Article Text

Abstract
Background In this study, we hypothesize that the ability of the protozoan Toxoplasma gondii to modulate immune response within the tumor might improve the therapeutic effect of immune checkpoint blockade. We examined the synergetic therapeutic activity of attenuated T. gondii RH ΔGRA17 strain and programmed death ligand-1 (PD-L1) treatment on both targeted and distal tumors in mice.
Methods The effects of administration of T. gondii RH ΔGRA17 strain on the tumor volume and survival rate of mice bearing flank B16-F10, MC38, or LLC tumors were studied. We characterized the effects of ΔGRA17 on tumor biomarkers’ expression, PD-L1 expression, immune cells infiltrating the tumors, and expression of immune-related genes by using immunohistochemistry, immunofluorescence, flow cytometry, NanoString platform, and real-time quantitative PCR, respectively. The role of immune cells in the efficacy of ΔGRA17 plus PD-L1 blockade therapy was determined via depletion of immune cell subtypes.
Results Treatment with T. gondii ΔGRA17 tachyzoites and anti-PD-L1 therapy significantly extended the survival of mice and suppressed tumor growth in preclinical mouse models of melanoma, Lewis lung carcinoma, and colon adenocarcinoma. Attenuation of the tumor growth was detected in the injected and distant tumors, which was associated with upregulation of innate and adaptive immune pathways. Complete regression of tumors was underpinned by late interferon-gamma-producing CD8+ cytotoxic T cells.
Conclusion The results from these models indicate that intratumoral injection of ΔGRA17 induced a systemic effect, improved mouse immune response, and sensitized immunologically ‘cold’ tumors and rendered them sensitive to immune checkpoint blockade therapy.
- antineoplastic protocols
- combined modality therapy
- immunotherapy
- melanoma
- programmed cell death 1 receptor
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. The remaining data are available within the Article, Supplementary Information or available from the authors upon request. Data are available upon reasonable request. Contact email: zhuyuchao9@qq.com.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- antineoplastic protocols
- combined modality therapy
- immunotherapy
- melanoma
- programmed cell death 1 receptor
Background
Immunotherapy has been a useful tool in the clinical management of cancer in addition to surgery, radiation, and chemotherapy.1 In particular, immune checkpoint blockade (ICB) therapies have been used in the treatment of multiple cancers (eg, hepatocarcinomas, gastric cancers, head and neck cancers, metastatic colorectal cancer and skin melanomas, squamous carcinoma of the lung, renal cell carcinoma, urothelial carcinoma, mismatch repair-deficient tumors, Hodgkin’s lymphoma and Merkel cell carcinomas) via antibodies inhibiting programmed death-1 (PD-1), programmed death ligand-1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) immune checkpoints.2 3 Unfortunately, the therapeutic effectiveness of immune checkpoint inhibitors can be compromised by factors related to tumor microenvironment and tumor genome, leading to ‘cold’ tumors that are tolerant to ICB therapy in some patients. These factors include, for example, PD-L1 expression level,4 5 tumor-infiltrating lymphocytes (TILs),6 mutational load and generation of neoantigens,7 increased infiltration of myeloid-derived suppressor cells (MDSCs),8–13 lack of major histocompatibility complex I (MHC-I) expression,14 and mutations in Janus kinase 1/2.15 These challenges increased the interest in the exploration of more innovative immuno-therapeutic approaches that bolster the antitumor immunity by switching non-infiltrated ‘cold’ tumors into more immunogenic, immune-infiltrated, ‘hot’ tumors.
Pathogens have been exploited as potential cancer immunotherapeutic agents, including tumor-targeting bacteria and oncolytic viruses (OVs),16–18 and some protozoan parasites, such as Trypanosoma cruzi and Plasmodium sp, can thwart the development of tumors in animal models.19 20 The OVs have been promoted as a therapeutic platform to deploy ICB or used in combination with ICB, due to their ability to augment the responses in some patients and tumor types that respond poorly to ICB.17 Despite the clinical utility of these OVs, particularly in combination with other chemotherapeutic and immunotherapeutic agents,21 the antitumor efficacy of OV monotherapy can be compromised in patients who are infected and/or harbor pre-existing neutralizing antiviral antibodies, activated complement or cytokines, which may attenuate the OV’s activity, and even clear these viruses.22 23 To overcome these limitations, exploring the potential of other pathogens as cancer immunotherapeutic agents is warranted.
The intracellular protist Toxoplasma gondii can invade nearly all nucleated cells and infect many warm-blooded animals.24 T. gondii is also widely prevalent in human population worldwide.25 The efficacy of fully attenuated, avirulent, T. gondii uracil auxotroph strains (ΔCPS and ΔOMPDCΔUP) as vaccine candidates has been shown.26–30 These strains can be grown normally in vitro with uracil supplementation; however, inside the mammalian hosts, these strains cannot replicate and are eliminated by the host immune system within ~5 days.30 ,31 In previous immunotherapy studies of established melanomas, and ovarian and pancreatic tumors, ΔCPS and ΔOMPDCΔUP strains increased the immunogenicity of the tumor microenvironment, reversed tumor-associated immune suppression, increased antigen processing, presentation, priming and activation of tumor antigen-specific CD8+ T cells, and completely regressed tumors or significantly extended mouse survival.30 32–38 The immunotherapeutic effect induced by T. gondii uracil auxotroph requires live parasites,30 38 active invasion,30 and secretion of parasite-derived rhoptry and dense granule (GRA) effector proteins into the host cells.30 A previous study showed that secretion of GRA24 into host cells by attenuated T. gondii strongly induces interleukin 12 (IL-12)-dependent CD8+ T cell activation and elicits protective immunity independent of MyD88 signaling and murine-specific toll-like receptors TLR11 and TLR12, which are absent in humans.39 Significantly, the efficacy of attenuated T. gondii immunotherapy was not diminished by either pre-existing chronic T. gondii infection26 or by pre-existing immunity to T. gondii,30 indicating that attenuated T. gondii therapy could be advantageous in comparison with OVs or other pathogen-based immunotherapies which are compromised by existing immune responses to the pathogen. These findings suggest that T. gondii can offer an effective antitumor therapeutic platform for cancer immunotherapy. However, the precise mechanisms used by attenuated T. gondii to transform ‘cold’ tumors into ‘hot’ tumors and whether tumor immunotherapy with attenuated T. gondii could sensitize the tumors to treatment with ICB are unknown.
The advent of the gene editing clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 (CRISPR-Cas9) system has facilitated the genetic manipulation of T. gondii.40 Some attenuated T. gondii strains have been constructed using CRISPR-Cas9 such as ΔGRA17, which induces a strong adaptive immune response, associated with CD8+ T cell maturation and expansion, and increased expression of IL-12 and interferon-gamma (IFN-γ).40 41 These immune responses can also potentiate an effective antitumor immunity, indicating that ΔGRA17 can be used to induce antitumor immunity to thwart the growth of tumors.
In this study, we show that the combination therapy using intratumoral administration of the avirulent ΔGRA17 mutant T. gondii strain and anti-PD-L1 therapy significantly arrests tumor growth compared with each treatment alone. We found that intratumoral injection of ΔGRA17 strain increased lymphocyte infiltration in local (parasite-injected) and distant (non-parasite-injected) tumors. Our data also revealed that intratumoral injection of ΔGRA17 sensitized the microenvironment of not only ΔGRA17-injected tumors but also the contralateral (distant) tumors to systemic PD-L1 blockade treatment, leading to reduction in tumor size and improved survival in most treated mice.
Methods
Sources of antibodies
The following antibodies were purchased from BD Biosciences (Waltham, Massachusetts, USA): anti-CD45 (AF700, clone: 30-F11), anti-CD3 (BV421, clone: 145-2 C11), anti-CD4 (BV510, clone: RM4-5), anti-CD8 (PE, clone: 53-6.7), anti-NK1.1 (PE-CY7, clone: PK136), anti-CD11b (BV605, clone: M1/70), anti-CD11c (APC, clone: HL3), anti-PD-1 (BV650, clone: J43), anti-PD-L1 (BV650, clone: MIH5), anti-FoxP3 (AF647, clone: MF23), anti-IFN-γ (AF647, clone: XMG1.2), anti-Ki-67 (AF488, clone: B56), anti-granzyme B (eFluor 660, clone: NGZB), anti-CD80 (BV421, clone: 16-10 A1), anti-CD86 (BV450, GL1), and anti-TNF-α (AF488, clone: MP6-XT22). Additionally, anti-PD-1 (clone: RMP1-14), anti-PD-L1 (clone: 10F.9G2), anti-CD4 (clone: GK1.5), anti-CD8 (clone:2.43), and anti-NK1.1 (clone: PK136) were purchased from BioXcell (Lebanon, USA).
