as shown in table 2. In 10 patients, the predominant T-cell subset was CD8+ (>50%), whereas the CD4+ subset dominated in 14 patients. There was no significant correlation to clinical efficacy, although there was a trend of higher proportion of CD8+ cells in the disease control group (table 2). One patient (#14) with choroidal melanoma had almost exclusively CD4−CD8− T cells, and further analysis revealed that they were gd T cells, which were not seen in the other patients (data not shown).
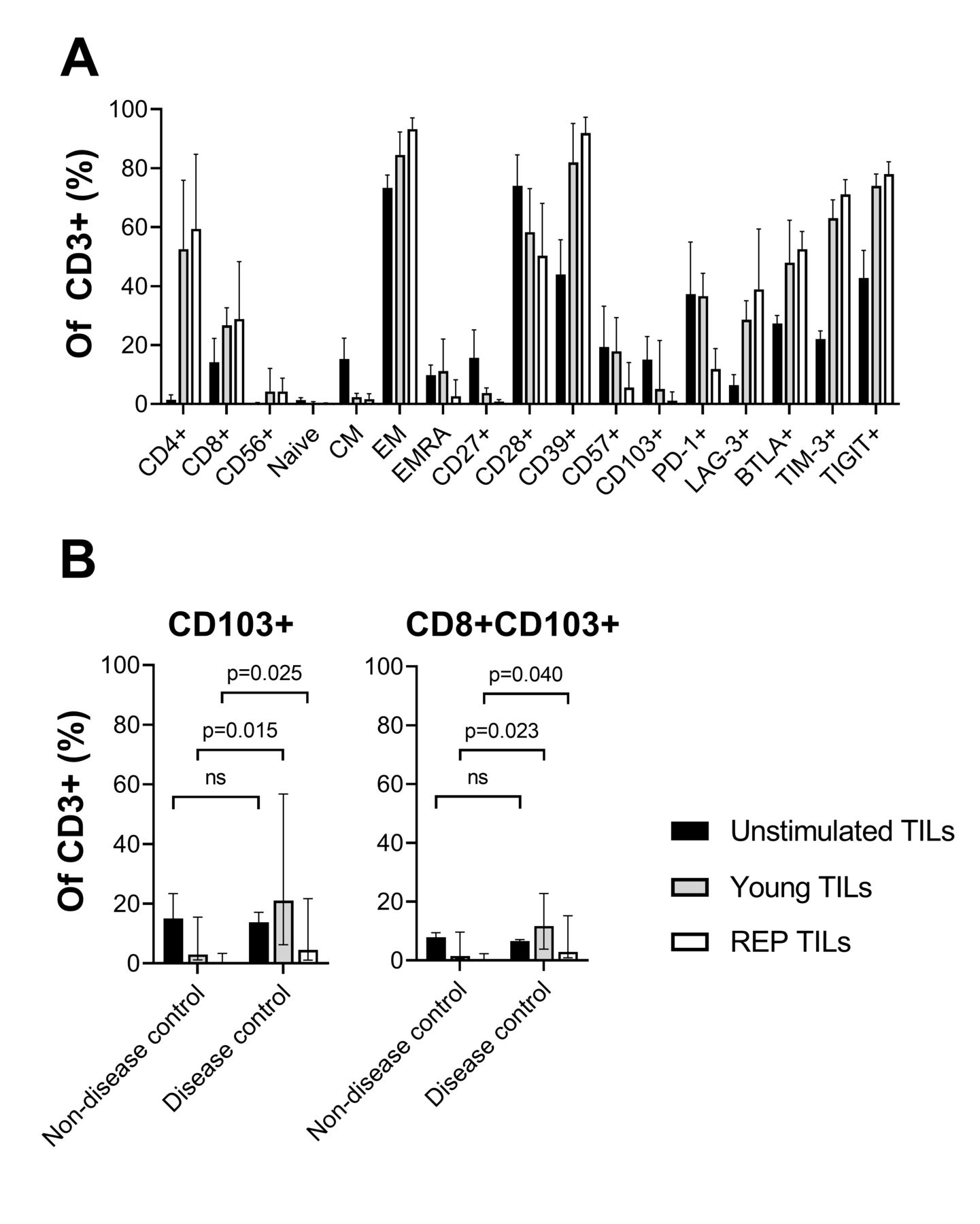
As shown in figure 4A, the REP TILs were largely CCR7−, indicating T cells with effector function (median of CCR7+ was 0.6%, min–max: 0.1–3.5). The dominating subset was CD45RA− effector memory T cells (median: 94.3%, min–max: 47.5%–98.9%), with a low fraction of CD45RA+CCR7− (Terminally differentiated effector memory, EMRA) T cells (median: 5.3%, min–max: 0.7%–52.4%). The REP TILs were high in CD39, CD28, and the immune regulatory checkpoints LAG-3, BTLA, TIM-3, and TIGIT, and low in CD27, CD57, CD103, and PD-1.