Cell lines and culture conditions
The cell lines were purchased from American Type Culture Collection (ATCC, Manassas, Virginia, USA), including murine B16-F10 melanoma cells (CRL-6475), murine Lewis lung carcinoma LLC (CRL-1642), murine colon adenocarcinoma cells MC38 (Kerafast ENH204-FP), and human foreskin fibroblasts (HFFs) (SCRC-1041). Murine melanoma B16-F10 labeled with luciferase (B16-F10-Luc) was generated as described previously.42 These cell lines were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS; Gibco, New York, USA) and 1% penicillin-streptomycin-glutamine 100x (Thermo, Waltham, Massachusetts, USA). HFFs were used to culture T. gondii ΔGRA17 tachyzoites. Lymphocytes were grown in RPMI 1640 medium containing 10% of HI-FBS, 10 mM HEPES, 1 mM sodium pyruvate, 0.05 mM β-mercaptoethanol, 1% penicillin–streptomycin–glutamine, and 1× minimal essential medium non-essential amino acids. Lymphocytes were used for flow cytometry analysis of tumor-infiltrating T cells and the immune status of the injected and uninjected tumors. Cultures of all above-mentioned cell lines and T. gondii were maintained in a humidified atmosphere at 37°C in 5% CO2.
Mice
Six to 8 week-old, female C57BL/6 mice and NOD/SCID/IL-2R g-chain knockout mice (NSG) were purchased from Zhejiang Laboratory Animal Center, Hangzhou, China. All mice were maintained on a standard commercial diet and sterile water ad libitum throughout the experiments. Mice were housed under specific-pathogen-free conditions and a 12 hour/12 hour light–dark cycle. Mice were randomly assigned to experimental groups. The study protocols were reviewed and approved by the Research Ethics Committee of Ningbo University (Permit No. SYXK (ZHE) 2019-0005). Mice were observed two times per day and were euthanized once they have reached their humane endpoint, including 20% loss of body weight from tumor induction, body condition score ≤2, or when the tumor size reaches ~1.5 cm.
Construction of T. gondii RH ΔGRA17 mutant strain
Type I T. gondii RH ΔGRA17 strain was constructed using CRISPR-Cas9 system as previously described.41 Deletion of GRA17 gene in the virulent RH strain diminishes the parasite’s ability to proliferate in vitro and partly attenuates its virulence in mice.41 Tachyzoites of ΔGRA17 strain were stored frozen at −80°C until analysis. Prior to administration of ΔGRA17, tachyzoites were purified by filtration using a 3.0 µm filter (Nuclepore; Sterlitech, Kent, Washington, USA) and washed with DMEM culture medium. The viability and number of ΔGRA17 tachyzoites were determined using trypan blue exclusion assay and cell counting with a hemocytometer, respectively.
Optimal dosing of T. gondii ΔGRA17
We performed a titration experiment to determine the optimal dose of tachyzoites for examining the therapeutic efficacy of intratumoral administration of ΔGRA17 strain. Briefly, mice bearing B16-F10 tumors were intratumorally injected with phosphate buffered saline (PBS) or serial doses of ΔGRA17 tachyzoites (5×103, 1×104, 5×104, or 1×105) at days 9, 11, and 13 after subcutaneous implantation of B16-F10 cells in the right flank of mice. A digital caliper was used to determine the volume of the subcutaneous tumors. The tumor volumes were calculated using the formula (width2 ×length)/2.
Efficacy of T. gondii ΔGRA17 against B16-F10 melanoma
We established a unilateral B16-F10 melanoma mouse model. Briefly, 1×106 to 3×106 B16-F10 cells were subcutaneously inoculated into the right flank of mice. When the tumor’s size has reached ~0.5 cm (at ~9 days), 100 µL of PBS or 5×104 ΔGRA17 tachyzoites were intratumorally injected on days 9, 11, and 13. We then determined the antitumor activity of ΔGRA17 treatment on the established unilateral B16-F10 melanoma by analyzing the tumor volume using a digital caliper, starting from day 9 and every other day until the end of the experiment (ie, 19 days after B16-F10 cell implantation). The tumors were dissected out after euthanasia of the mice and the tumor’s size was compared between ΔGRA17-treated and PBS-treated (control) mice. The body weight of ΔGRA17-treated and control mice was monitored two times per day up to 30 days.
The expression of mouse melanoma markers, CD63, S100β, and Ki67,43 was studied using immunohistochemistry analysis. Briefly, following deparaffinization and rehydration of the paraffin-embedded tumor tissue sections, the antigen was retrieved with high temperature in sodium citrate solution 0.01 M, pH 6.0. Then, histological sections were incubated with diaminobenzidine and counterstained with hematoxylin (Solarbio, Beijing, China), followed by incubation with anti-CD63, anti-S100β, and anti-Ki67 and antibodies (Servicebio, Wuhan, China), and secondary antibodies (Servicebio, Wuhan, China). Images were acquired using a microscope with ×200 magnification (Olympus, Tokyo, Japan).
Immunohistochemical (IHC) staining of treated and untreated tumor tissue was scored by two independent pathologists blinded to the mouse data according to the semiquantitative Immunoreactivity Score (IRS). Category A documented the percentage of stained area with immunoreactive cells as 0 (0%–5%), 1 (6%–25%), 2 (26%–50%), 3 (51%–75%), and 4 (76%–100%). Category B documented the intensity of immunostaining as 0 (negative), 1 (weak), 2 (moderate), and 3 (strong). Multiplication of category A and B resulted in an IRS ranging from 0 to 12.
The survival of ΔGRA17-inoculated and PBS-inoculated (control) mice was monitored for up to 30 days. Mice that remained tumor-free after 30 days from B16-F10 induction were rechallenged with B16-F10 cells in the opposite (left) flank. These mice were monitored for additional 60 days (ie, a total of 90 days after tumor implantation) for the development of the reimplanted tumors.
Analysis of key parameters of the biologic feasibility of ΔGRA17
To test the therapeutic efficacy of ΔGRA17 injected into extratumoral sites, we injected mice with 100 µL of PBS or 5×104 ΔGRA17 intratumorally, intraperitoneally, or intravenously. The tumor volume in mice inoculated by each of these three different inoculation routes was determined up to 19 days and mouse survival was determined up to 30 days.
To determine whether latent toxoplasmosis interferes with treatment efficacy, we tested the efficacy of ΔGRA17 against B16-F10 melanoma in mice chronically infected via intragastric administration with 10 cysts of T. gondii (Type II) PRU strain. The tumor volume was determined up to 17 days and mouse survival was determined up to 30 days.
We investigated whether live ΔGRA17 tachyzoites are necessary to achieve the required antitumor efficacy or ΔGRA17-derived constituents are sufficient to elicit an adequate immune response via recognition of the parasite’s molecular patterns. Briefly, tachyzoites obtained from viable ΔGRA17 strain or inactivated ΔGRA17 strain, prepared by heating to 60℃ for 3 min, were inoculated into B16-F10 melanoma-bearing mice as described above. The tumor volume in mice inoculated by viable ΔGRA17 tachyzoites, inactivated ΔGRA17 tachyzoites, or PBS was determined up to 19 days, and mouse survival was determined up to 30 days.
We further used immunocompromised NSG mice bearing B16-F10 tumors to determine whether an active immune response is necessary for ΔGRA17-mediated clearance of pre-existing B16-F10 tumors. The tumor volume in NSG mice inoculated by ΔGRA17 tachyzoites or PBS was determined up to 16 days, and mouse survival was determined up to 20 days.
Tumor-infiltrating T cells (TILs) after administration of ΔGRA17
The levels of expression of key proinflammatory cytokines IFN-γ (70-EK280HS), interleukin (IL)−12p40 (70-EK2183), and IL-12p70 (70-EK212HS) in the tumor tissue of ΔGRA17-inoculated mice 24 hours after the second treatment (ie, on day 12) were determined using ELISA kits as per the manufacturer’s instructions (Multisciences, Hangzhou, China). We also analyzed intratumoral immune cell infiltration by using flow cytometry. Briefly, tumors were dissected out of the mouse tissue and digested with 2% FBS, collagenase type I (Clone: SCR103, Sigma), and DNase I (Clone: H6254, Sigma) in RPMI 1640 medium for 1 hour at 37°C. The tumor tissues were homogenized and filtered through a 70 µm nylon strainer to obtain single cell suspensions. The levels of immune cells expressing CD45+, CD3+, CD4+, CD8+, NK1.1, CD11b+, CD11c+, IFN-γ, TNF-α, Ki-67, and granzyme B were analyzed using Beckman CytoFLEX S and FlowJo software (Tree Star, Ashland, Oregon, USA).
Mouse (IFN-γ enzyme-linked immune absorbent spot (ELISPOT) assay was used to monitor IFN-γ secretion by tumor-infiltrating T cells as described previously.26 Briefly, CD8+ T cells were isolated from spleen or draining inguinal lymph nodes and purified using anti-CD8 MACS magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany). CD8+ T cells were cocultured at 10:1 ratio with cell targets (irradiated EL-4 thymoma cell) for 48 hours in coated and blocked ELISpot plates. Cell targets were previously pulsed overnight with 1 mg/mL MHC-I–restricted peptide epitopes TRP-2 (melanoma Ag), ovalbumin (OVA), or heat-killed T. gondii ΔGRA17 Ag. Control samples were pulsed with culture media. IFN-γ–expressing T cells were detected and analyzed according to the manufacturer’s protocol (BD Bioscience).
We examined whether treatment efficacy could be extended to other tumor types via intratumoral administration of ΔGRA17 in mouse MC38 colon carcinoma and LLC lung carcinoma models. Briefly, a similar treatment schedule was followed as described above, except that MC38 or LLC tumor cells, instead of B16-F10 tumor cells, were injected in the right flank of mice. MC38 and LLC tumor volume was determined. We also studied the intratumoral immune infiltrates in LLC or MC38 tumor tissues. We determined the percentages of CD45+ immune cells, CD3+ T cells, CD4+ T cells, CD8+ T cells, natural killer (NK) cells, natural killer T (NKT) cells, macrophages and DCs in PBS-treated or ΔGRA17-treated LLC tumor cells and PBS-treated or ΔGRA17-treated MC38 tumor cells.
Systemic effects of ΔGRA17 on distant tumors
We investigated whether ΔGRA17 treatment has a systemic immune-enhancing effect on non-injected (distant) tumors. To establish a bilateral tumor model, 1×106 to 3×106 of B16-F10, LLC or MC38 cells were subcutaneously inoculated into the right flank of mice. Three days later, 5×105 to 8×105 cells of each cell line were subcutaneously inoculated into the left flank. Then, 5×104 ΔGRA17 tachyzoites were inoculated inside the right flak tumor as described above. We determined the volume of the inoculated and non-inoculated (distant) tumors over 29–31 days and monitored the overall mouse survival in the bilateral B16-F10, LLC, or MC38 tumor models over 40–50 days.
To detect the tumorigenesis of mouse melanoma, B16-F10-Luc cells were used to establish B16-F10-Luc tumors, and the tumor size was monitored by bioluminescence imaging using PerkinElmer IVIS Lumina XRMS instrument (PerkinElmer, Waltham, USA). Immediately before the in vivo imaging, mice received intraperitoneally 200 µL of VivoGlo fluorescein (15 mg/mL) (Promega, Madison, USA). Additionally, IHC staining and IRS analysis of mouse tumor tissues were performed as described above to determine the expression of CD63, S100β, and Ki67 in melanoma tissue. Furthermore, systemic therapeutic efficacy of ΔGRA17 was determined by comparing the volume of the injected and distant tumor (ie, MC38 colon carcinoma and the LLC lung carcinoma) and by monitoring the survival of mice.
Analysis of the immune status of the injected and distant tumors
We examined whether intratumoral administration of ΔGRA17 changes the immune status of non-injected (distant) tumors. A treatment schedule was followed as described in figure 1A, and B16-F10 tumors were harvested 24 hours after the second treatment (ie, on day 12). Dissected tumors were digested for preparation of single-cell suspensions followed by antibody staining for CD45+, CD3+, CD4+, CD8+, NK1.1, CD11b+, CD11c+, IFN-γ, TNF-α, Ki67, and granzyme B. Cytometry analysis and IFN-γ ELISPOT assay were performed as described above. The numbers of infiltrating immune cells in treated tumors and distant, untreated, tumors were determined and compared.
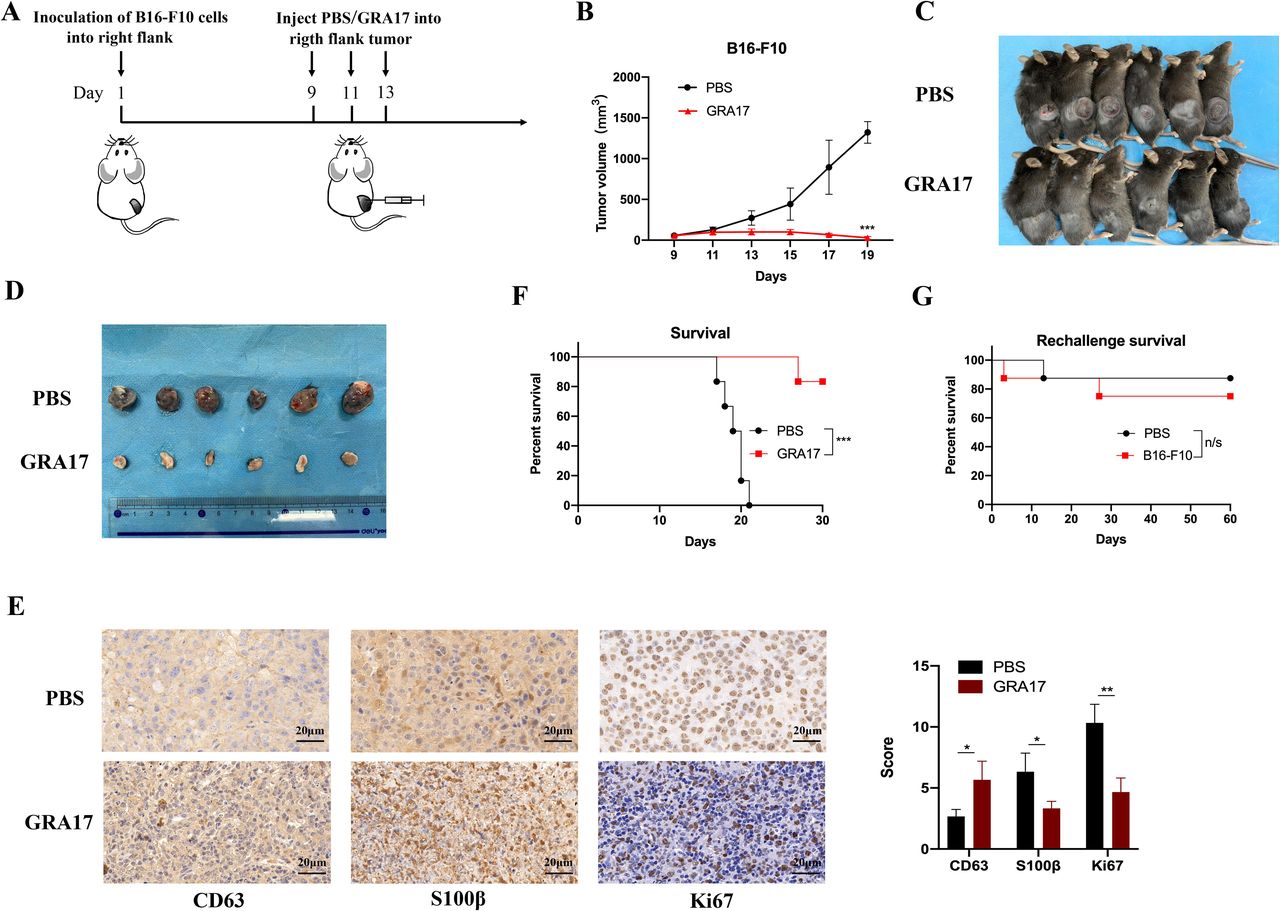
Toxoplasma gondii ΔGRA17 strain significantly reduces the growth of melanoma in mice. (A) Schematic representation of the treatment regimen and timeline for mice inoculated with B16-F10 cells in the right flank. Once the B16-F10 tumor size has reached 5 mm at ~9 day, the tumor was injected with 100 µL of phosphate buffered saline (PBS) (control) or 5×104 ΔGRA17 tachyzoites and then again at days 11 and 13 after implantation of B16-F10 cells. Using a vernier caliper, the tumor size was measured, starting from day 9 and every other day until 19 days after B16-F10 cells’ implantation. When the tumor size has reached ~15 mm, mice were humanely euthanized. (B) ΔGRA17 administration significantly reduced the tumor size compared with control mice treated with PBS only. (C) The size and location of the tumors detected in mice euthanized on day 19. (D) The tumors dissected from mice shown in (C), showing a significant reduction in the tumor size of ΔGRA17-injected mice compared with control mice. (E) Immunohistochemical analysis and the corresponding Immunoreactivity Score (IRS) of mouse melanoma markers CD63, S100β, and Ki67. (F) Survival curve of B16-F10-bearing mice shows a significant improvement in the survival of ΔGRA17-inoculated mice compared with control mice. (G) Tumor-free mice were reinoculated in the left flank with 1×106 to 3×106 B16-F10 cells at 30 days and their survival was monitored for 60 days (ie, 90 days after tumor implantation). The survival of tumor-bearing mice was presented as Kaplan-Meier plots and tested for significance using log-rank tests. Data are mean±SD (n=6 mice/group) of three independent experiments; unpaired Student’s t-test, *p<0.05, **p<0.01, ***p<0.001, n/s, not significant, compared with the indicated control.