Phenotype analysis of TILs throughout expansion. (A) Single marker expression analysis of the unstimulated TILs before (unstimulated TILs), after initial in vitro expansion (young TILs) and after the REP TILs. Expression is shown as the fraction of CD3+ cells. (B) Comparison of CD103+ and CD8+CD103+TILs during the in vitro expansion according to disease control (partial response and/or stable disease >4.5 months). The difference was tested for significance with a non-parametric Mann-Whitney U test. CD4 expression is low in unstimulated TILs as the dissociation agent (collagenase IV) is known to cleave CD4. CM, central memory; EM, effector memory; EMRA, terminally differentiated effector memory; ns, not significant; REP, rapid expansion protocol; TIL, tumor-infiltrating lymphocyte.
In all patients, a marked increase in CD39 expression and other immune regulatory checkpoints was seen throughout the in vitro expansion with the notable exception of PD-1, which decreased significantly during the REP. The differentiation marker CD27 that has been linked to better in vivo efficacy of TILs also decreased during the REP.
A phenotype comparison of REP TILs from patients in the disease control group to the non-control group revealed significantly increased expression of an alpha-integrin CD103 associated with tissue migration and residency (figure 4B, p=0.025). The higher expression of CD103 was also seen in CD8 T cells (figure 4C, p=0.040). As also shown in figure 4B,C, the overall expression of CD103 generally decreased during the in vitro expansion especially after the initial young TIL expansion with IL-2 but to a lesser extent in the disease control group. CD103 was present (>1% of CD3) in all but one patient in the disease control group, including the two patients with an objective response (3.9% and 5.7%), and in less than half of the patients in the non-disease control group.
Antitumor reactivity in vitro
Reactivity against autologous tumor cells was demonstrated in REP TILs from eight patients and in up to 12.4% of infused T cells (figure 5). Three of the reactive REP TILs were derived from colon cancer. Reactivity was seen in both CD4+ and CD8+ T cells, but primarily in the subset that dominated the REP TILs of the individual patient. Two of the REP TILs (patients 2 and 11) were reactive against TC, while six were reactive against TD (patients 1, 3, 8, 17, 19 and 20). Of note, we also observed very high expression of all reactivity markers in the CD4−CD8− gd T cells of patient 14. Although it increased when incubated with TCs, the background expression of TNF-α, IFN-γ, CD107a, and CD137 (6.8%, 5.2%, 7.16%, and 28.4% of CD3, respectively) was so high that specific tumor reactivity could not be determined. The reactivity data from all patients are shown in online supplemental figure 1.