To determine whether the regression of distant tumors was related to the immune responses evoked by TILs or to the systemic dissemination of ΔGRA17, we analyzed T. gondii-specific B1 gene expression of ΔGRA17 tachyzoites using quantitative PCR (qPCR). Briefly, tumor cells were injected into both flanks of wild type (WT) mice and NSG mice. On day 9, the right flank was injected with 5×104 ΔGRA17 tachyzoites, and 5 days later (ie, on day 14), the injected and the distal tumors were harvested from WT and NSG mice. Tumors were homogenized using the gentle MACS Dissociator (Miltenyi Biotec, Bergisch Bladbach, Germany) and genomic DNA was isolated using a DNA extraction Kit (TIANGEN, Beijing, China). The isolated DNA was used as a template to amplify T. gondii B1 gene. qPCR was performed using a SYBR Premix Ex Taq II (Takara, Tokoyo, Japan) on a Mx3005P real-time PCR System (Stratagene, Waltham, Massachusetts, USA) as described previously.44
We established a standard curve to quantify the number of ΔGRA17 tachyzoites from the Ct values obtained from qPCR analysis of tumor tissues. Briefly, DNA was isolated from a 10-fold dilution series of ΔGRA17 tachyzoites (106, 105, 104, 103). qPCR was carried out as described above. This experiment was repeated three times, and mean Ct values were used to generate a standard curve (R2=0.99, E=98%, y=−2.252x+31.02) by plotting the Ct values against the logarithm of the numbers of tachyzoites in each dilution. Linear regression analysis was used to fit the curve to the data. PCR amplification efficiency (E) was calculated from the slope of the standard curve using the equation E = [10−1/slope−1].
Profiling immune-related genes in treated and distant tumors of ΔGRA17-treated mice
We further examined whether intratumoral administration of ΔGRA17 changes the immune status of the distant tumors in B16-F10-bearing mice. NanoString platform (WuXi AppTec, Shanghai, China) was used to study the gene expression of the injected tumors, distant tumors, and control mouse groups. Briefly, B16-F10 tumor-bearing mice were treated with PBS or ΔGRA17 as described above. Twenty-four hours after the second treatment (ie, on day 12), RNA was isolated from the dissected tumors and hybridized with the PanCancer Mouse Immune Profiling panel and quantified using the digital analyzer (nCounter). Data were processed and normalized using nSolver Analysis Software and the Advanced Analysis module (NanoString Technologies, Waltham, Massachusetts, USA). The expression profile of immune-related pathways was determined in the injected and distant tumors. The ggpubr R package was used to construct the volcano plots and pheatmap R package was used to perform hierarchical clustering of tissue gene expression.
The expression levels of mRNA of genes (H2-K1, CD74, H2-Eb1, H2-T23, H2-Aa, Psmb8, CCL3, IL1b, CD80, Fas, IL2RA, and JAK3) involved in antigen processing and T cell function in the injected and distant tumors were determined by quantitative reverse transcriptase PCR (qRT-PCR). Briefly, total RNA was isolated from B16-F10 tumor tissue using Trizol reagent (Invitrogen, Waltham, Massachusetts, USA). DeNovix DS-11 spectrophotometer (DeNovix, Waltham, Massachusetts, USA) was used to detect the purity and concentration of RNA. qRT-PCR analysis was performed using SYBR Premix Ex Taq II (Takara, Tokyo, Japan) with a Mx3005P real-time PCR System (Stratagene, La Jolla, California, USA). The primer sequences are provided in the online supplemental material.
Supplemental material
Immunofluorescence analysis was also used to quantify the level of expression of PD-L1 in injected and distant tumors. Briefly, formalin-fixed, paraffin-embedded B16-F10 tumor tissue was cut into 6 µm. The sections were deparaffinized in xylene and rehydrated in a graded series of ethanol. For antigen retrieval, the sections were immersed in 10 mM sodium citrate (pH 6.0), heated in a water bath at 95°C–99°C for 20 min, and then cooled to room temperature. Then, slides containing B16-F10 tumor tissue were fixed for 10 min at 4°C in 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, USA) for 10 min at room temperature, and blocked for 1 hour in PBS with 1% horse serum (Sigma-Aldrich, Saint Louis, USA) overnight at 4°C and washed three times for 5 min with PBS. Slides were subsequently incubated with primary antibodies: anti-PD-L1 (1:100; clone: ab213524, Abcam, Cambridge, UK) and anti-Firefly-Luciferase (1:100, clone: ab21176, Abcam, Cambridge, UK). Alexa Fluor secondary antibody (1:500; ab150077, Abcam, Cambridge, UK) was added for 1 hour at room temperature in the dark. The slides were washed three times with PBS and DAPI (Sigma-Aldrich, Saint Louis, USA) was added to the first wash to stain the nuclei. Images were acquired using a Confocal Zeiss LSM880. Intensity analysis was performed using ImageJ V.1.51j8 (National Institutes of Health, USA).
To further examine how PD-L1 expression is influenced in B16-F10 cells following ΔGRA17 infection, B16-F10 cells cultured in vitro were PBS treated (control) or infected with ΔGRA17 tachyzoites and harvested after 24 hours. The cells were then blocked with a-CD16/32 Ab (clone 93, eBioscience; 1:1000) and stained with antibodies against mouse PD-L1. The cell surface expression levels of PD-L1 were shown as mean fluorescence intensity (MFI) values.
We examined whether T. gondiiΔGRA17 treatment increased expression of CD80 in the tumor microenvironment. CD45+CD11c+ cells from ΔGRA17-injected and distant B16-F10 tumor-bearing mice were analyzed for the expression CD80+ using flow cytometry.
Combination therapy using ΔGRA17 and PD-L1 blockade
To examine the efficacy of intratumoral therapy using ΔGRA17 and PD-L1 on bilateral (injected and distant) melanomas, B16-F10-bearing mice were treated with ΔGRA17 on one side in addition to intraperitoneal injection with or without 250 µg anti-PD-L1 antibody on days 9, 11, and 13. Control mice included age-matched B16-F10-bearing mice treated with PBS. The tumorigenesis or regression of mouse B16-F10-Luc cells was determined using bioluminescence imaging as described above. The tumor volume (up to 30 days) and survival (up to 60 days) of mice receiving monotherapy or dual therapy was determined. Body weight in each mouse group was also determined. Mice were deemed to be completely cured when no tumor was detected by palpation. Mice cured of B16-F10 tumors and age-matched tumor-naive (control) mice were rechallenged with the same tumors within 25 days. The previously cured mice were treated with a combination therapy (ΔGRA17 and PD-L1 blockade) in the right flank on day 60 to determine the long-term persistence of antitumor memory, while control mice received no treatment.
Role of immune cells in the efficacy of the dual ΔGRA17 plus PD-L1 blockade therapy
We determined which cellular subsets underlay the observed therapeutic effect of the combined therapy and investigated the role of cellular subsets during early and late stage of dual therapy. Briefly, mice were injected intraperitoneally with 500 µg monoclonal antibodies (Ab) to CD8+, CD4+, or NK1.1, either the day before tumor implantation (day –1) or on the first day of treatment (day 0), followed by injections of 250 µg every 5 days until mice have reached their humane endpoint. Animals that did not receive any depleting Ab for CD4+, CD8+, and NK cells were used as controls. The survival of mice was monitored two times per day.
Statistical analysis
Statistical analysis was performed using GraphPad Prism V.8 (GraphPad Software, California, USA). Two-tailed Student’s t-test was used to derive the significance of the differences between the two groups. Differences were considered significant when p<0.05. We used log-rank tests for the Kaplan-Meier survival analysis. The numbers of mice used in each experiment are provided in the figure legends. Data are presented as mean±SD. All experiments were performed in triplicate.
Results
T. gondii ΔGRA17 strain regresses B16-F10 melanoma
The dose-dependent tumor growth regression analysis identified 5×104 ΔGRA17 tachyzoites as the optimal dose to examine the efficacy of intratumoral administration of ΔGRA17 on the tumor growth (online supplemental figure S1). We evaluated the antitumor effects of ΔGRA17 on a unilateral flank B16-F10 tumor model in immunocompetent mice (figure 1A). In agreement with a previous study,26 intratumoral injection of ΔGRA17 resulted in growth arrest of the unilateral melanoma and a regression in the size of parasite-injected tumors (figure 1B, C and D) without any impact on the body weight of mice (online supplemental figure S2). To characterize the tumor growth, mice were killed on day 19 and B16-F10 melanoma tissue samples were collected, and the level of expression of three melanoma markers42 was determined using immunohistochemistry. Mice injected with ΔGRA17 exhibited a lower malignancy as shown by increased expression of CD63 tetraspanin, and lower metastatic burden as indicated by decreased expression of S100β and Ki67 (figure 1E). We also detected a significant increase in the survival of mice bearing B16-F10 melanoma (figure 1F). After 30 days, tumor-free mice were rechallenged with B16-F10 cells in the left flank. Mice were monitored for additional 60 days (ie, 90 days after tumor implantation). Approximately 70% of the rechallenged mice rejected the reimplanted tumors (figure 1G).
Supplemental material
Supplemental material
To test the therapeutic potential of ΔGRA17 monotherapy injected into non-tumor sites, we infected mice with ΔGRA17 intratumorally, intraperitoneally, or intravenously. As expected, intravenous and intraperitoneal injection of ΔGRA17 did not attenuate the tumor growth, nor did it improve the survival of mice with an established B16-F10 melanoma (figure 2A,B). This suggests that ΔGRA17 tachyzoites must be in the tumor microenvironment to induce protective immunity against a primary tumor.

Biological feasibility and characteristics of Toxoplasma gondii ΔGRA17 strain. (A) Changes in the tumor volume of mice injected with ΔGRA17 using the three indicated inoculation methods compared with phosphate buffered saline (PBS)-treated (control) mice. (B) Survival of B16-F10 tumor-bearing mice injected using the shown three different inoculation methods. (C) Tumor volume and survival of B16-F10 tumor-bearing mice treated with PBS or ΔGRA17 tachyzoites after establishment of a latent infection by T. gondii PRU strain. (D) Tumor size and survival of mice following inoculation with heat-inactivated ΔGRA17 tachyzoites. (E) Tumor size and survival of B16-F10 tumors in NOD/SCID/IL-2gR knockout (KO) mice. The survival of tumor-bearing mice was presented as Kaplan-Meier plots and tested for significance using log-rank tests. Data are mean±SD (n=5 mice/group) of three independent experiments; Student’s t-test, ***p<0.001, n/s, not significant, compared with the indicated control.
Latent infection is often accompanied with a pre-existing immunity that attenuates oncolytic virus activity and reduces treatment efficacy against tumors.16 Latent T. gondii infection is widespread worldwide,25 and was previously shown to not interfere with T. gondii uracil auxotroph immunotherapy of B16F10 tumors.26 Therefore, we determined if chronic toxoplasmosis interferes with the treatment efficacy by testing the efficacy of ΔGRA17 against B16-F10 melanoma in mice infected by T. gondii PRU strain, which produces a latent infection. Chronic infection did not influence the antitumor efficacy induced by ΔGRA17 (figure 2C), indicating that pre-existing T. gondii infection is unlikely to interfere with the antitumor efficacy of ΔGRA17 treatment.
Previous studies have demonstrated the necessity for live pathogens to achieve antitumor efficacy.26 On the other hand, T. gondii components can be effective in the regression of tumor outgrowth.45 To establish if live ΔGRA17 tachyzoites are necessary for antitumor efficacy, established tumors were inoculated with viable or heat-inactivated ΔGRA17 tachyzoites. Treatment with inactivated ΔGRA17 tachyzoites resulted in a major failure to delay tumor growth or to improve survival time in all treated mice (figure 2D), demonstrating the need for live pathogens to obtain antitumor efficacy.
Antitumor efficacy of intratumorally administrated pathogens often depends on an active host immune response being induced by their invasion into tumors.16 To reveal the requirement of an effector immune response for ΔGRA17-mediated clearance of pre-existing B16-F10 tumors, we examined the therapeutic efficacy of ΔGRA17 treatment in NSG mice-bearing B16-F10 tumors. No protection was observed in these immunocompromised mice (figure 2E), demonstrating that antitumor effect of ΔGRA17 is immune-mediated.
Increased TILs after administration of ΔGRA17
Although in vitro cytotoxicity against tumor cell lines has been reported in previous studies,46 an activated immune mechanism in the tumor microenvironment is the main indicator of antitumor efficacy in in vivo mouse models following therapy with T. gondii.37 IL-12 is required for CD8+ T cell-dependent immunity against T. gondii, and also IL-12 and IFN-γ production are indispensable for the therapeutic efficacy of non-replicating completely attenuated T. gondii CPS strain.34 47 48 In line with previous studies, we detected significant increase in the expression of IL12p40, IL12p70, and IFN-γ in the ΔGRA17-inoculated tumors (online supplemental figure S3). Therefore, we further analyzed intratumoral inflammatory cell infiltration by flow cytometry. Examination of the parasite-injected tumors revealed an increased intratumoral infiltration with immune cells expressing leukocyte common antigen CD45 (figure 3A). The intratumoral immune cell infiltrates were characterized by an increase in innate and adaptive immune components, including CD3+ T cells, CD4+ T cells, CD8+ T cells, NK cells, NKT cells, CD11b+ (macrophages), and CD11b+CD11c+ (dendritic cells, DCs) (figure 3A,B). Also, effector T cells expressing increased effector, proliferation, and lytic markers were detected in the injected tumors, including tumor necrosis factor-α (TNF-α) and IFN-γ, Ki-67, and granzyme B (figure 3C). Additionally, IFN-γ ELISPOT showed a significantly increased melanoma Ag (TRP-2)-specific CD8+ T cells after ΔGRA17 treatment (figure 3D). These data demonstrated that tumor specific CD8+ T cells response was elicited after ΔGRA17 inoculation.
Supplemental material

Toxoplasma gondii ΔGRA17 increases infiltration of tumors with lymphocytes. A treatment schedule shown in figure 1A was followed, B16-F10 tumors were harvested 24 hours after the second treatment (ie, on the 12th day), and digested for preparation of single-cell suspensions followed by antibody staining for CD45, CD3, CD4, CD8, NK1.1, CD11b, CD11c, interferon-gamma (IFN-γ), tumor necrosis factor-α (TNF-α), Ki67, and granzyme B. (A) Representative plots of the percentages of CD45+ cells, CD3 T cells, CD4+ T cells, CD8+ T cells, natural killer (NK) cells, macrophages, and dendritic cells (DCs) in phosphate buffered saline (PBS)-treated or ΔGRA17-treated tumors. (B) Percentages of CD45+ cells, CD3 T cells, CD4+ T cells, CD8+ T cells, NK cells, natural killer T (NKT) cells, macrophages, and DCs in PBS-treated or ΔGRA17-treated tumors. (C) Percentages of interferon-gamma (IFN-γ), tumor necrosis factor-α (TNF-α), ki67, and granzyme B in CD8+ T cells. (D) IFN-γ ELISPOT showing that tumor Ag TRP-2-specific CD8+ T cells in spleen are enriched in infiltrated lymphocytes. (E– G) A similar treatment schedule was followed as in figure 1A, except that LLC or MC38 tumor cells were used to inject the mice, instead of B16-F10 tumor cells. (E) LLC and MC38 tumor volume in mice. (F) Percentages of CD45+ immune cells, CD3 T cells, CD4+ T cells, CD8+ T cells, NK cells, NKT cells, macrophages, and DCs in phosphate buffered saline (PBS)-treated or ΔGRA17-treated LLC tumors. (G) Percentages of the CD45+ immune cells, CD3 T cells, CD4+ T cells, CD8+ T cells, NK cells, NKT cells, macrophages, and DCs in PBS-treated or ΔGRA17-treated MC38 tumors. Data are mean±SD (n=6 mice/group) of three independent experiments; Student’s t-test, **p<0.01, ***p<0.001 compared with the indicated control.
We examined the effect of intratumoral administration of ΔGRA17 in MC38 colon carcinoma and LLC lung carcinoma models to determine if treatment efficacy could be extended to other tumor types. As with the B16-F10 model, this treatment strategy also regressed growth of the injected tumor, which accords with intratumoral immune infiltrates in these two models (figure 3E, F and G).
Effects on non-injected distant tumors
To test if intratumoral injection with ΔGRA17 is able to provoke systemic antitumor activity, we evaluated antitumor activity using a bilateral B16-F10 melanoma model, where mice had subcutaneous B16-F10 melanoma inoculated into both flanks (figure 4A). Intratumoral therapy with ΔGRA17 delayed growth of the inoculated tumor and the distant tumors, in addition to increasing the mouse survival (figure 4B). This finding supports the potential efficacy of this parasite therapy against distant metastatic disease.

Intratumoral administration of Toxoplasma gondii ΔGRA17 inhibits the growth of non-injected (distant) tumors. (A) Schematic of treatment regimen and timeline for mice inoculated with B16-F10, LLC, or MC38 cells. (B) Growth kinetic of injected tumors and distant tumors, and the overall animal survival in a bilateral B16-F10 melanoma mouse model. (C) Representative luminescence images of mice injected with melanoma B16-F10- Luc cells and treated with ΔGRA17 compared with control mice treated with phosphate buffered saline (PBS). (D) Representative photos of injected and distant melanoma tumors. (E) The size of the injected and distant melanoma tumors, corresponding to the mice in (D). (F) Immunohistochemical staining analysis of melanoma malignancy and the corresponding Immunoreactivity Score (IRS) of CD63, S100β, and Ki-67 expression. (G) Tumor size and mouse survival in bilateral LLC tumor model. (H Tumor size and mouse survival in bilateral MC38 tumor model. The survival of tumor-bearing mice was monitored by Kaplan-Meier analysis, and statistical analyses were performed with log-rank test. Data are shown as mean±SD (n=6 mice/group) of three independent experiments; Student’s t-test, *p<0.05, **p<0.01, ***p<0.001, n/s, not significant compared with the indicated control.
Remarkably, luminescence images from mice treated with ΔGRA17 showed a reduced value of the luminescence color in the inoculated and distant tumors compared with PBS-injected controls, and even no value of the luminescence color was observed in some ΔGRA17-injected tumors compared with that in controls, which may indicate a tumor free by luminescence image analysis. However, these injected right-sided tumors with no value of the luminescence color could still be observed, but appeared as degraded white tissue (figure 4C, D and E). Therefore, we characterized the degree of malignancy in the bilateral tumors using IHC staining. Our findings showed a lower degree of malignancy with increased expression of CD63, and lower tumor proliferation and metastasis with decreased expression of S100β and Ki67 in the ΔGRA17-injected tumors (figure 4F). Systemic therapeutic efficacy of ΔGRA17 may also extend to other tumor types, including MC38 colon carcinoma and the LLC lung carcinoma models (figure 4G,H).