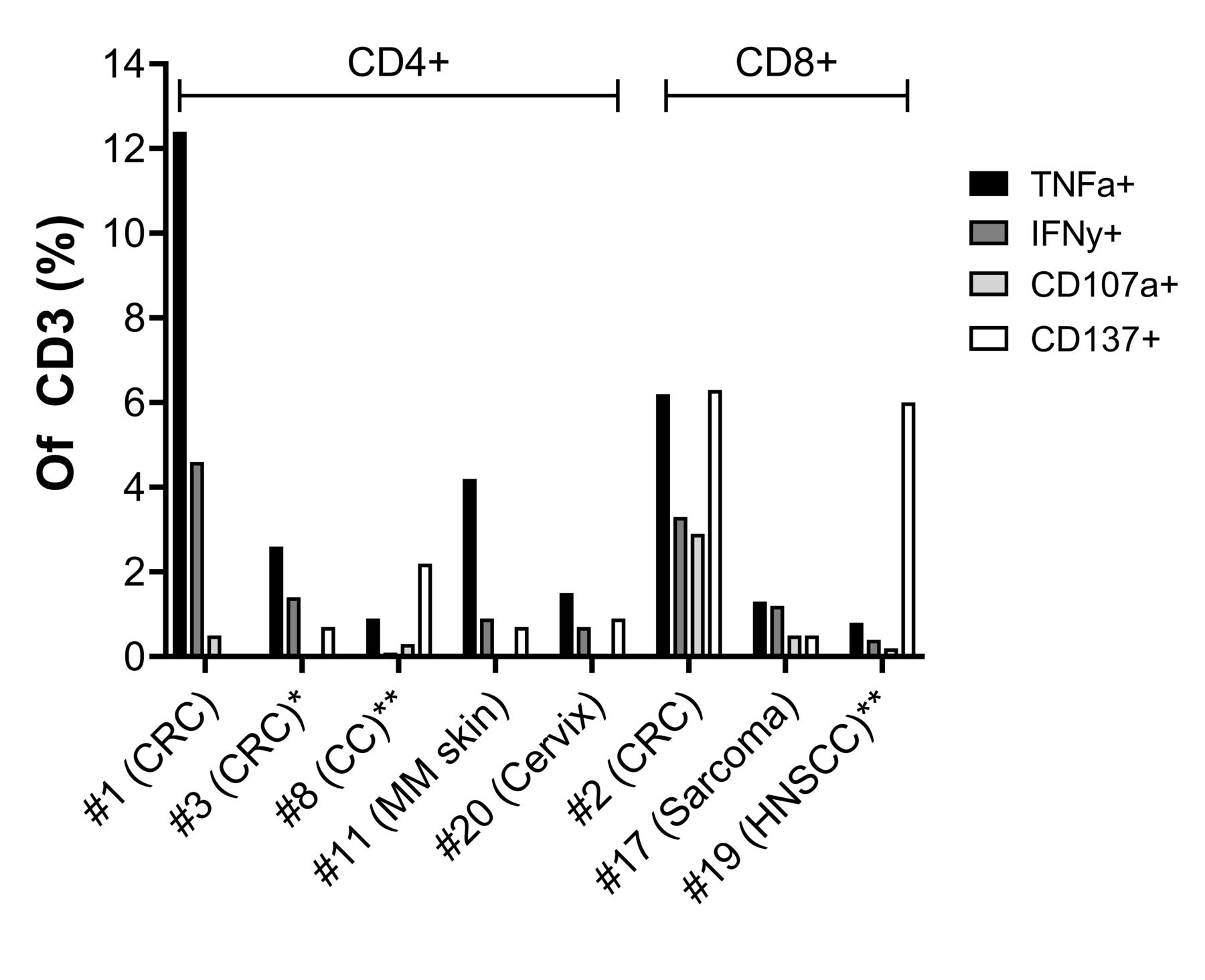
In vitro antitumor reactivity of infusion product. Expression of reactivity markers in REP TILs from eight patients where antitumor in vitro reactivity was seen in more than 0.5% of CD3+ cells. The REP TILs are divided according to the dominant T-cell subset: either CD4 or CD8. Reactivity was assessed with intracellular flow cytometry after coculture with unstimulated autologous tumor material. REP TILs with expression of at least two markers (TNF-α, IFN-γ, CD107a or CD137) in more than 0.5% of all CD3+ cells were considered reactive. *Patients in the disease control group. **Patients in the disease control group with an objective response. CC, cholangiocarcinoma; CRC, colorectal cancer; HNSCC, head-and-neck squamous cell cancer; IFN-γ, interferon gamma; MM, malignant melanoma; REP, rapid expansion protocol; TIL, tumor-infiltrating lymphocyte; TNF-α, tumor necrosis factor-alpha.
In vitro antitumor reactivity of REP TILs was only seen in 3 (out of 8) in the disease control group but in 6 out of 10 patients with tumor size regressions (figure 2, p=0.028), including the two patients with confirmed PR.