Analysis of the uninjected tumors revealed significant changes in immune status, which was identical to the infiltration pattern of immune cells observed in the injected left flank tumors, characterized by increased numbers of innate immune cells and adaptive T cells (figure 5A,B). Notably, characterization of CD4+ and CD8+ TILs from mice receiving the ΔGRA17 therapy demonstrated downregulation of Foxp3-expressing CD4+ cells and PD-1-expressing CD8+ cells with a larger percentage of IFN-γ and TFN-α-expressing CD8+ cells in local and distal tumors (figure 5B,C). These data suggest that ΔGRA17 therapy can modulate the immunosuppressive tumor microenvironment by enhancing the proliferation and activation of the tumor-infiltrating cytotoxic T cells, and reduction of immunosuppression.

Intratumoral administration of Toxoplasma gondii ΔGRA17 strain the immune status of non-injected distant tumors. The treatment schedule shown in figure 4A was followed, B16-F10 tumors were harvested 24 hours after the second treatment (ie, on day 12), and digested for preparation of single-cell suspensions followed by antibody staining for CD45+, CD3+, CD4+, CD8+, NK1.1, CD11b+, CD11c+, interferon-gamma (IFN-γ), tumor necrosis factor-α (TNF-α), Ki67, and granzyme B. (A) The numbers of infiltrating CD45+ cells, dendritic cells (DCs), natural killer (NK), natural killer T (NKT), CD4+, and CD8+ T cells in treated tumors and distant, untreated, tumors. (B) Representative plots of IFN-γ, TNF-α, programmed death-1 (PD-1), Ki-67, and granzyme B of tumor-infiltrating CD8+ T cells and FoxP3 of tumor-infiltrating CD4+ T cells in injected tumors and distant tumors. (C) Concentrations of IFN-γ, TNF-α, PD-1, Ki-67, granzyme B, and regulatory T cells (Tregs). (D) IFN-γ ELISPOT showing tumor Ag TRP-2-specific CD8+ T cells enriched in intratumoral infiltrated lymphocytes. Data are shown as mean±SD (n=6 mice/group) from three independent experiments; two-tailed Student’s t-test, ***p<0.001, n/s, not significant, compared with the indicated control.
IFN-γ ELISPOT revealed that tumor Ag TRP-2-specific CD8+ T cells are enriched in the infiltrated lymphocytes in the injected and distal tumors after ΔGRA17 immunotherapy (figure 5D). We investigated whether the regression of contralateral tumors was directly due to the immune responses evoked by TILs or systemic spread of the parasite. We used qPCR to establish a standard curve so that Ct values of T. gondii-specific B1 gene expression could be used to determine the number of ΔGRA17 tachyzoites and the level of infection in injected and distant tumors harvested from WT and NSG mice. Intratumoral administration of ΔGRA17 into the right flank tumor resulted in parasite replication within the injected tumors of WT and NSG mice. However, no tachyzoites were detected in the distant left flank tumor of WT mice (online supplemental figure S4). Interestingly, tachyzoites were detected in the distant tumors of the NGS mice. These findings suggest that T. gondii infection in WT mice was largely limited to the injected tumors, and that therapeutic efficacy against contralateral tumors was mediated via immune responses induced by TILs following the treatment with ΔGRA17 and not due to the systematic distribution of ΔGRA17 parasites.
Supplemental material
ΔGRA17 therapy induces immune changes in treated and distant tumors
Although intratumoral injection of ΔGRA17 significantly reduced the growth of injected tumors, only moderate or low therapeutic efficacy was observed in distant tumors. This may have been due to insufficient inflammatory efficacy in driving complete tumor rejection in distant tumors. Thus, the potential inhibitory mechanisms of this therapy, which has been previously characterized by local injection with oncolytic virus,49 50 could be involved in dampening the immune response in the microenvironment of ΔGRA17-injected and distant tumors. To reveal any potential changes in immune pathways, NanoString was used to analyze the expression level of immune-related genes in injected and distant tumors of ΔGRA17-injected mice. We detected an increase in the expression in a range of immune checkpoint genes, including PD-L1 (figure 6A), a few of these are being explored as targets for cancer immunotherapy.51 A considerable number of immune response-related pathways in injected and distant tumors were also changed after treatment with ΔGRA17 (figure 6B).

Toxoplasma gondii ΔGRA17 changes immune status inside the tumors. (A) Volcano plots show overall gene expression levels of the injected (T2), distant tumors (T1) and control group (C) as determined by using a NanoString platform. (B) Heatmap shows the expression profile of immune-related pathways in B16-F10-bearing mice (n=4 mice per group) treated with phosphate buffered saline (PBS) or 5×104 ΔGRA17 tachyzoites. Red and blue color indicate high and low expression, respectively. (C) Upregulation of programmed death ligand-1 (PD-L1) in tumor cells in injected tumors and distant tumors as revealed by immunofluorescence analysis. (D) Expression profile of antigen processing genes of tumors from B16-F10-bearing mice (n=8 mice/group) treated with PBS or ΔGRA17. Red indicates high expression, and blue indicates low expression. (E, F) Expression of representative antigen processing-related genes, chemokine genes and T cell response-related genes in the injected and distant tumors was determined by quantitative reverse transcriptase PCR. Data are shown as mean±SD of three independent experiments; Student’s t-test, *p<0.05, **p<0.01, **p<0.001, n/s, not significant, compared with the indicated control.
Among these upregulated immune-inhibitory targets, we focused on the PD-1/PD-L1 pathway, given its promising role in T cell exhaustion during chronic viral infection by hepatitis B virus (HBV), hepatitis C virus (HCV), and HIV, as well as in anticancer therapies targeting these checkpoints and OV in combination with systemic PD-1 blockade to treat cancer.52 53 Immunofluorescence analysis showed a significant increase in the expression of PD-L1 in tumor cells in the injected and distant tumors in response to ΔGRA17 treatment, while non-tumor cells did not contribute to PD-L1 expression (figure 6C). To further confirm that PD-L1 expression was upregulated in B16-F10 cells following ΔGRA17 infection, cultured B16-F10 cells were either sham-treated with PBS (control) or infected by ΔGRA17, and harvested after 24 hours. Flow cytometry revealed that PD-L1 expression in ΔGRA17-infected cells was upregulated (online supplemental figure S5). These results showed increase in PD-L1 expression following ΔGRA17 infection both in vivo and in vitro.
Supplemental material
Gene expression analysis revealed an increased expression of genes related to T cell function, immune pathways involved in antigen processing, interferon, inflammation, chemokines, and receptors, in ΔGRA17-injected and distant tumors (figure 6D and online supplemental figures S6,S7,S8,S9). Using qRT-PCR, we detected a significant increase in the expression of mRNA for genes involved in antigen processing and increased T cell function (figure 6E,F). We detected increased expression level of CD80+ in CD45+CD11c+ cells in injected and distant tumor tissues compared with that in control (online supplemental figure S10), indicating a comparative antigen processing in both ΔGRA17-injected and distant tumors. Results demonstrated a more intense T cell function in ΔGRA17-injected tumors than in distant tumors.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
ΔGRA17 and PD-L1 blockade synergize to suppress local and distant tumors
We hypothesized that intratumoral administration of ΔGRA17 induced changes in immune status, with enhanced TILs and the elevated PD-L1 expression by cancer cells in the ΔGRA17-injected and distant tumors, which would sensitize tumors to PD-L1 blockade therapy, as has been shown in patients with pre-existing tumor inflammatory infiltrates.54
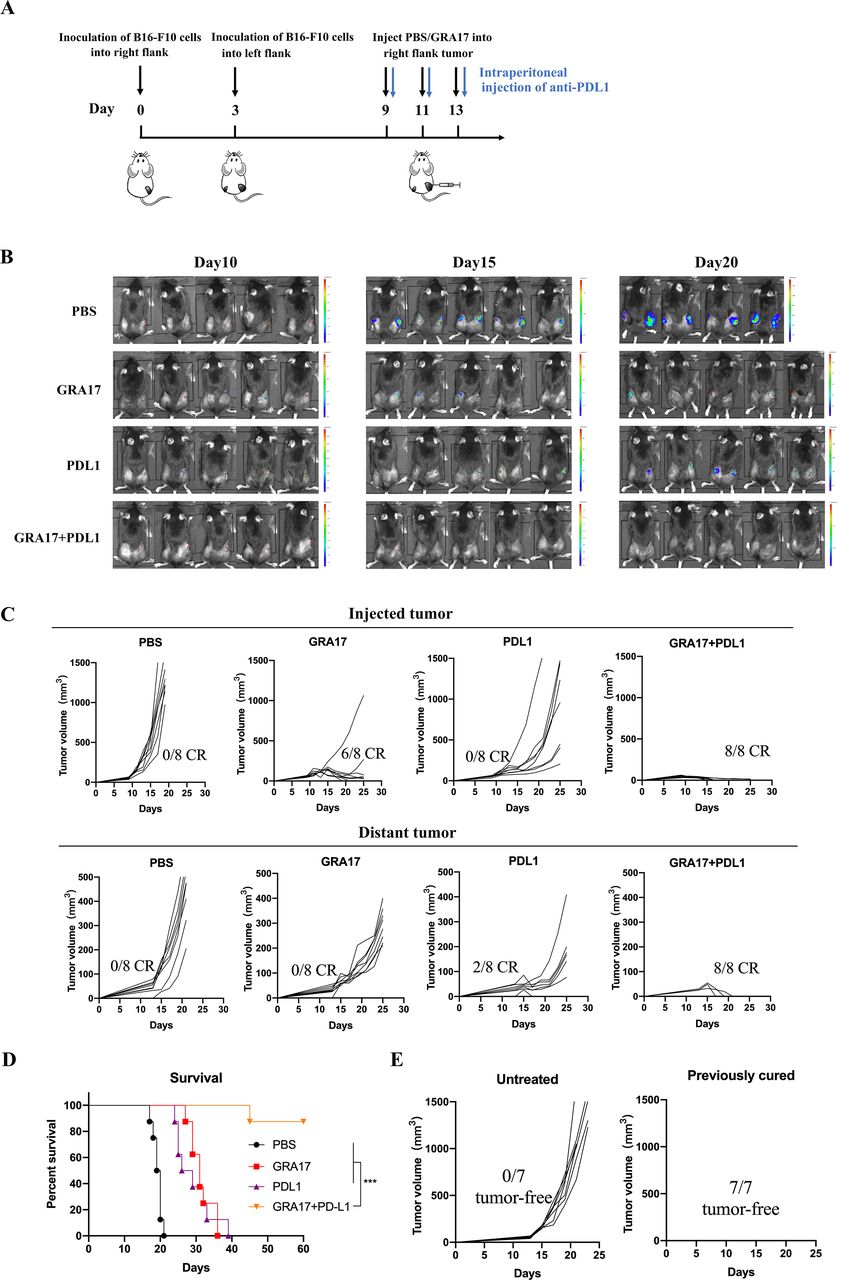
To analyze the effects of combined ΔGRA17 and PD-L1 blockade treatment on the bilateral melanoma, ΔGRA17 treatment was administered with or without anti-PD-L1 antibody into tumors on one side (figure 7A). Monotherapy with ΔGRA17 or anti-PD-L1 antibody showed modest efficacy (figure 7B) and induced no or 2/8 regression in the distant tumors in mice (figure 7C). In contrast, a combination of ΔGRA17 and PD-L1 blockade induced a much earlier tumor complete regression than ΔGRA17 monotherapy (figure 7B,C), prolonged survival of mice (figure 7D), did not influence the mouse body weight (online supplemental figure S11), and resulted in complete regression of bilateral tumors (online supplemental figure S12).
Supplemental material
Supplemental material

Local and abscopal effects of intratumoral Toxoplasma gondii ΔGRA17 plus programmed death ligand-1 (PD-L1) blockade. (A) Schematic of treatment regimen and timeline for mice inoculated with B16-F10 cells. (B) Bioluminescence monitoring of B16-F10-Luc cells in live mice at days 10, 15, and 20. (C) Mice that achieved complete regression against B16-F10 for about 3 months after treatment with ΔGRA17 and/or PD-L1 blockade and age-matched treatment-naive mice were subcutaneously inoculated with B16-F10 cells. Tumor growth for individual mice is shown (n=8 mice/group). (D) Survival of B16-F10 rechallenged mice which achieved complete regression of melanoma after treatment. (E) Mice that achieved complete regression against B16-F10 rechallenge after treatment with ΔGRA17 in local and abscopal tumor models. The survival of tumor-bearing mice (n=8 mice/group) was presented as Kaplan-Meier plots and tested for significance using log-rank tests. Student’s t-test, ***p<0.001 compared with the indicated control.
We determined the long-term persistence of antitumor memory. Mice cured of B16-F10 tumors and control mice were rechallenged with B16-F10 tumor cells within 25 days, then treated with ΔGRA17 and PD-L1 blockade in the right flank on day 60, while control mice received no treatment. B16-F10 tumors in the previously cured mice showed complete regression against rechallenge of the same tumors within 25 days, whereas all age-matched tumor-naive control mice developed tumors (figure 7E).
Role of immune cells in the efficacy of ΔGRA17 plus PD-L1 blockade therapy
We determined which cellular subsets underlay the efficacy of the dual therapy and whether specific cellular subsets play a role during the early or late stage of the therapeutic effect. An immune cell depletion experiment was performed to investigate the contribution of the infiltrated CD4+, CD8+, and NK cells to the therapeutic efficacy of the combined treatment. B16-F10 tumor-bearing mice receiving the combination therapy (ΔGRA17 and PD-L1 blockade) were injected with depletion Ab at the timepoints shown (figure 8A). Depletion of either NK or CD8+ cells led to abrogation of the therapeutic effect, in both ΔGRA17-injected and distant tumors, with a significant reduction in survival (figure 8B,C). These results showed the essential roles played by NK and CD8+ cells in the therapeutic efficacy of the dual therapy. Remarkably, NK cells were essential for therapeutic efficacy earlier than CD8+ cells, suggesting NK cells may be immediate immune responders to therapeutic treatment with ΔGRA17. On the contrary, depletion of CD4+ cells did not cause any significant alteration in the treatment outcome.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Antitumor activity of Toxoplasma gondii ΔGRA17 and programmed death ligand-1 (PD-L1) blockade therapy depends on CD8+ and natural killer (NK) cells. (A) Schematic showing the experimental protocol, including timeline for inoculation of B16-F10 cells, treatment with phosphate buffered saline (PBS) or ΔGRA17, and depletion of CD4+, CD8+, and NK cells using monoclonal antibodies. (B) Survival plot of mice that received the depleting antibodies before tumor implantation. (C) Survival plot of mice that received the depleting antibodies on day 0 of therapy. The survival of tumor-bearing mice (n=8 mice/group) was presented as Kaplan-Meier plots and tested for significance using log-rank tests. Student’s t-test, ***p<0.001, n/s, not significant compared with the indicated control.
Discussion
Recent clinical trials using immunotherapy in the treatment of solid tumors, including immune checkpoint-targeted therapies, have produced durable therapeutic effects against advanced metastatic cancers, such as melanoma, metastatic lung, kidney, and bladder carcinoma.55 56 However, the most common cancers are resistant to anti-PD-1/PD-L1, anti-CTLA-4, and their combination, and are referred to as ‘cold’ tumors. This failure of immunotherapy may be attributed to their low or no mutational burden and neoantigen load, poor MHC presentation, and insufficient T cell infiltrates, and insensitivity to ICB.5 57–61 Improvement in the treatment efficacy against poorly immunogenic tumors requires new treatment interventions that can induce a robust and durable antitumor immune response capable of markedly thwarting the tumor progression and prolonging patient survival.
The present study explored whether a combined treatment approach involving attenuated T. gondii ΔGRA17 strain and anti-PD-L1 therapy would improve the tumor immunogenicity and overcome resistance to ICB-based therapies. Our results showed that intratumoral administration of ΔGRA17 tachyzoites not only changed the immune status in the tumor microenvironment (leading to regression of local and distant tumors in B16-F10 melanoma, MC38 colon carcinoma, and LLC lung carcinoma mouse models), but also sensitized B16-F10 melanoma to ICB, which is refractory to treatment with anti-PD-L1 immunotherapy.