Tumor mutational burden
The TMB analysis (figure 6) showed that four patients were TMB-high defined as >10 mutations per megabase.39 Two TMB-high patients, both patients with CRC, were in the disease control group. The human papilloma virus (HPV)-associated cancers (patients 19 and 20) also presented with a high TMB. There was a trend towards greater TMB in patients with disease control, but the difference did not reach statistical significance. There was no significant correlation between high TMB and in vitro TIL expansion feasibility or in vitro antitumor reactivity. Four patients had TMB <1, indicating either a high proportion of non-tumor tissue or low-frequency mutations. In one patient, the TMB could not be determined due to a low-frequency variant (detection limit of 5%).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TMB versus disease control of ACT. The resected tumor sample used for in vitro TIL expansion was examined for TMB. The TMB of 24/25 of the patients treated with ACT is shown together with the best overall response and the patients are divided according to disease control rate (PR and/or SD >4.5 months). Non-PR indicates SD or PD. ACC, adrenocorticocarcinoma; ACT, adoptive cell therapy; CC, cholangiocarcinoma; CRC, colorectal cancer; HNSCC, head-and-neck squamous cell carcinoma; MM, malignant melanoma; NCSLC, non-small cell lung cancer; PD, progressive disease; PR, partial response; SD, stable disease; TIL, tumor-infiltrating lymphocyte; TMB, tumor mutational burden.
Discussion
The results from this clinical trial demonstrate the safety and feasibility of TIL-based ACT in a wide range of solid cancer types. Despite many patients being heavily pretreated with chemotherapy and/or immune therapy, TIL expansion and therapy could be completed for the great majority. Furthermore, we show that the addition of CPIs both before and during TIL infusion with low-dose IL-2 only adds manageable and expected toxicity comparable to CPI monotherapy.
In many patients, the number and degree of adverse events from low dose IL-2 were more pronounced than what has been reported with low-dose IL-2 in MM.40 41 Retrospectively, it was evident that IL-2 was not well tolerated in the non-benefit group and was often prematurely discontinued. These patients generally had a very poor survival (median OS of 159 days) indicative of a fragile clinical state.
Objective responses were limited to a subset of patients but do provide additional evidence of the clinical potential of ACT in patients outside MM, specifically CC and HNSCC. In CC, an earlier case report of ACT with selected TILs demonstrated clinical efficacy.6 Clinical efficacy of CPI monotherapy is modest in CC, and it is possible that combinational regime with, for example, ACT is needed to achieve clinical efficacy in this cancer type.42 In HNSCC, preclinical data from our center and others have already suggested a potential for TIL therapy, and a recent trial has also demonstrated clinical efficacy of HPV-reactive TILs in HPV-associated HNSCC.10 15 43 44 The overall potential of immune therapy in HNSCC is also evident by the recent approval of CPIs as first-line therapy in these patients.45
With multiple drugs included in the study design, it is difficult to distinguish whether the TILs, chemotherapy, CPIs, IL-2, or a combination was the main driver of clinical response. However, in vitro antitumor reactivity and clinical efficacy in chemotherapy—and CPI-resistant patients, as seen in the patient with HNSCC, is suggestive of a beneficial role of the TILs. We also observed more pronounced tumor regressions in patients treated with REP TILs that also showed antitumor reactivity in vitro.
Nivolumab was given together with the TIL infusion, and with a proportion of the TILs expressing PD-1 (median 11.9%, min–max: 4.5%–45.1%), an impact on these cells is possible. Recently, a case report of a complete and durable response was demonstrated with the TIL and anti-PD-1 combination in a patient with breast cancer.8
The success rate of the in vitro TIL expansion was indeed comparable to MM but with a slightly longer median young TIL expansion time of 23 days as compared to 17–21 days for MM; also the median total number of cells produced was considerably lower.22 23 41 We noted a trend towards shorter expansion time and larger REP TIL products in responding patients, similar to what was described in MM, but this difference did not reach statistical significance.46 The specific effect of adding ipilimumab before tumor resection is difficult to dissect. However, based on earlier data, including clinical evidence in patients with MM,28 29 it could be argued that the ipilimumab dosing prior to tumor harvest contributed to the high success rate of the in vitro TIL expansion across tumor types.
The phenotype analysis of the REP TILs suggested an importance of maintaining CD103+ CD8 T cells throughout the in vitro expansion. These cells have been associated with antitumor specificity and reactivity in several cancers, and their presence has been associated with improved survival in several cancers.47–51 Further, the TILs showed high expression of CD39—a marker associated with tumor reactivity but also activation and exhaustion.49 52–54 CD39 expression increased during the in vitro expansion and was highly expressed on the REP TILs. Our data indicated that the high CD39 expression in REP TILs was more likely a result of in vitro stimulation than being directly linked to antitumor reactivity. Similarly, many other immune regulatory checkpoints such as LAG-3, BTLA, TIM-3, and TIGIT also increased during the expansion.
Recently, the size of a CD39−CD69− stem-like subset within the REP TILs was retrospectively linked to durable responses to TIL-based ACT in MM, while high CD27 expression associated to a ‘young’ phenotype has been linked to better in vivo TIL efficacy.2 54 We observed no correlation to CD27+ or CD39− cells to clinical efficacy.
The high expression of several immune regulatory checkpoints on REP TILs besides PD-1 indicates that other targets may be suited for combinational therapy, including anti-CD39 drugs or anti-LAG-3, which is already in clinical development.55 56 To this end, we are currently undertaking a clinical trial with TIL-based ACT in combination with the anti-LAG-3 antibody relatlimab based on prior observations of high LAG-3 expression in REP TILs of patients with ovarian cancer.9
TMB has been established as an independent prognostic factor of response to CPI therapy, and we therefore wanted to investigate TMB as a common denominator of various cancer types in the setting of TIL-based ACT.57 58 We observed a trend towards higher TMB in the benefit group including the two patients with an objective response; however, this was not statistically significant, which indicates that other mechanisms are involved. Interestingly, there was no significant correlation between TMB and TIL expansion feasibility despite high TMB being a driver of immunogenicity and immune infiltration.59 60 The patient with the highest TMB was a patient with adrenocorticocarcinoma (ACC) who did not respond or benefit from therapy. ACC has generally not been responsive to immune therapy, but a recent subgroup analysis linked clinical efficacy of CPIs to high TMB in this diagnosis.61 62
Another interesting finding was the clear feasibility of TIL production in two HPV-associated cancers (patients 19 and 20). TILs from both patients showed remarkably high fold expansions in the REP (4748 and 4960), had a high percentage of CD103+ cells (4.9% and 8.0%) and demonstrated antitumor reactivity in vitro. The patient with HNSCC (patient 19) who achieved a PR was also HPV-associated, and the clinical potential of TIL expansion in HPV-associated epithelial cancer is supported by clinical results with HPV-reactive TILs from the National Cancer Institute.7 10
Our results are limited by the complex therapy regimen, the heterogenous and advanced cancer disease that affected all patients, and the small sample size. The number of objective responses in this phase I/II clinical trial is low but still difficult to compare to the success of TIL-based ACT in MM. MM is highly susceptible to immune therapy, and TIL-based ACT is usually tested after fewer lines of therapy. Up to now TIL-based ACT has almost exclusively been tested in patients with MM and the few patients outside MM have primarily been treated with selected TILs. Our results show that the TIL-based ACT platform can be combined with CPIs and applied to a much broader spectrum of cancer indications than solely MM but also indicate that more work is needed to optimize the clinical efficacy before it can be considered a viable therapeutic option.
Conclusion
We demonstrate that TIL-based ACT with in vitro expanded REP TILs in combination with CPIs is feasible in multiple solid cancers besides MM. Clinical efficacy and tumor regressions are demonstrated in a subset of patients and supported by in vitro antitumor reactivity of the REP TILs. Our data suggest that the phenotype of the REP TILs, specifically CD103 expression and possibly also TMB, is linked to clinical efficacy.
Data availability statement
Data are available upon reasonable request. Flow cytometry data and genetic data from the tumor material is avaliable to share upon reasonable request within the national legal framework of data sharing and GDPR.
Ethics statements
Patient consent for publication
Ethics approval
The clinical trial was approved by the National Ethics Committee and the Danish Medicines Agency. Oral and written informed consent were obtained prior to recruitment in accordance with the Helsinki Declaration.
Acknowledgments
We are deeply grateful for the patients’ willingness to participate and the support of their families. A special thanks to the technicians responsible for tumor-infiltrating lymphocyte expansion at the GMP facility at Herlev Hospital and to the Department of Oncology, including attending physicians and nurses. Thanks to Morten Hansen at the National Center for Cancer Immune Therapy for support and assistance regarding the flow cytometry. This research was funded by the Danish Cancer Society and Herlev Hospital. The funders played no role in study design, data collection, analysis, or preparation of this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Study design: AHK, ÖM, IMS, MD, and THB. Patient therapy and clinical data collection: AHK, THB, MN, SKM, JWK, and CLL. Translational analysis: AHK, CAC, LPJ, LBR, and CWY. Data analysis: AHK. Manuscript writing: AHK and IMS. Manuscript proofs: AHK, CAC, THB, MN, JWK, CLL, LPJ, LBR, CWY, ÖM, MD, and IMS.
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.