In clinical trials, the presence of TILs, especially high baseline levels of innate and adaptive immune responses in tumor, indicates they are potentially responsive to immunotherapy.3 62 In our study, to trigger an inflammatory response in tumors, we used avirulent T. gondii RH ΔGRA17 strain, which is a potent inducer of T cell maturation and expansion as shown by the increased concentrations of IL-12 and IFN-γ41 that also have an effective antitumor immunity.17 37 In our studies, we chose a bilateral flank melanoma model and poorly immunogenic MC38 colon carcinoma and LLC lung carcinoma model, which have been previously recognized to be unaffected by concurrent immunity. We found that intratumoral inoculation of ΔGRA17 led to an immune infiltration, with activation and expansion of DCs, CD4+ T cells, CD8+ T cells, NK cells, and NKT cells in injected and distant tumors, suggesting a favorable immunological response to therapy.63 64
In addition to the recruitment of TILs to lymphocyte-deficient sites, the goal of immunotherapy is to reverse the suppression of immune effector functions.65 Regulatory T cells (Tregs) and MDSCs can inhibit immune lymphoid function via local immune counter-regulatory pathways, leading to immunosuppression.66–68 Other immunosuppressive pathways, including the upregulation of inhibitory CTLA-4 and PD-1 receptors on lymphocytes and direct tumor expression of inhibitory ligands, such as PD-L1, B7-H3, and B7x, can be used by tumors to limit T-cell responses.69 We found that intratumoral administration of ΔGRA17 increased the infiltration of CD8+ cytotoxic T cells, increased expression of IFN-γ and TNF-α while reversing immunosuppressive pathways and decreased the expression of Foxp3 in CD4+ T cells and PD-1 in CD8+ T cells. It remains unknown what causes the relative reduction in Tregs, but parasite-relevant inflammatory cytokines have been shown to counter Tregs activity in Newcastle disease virus-injected tumors.70
As biotherapeutics, OVs and tumor-targeting bacteria can act at primary and metastatic tumor sites and this can be exploited when determining a locoregional or systemic delivery strategy.16 17 In our study, we found that local injection of ΔGRA17 regressed tumors, including non-injected distant tumors. This regression was associated with increased infiltration of immune cells, upregulated immune-related pathways and enhanced innate and adaptive immune responses (eg, enhanced antigen processing, chemokines, T cell responses). Interestingly, the ΔGRA17 strain was not detected in non-injected distant tumors of WT C57BL/6 mice. It is unclear whether ΔGRA17 tachyzoites have spread to the distant site, its draining lymph node, or another location, or at a time point not assessed in the present study. These findings suggest that local injection of ΔGRA17 activated immune cells in the treated tumors, and these immune cells were subsequently recruited to distant tumors possibly via chemokines expressed in different tissue sites, which contributed to the clearance of the parasite in non-injected distant tumors. This mechanism underlies intratumoral oncolytic virotherapy, which can drive the activation of both antivirus and antitumor immunity. However, this vaccination strategy alone is of limited therapeutic value.16 17
The therapeutic efficacy against distant tumors was weak using ΔGRA17 monotherapy, although the significant increase of TILs in tumors, suggesting the immunosuppressive nature of the tumor microenvironment.60 In this study, we revealed the potential resistance mechanisms to ΔGRA17-induced immune activation. We detected that ΔGRA17 monotherapy up-regulated a few immune-inhibitory pathways, including PD-L1, in both injected tumors and distant tumors not injected by the parasite. This upregulation was most likely induced by intratumoral IFN-γ.71 72 Although PD-L1 generally down-regulates, or even dampens, T cell activity in tumor microenvironment,73 74 recent clinical trials have shown that high tumor positivity for PD-L1 is common in patients who preferentially respond to PD-1/PD-L1 and CTLA-4 blockade.3 5 75 76 Even though these immune-inhibitory mechanisms can reduce the efficacy of immunotherapy in the tumor microenvironment, the blockade of some of these mechanisms, using combination therapies, can lead to better therapeutic effects, for example, using local immunotherapies with OVs to prime ‘cold’ tumors for systemic ICB.16 17 In line with this assertion, our findings showed that a combination of intratumoral ΔGRA17 and systemic anti–PD-L1 antibodies led to the regression of distant B16-F10 tumors, together with improved mouse survival. The completely cured mice were also protected against tumor rechallenge, suggesting an establishment of a persistent immunity, although we did not determine whether memory lymphocytes resided in the bone marrow or the lymphoid organs.77 Infiltrated T lymphocytes with potent antitumor memory in the tumor microenvironment are known to correlate with a more favorable outcome and better survival in clinical immunotherapy studies.78–80 Further clinical studies are needed to reveal specific immune cells responsible for the persistent antitumor immunity observed in the present study. We also detected therapeutic efficacy in MC38 and LLC tumor models, which have previously exhibited poor sensitivity to immunotherapy. These results highlight that intratumoral inoculation with ΔGRA17 turned ‘cold’ tumors into ‘hot’ tumors and primed the tumors to PD-L1 inhibition in tumor models that were unresponsive to such treatment strategy. These results further support the concept that activation of immune responses and prevention of immuno-suppressive responses are needed for an effective antitumor therapeutic outcome.55
We detected an increase in immune cell infiltrates and regression of distant tumors together with better survival, without evidence of disease recurrence in the lungs. B16-F10 melanomas have the propensity for spontaneous metastases in the lungs, which can be mitigated by intratumoral oncolytic virotherapy.81 It is interesting to determine whether a similar effect exists in animal models with pre-established metastases. The present study demonstrated that inflammatory responses and PD-L1 upregulation in tumors treated with ΔGRA17 can sensitize these tumors to PD-L1 inhibition; however, we did not monitor the inflammatory response in tumors. In one clinical trial, intratumoral administration of an oncolytic herpes virus, HSV1716, induced transient increases of fluorodeoxyglucose-positron emission tomography activity in some patients, which is consistent with a tumor inflammatory reaction.82 A recent study demonstrated that in vivo monitoring of tumor infiltration by CD8+ T cells could predict the therapeutic efficacy of anti-CTLA-4 therapy using a Zr-labeled anti-CD8 antibody fragment in animal models.83 These studies support in vivo monitoring of tumor infiltration, possibly by imaging CD8+ lymphocytes. Our study primarily focused on characterizing the adaptive immune response; however, it is important to determine the role of monocytes and NK cells, which are also upregulated in distant tumors in ΔGRA17-injected mice. Our findings showed that NK cells may contribute to early regression of tumor, but CD8+ T cells are required for long-term tumor control, suggesting that further experiments are warranted to clarify and confirm this observation.
Intratumoral administration of reovirus, in combination with systemic PD-1 blockade, led to a synergistic tumor growth regression, which was dependent on NK and CD8+ T cells.84 In contrast to intratumoral injection of ΔGRA17, no significant PD-L1 upregulation was observed in response to administration of reovirus alone, with an increased expression of PD-L1 being ascribed to the tumor-infiltrating NK cells. Therapeutic efficacy of oncolytic herpes simplex virus expressing IL-12, in combination with PD-1 inhibition, in a glioma model is dependent on CD4+ T cells, CD8+ T cells, and macrophages.85 86 Notably, the combination of vesicular stomatitis virus and adoptive cell therapy expended the PD-1+CD8+ T cells, but an addition of anti-PD-1 antibody failed to improve their therapeutic efficacy.87 88 The differences between these studies may probably be attributed to intrinsic differences between the different organisms and indicate that results with one pathogen should not be extrapolated to all pathogens.
A previous study showed that deletion of GRA17 in T. gondii type II ME49 strain resulted in a partial loss of the parasite virulence where immunocompetent mice challenged intraperitoneally with ME49 ΔGRA17 survived infection doses up to 1×105 parasites, but a dose of 1×106 tachyzoites caused a 100% death rate.89 The ΔGRA17 strain is a replicating, partially attenuated strain, as shown in the present and previous study.41 This raises safety concerns regarding the virulence of ΔGRA17 strain in any immuno-therapeutic intervention particularly immunocompromised hosts like patients with cancer. ΔGRA17 tachyzoites were detected in the distant tumors of NOD/SCID/IL-2R g-chain knockout mice, showing that systematic dissemination of tachyzoites can occur from the inoculated tumors to a contralateral site in immunodeficient mice. Given the virulence of ΔGRA17 strain at moderate infection doses and its tolerance to pyrimethamine, the frontline therapeutic agent currently used to treat T. gondii infection in humans, and the inability of immunocompromised hosts to clear a replicating strain, the use of ΔGRA17 should be only limited to biological discovery. In the present study, ΔGRA17 strain was used as a model to test whether parasite therapy can make unresponsive melanoma tumors responsive to immunotherapy with anti-PD-1 blockade.
In summary, we have shown that intratumoral administration of T. gondii RH ΔGRA17 tachyzoites induces systemic inflammatory responses in injected and non-injected distant tumors. We have also shown that localized ΔGRA17 therapy was able to sensitize poorly immunogenic tumors to ICB treatment. Combined treatment with ΔGRA17 tachyzoites and anti-PD-L1 therapy significantly extended the survival of mice and suppressed tumor growth in preclinical mouse models of melanoma, Lewis lung carcinoma, and colon adenocarcinoma. Attenuation of the tumor growth was detected in the injected and distant melanoma tumors, which was associated with upregulation of innate and adaptive immune pathways. Complete regression of tumors was underpinned by late IFN-γ-producing CD8+ cytotoxic T cells. The lack of interference of immunity to T. gondii with the antitumor immunogenic effect of ΔGRA17 is an added advantage over the approach using OVs. Our data showed that T. gondii-induced immunotherapy improves the responsiveness of melanoma tumors to immunotherapy with PD-L1 blockade, as shown by the better treatment outcome compared with monotherapy. T. gondii ΔGRA17 should be used in further studies to identify the correlates of protection and reveal the molecular and immunological mechanisms needed to augment the efficacy of anti-PD-L1 therapy.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. The remaining data are available within the Article, Supplementary Information or available from the authors upon request. Data are available upon reasonable request. Contact email: zhuyuchao9@qq.com.
Ethics statements
Patient consent for publication
Ethics approval
All the experimental protocols were reviewed and approved by the Research Ethics Committee of Ningbo University (Permit No. SYXK (ZHE) 2019-0005).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JC, HME and X-QZ conceived and designed the study. YZ, SF and J-HW performed the experiments and assisted in the preparation of the early draft of the manuscript. YZ, JH, SF and JC analysed the data. YZ and JC prepared the initial draft of the manuscript. HME and X-QZ contributed significantly to the critical analysis and interpretation of the data, and preparation and revision of the manuscript. JC is the guarantor.
Funding This research was funded by the National Key Research and Development Program of China (Grant No. SQ2021YFC2300162), the Fund for Shanxi “1331 Project” (Grant No. 20211331-13), the State Key Laboratory of Veterinary Etiological Biology (Grant no. SKLVEB2019KFKT017), the National Natural Science Foundation of China (Grant No. 8217071207) and Yunnan Expert Workstation (Grant No. 202005AF150041). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.