Article Text

Abstract
Background No standard therapies beyond first line are established for advanced squamous cell anal carcinoma (aSCAC). Earlier preliminary data suggest activity of epidermal growth factor receptor (EGFR) inhibition and programmed cell death ligand (PD-(L))1 blockade in patients with previously treated disease. Aim of this study was to explore activity and safety of avelumab with/without cetuximab in patients with aSCAC.
Methods In this open-label, non-comparative, ‘pick the winner’, multicenter randomized phase II trial (NCT03944252), patients with aSCAC progressing after one or more lines of treatment were randomized 1:1 to the anti-PD-L1 agent avelumab alone (arm A) or combined with cetuximab (arm B). Overall response rate (ORR) was the primary endpoint. With one-sided α error set at 0.05 and power of 80%, at least 4 responses out of 27 patients per arm had to be observed to declare the study positive. Secondary endpoints were progression free survival (PFS), overall survival (OS), and safety.
Results Thirty patients per arm were enrolled. Three patients in arm A and five in arm B achieved partial response: primary endpoint was reached in combination arm. ORR was 10% (95% CI 2.1 to 26.5) and 17% (95% CI 5.6 to 34.7) in arms A and B; disease control rate was 50% (95% CI 31.3 to 68.7) in arm A and 57 (95% CI 37.4–74.5) in arm B. At a median follow-up of 26.7 months (IQR 26.5–26.9), median PFS was 2.0 months (95% CI 1.8 to 4.0) in arm A and 3.9 (95% CI 2.1 to 5.6) in arm B. Median OS was 13.9 months (95% CI 7.7 to 19.4) in arm A and 7.8 (95% CI 6.2 to 11.2) in arm B. Acceptable safety profile was observed in both arms.
Conclusions CARACAS study met its primary endpoint in arm B, documenting promising activity of dual EGFR and PD-L1 blockade in aSCAC.
- clinical trials
- phase II as topic
- immunotherapy
- gastrointestinal neoplasms
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Although squamous cell anal carcinoma (SCAC) is a rare malignancy and accounts for around 2% of all gastrointestinal tumors,1 its annual incidence has steadily risen over the past 15 years.2 About 90% of cases are linked to human papillomavirus (HPV) infection and additional risk factors include smoking, HIV or other causes of immunosuppression.
The management of patients with advanced disease has historically consisted of palliative chemotherapy, mainly cisplatin and 5-fluorouracil (5-FU) combination, based on the limited evidence of retrospective case series.3 More recently, the randomized phase II InterAACT study of initial therapy with either cisplatin/5-FU or carboplatin/paclitaxel established the latter regimen as another standard treatment option.4 The DCF (docetaxel, cisplatin and 5-FU) regimen at standard or modified dosage was recently tested in chemotherapy-naïve patients with advanced SCAC in a single-arm, phase II trial. With both dosages, DCF showed remarkable disease control rate (DCR) and durable progression-free survival (PFS) and is nowadays another option in first-line treatments for patients in good clinical conditions.5
Nevertheless, the outcome of patients with advanced SCAC is poor and no evidence-based options are currently available after failure of first-line chemotherapy.
The sensitivity of HPV-associated cancers to immune checkpoint inhibitors may be sustained by the expression of highly antigenic viral proteins and the upregulation of programmed cell death protein 1 (PD)-1/programmed cell death protein ligand 1 (PD-L1) axis, which is responsible of immune-escape, establishment of chronic HPV infection and cancer initiation.6 7 Even though tumor mutational burden (TMB) is low in the majority of SCACs, some tumors—mainly the HPV-negative ones—may be hypermutated.8 9 Based on these considerations, anti-PD-(L)1 agents are under clinical investigation in patients with SCAC. PD-1 blockade with either nivolumab or pembrolizumab achieved modest activity in patients with chemorefractory and advanced disease, but tumor responses were durable and independent from tumor PD-L1 expression. The safety profile of these agents was consistent with the literature and apparently not impacted by HIV infection.10–12
The antiepidermal growth factor receptor (EGFR) agents cetuximab or panitumumab achieved responses in small retrospective series of patients with advanced SCAC.13 14 The rationale for investigating EGFR inhibition in SCAC is based on the broad expression of EGFR15 which is activated by the HPV-associated E5 protein16 and the rarity of KRAS mutations in this tumor type (about 5%).17
Cetuximab exerts antibody-dependent cytotoxicity (ADCC) on tumor cells mediated by natural killer (NK) cells.18 The secretion of interferon gamma by NK cells may induce tumor PD-L1 expression,19 leading to potential synergistic activity between PD-L1 and cetuximab. Avelumab, a fully human IgG1 anti-PD-L1 antibody, has itself ADCC properties and has been approved for the treatment of advanced Merkel cell carcinoma,20 by the European Medicines Agency for advanced renal cell carcinoma21 and by Food and Drug Administration (FDA) as maintenance therapy after chemotherapy in metastatic urothelial carcinoma.22
Recently, avelumab plus cetuximab combination showed a manageable safety profile as third-line therapy of anti-EGFR-experienced patients with RAS wild-type metastatic colorectal cancer.23 Moving from this background, we designed a randomized phase II trial of avelumab or avelumab plus cetuximab for patients with previously treated, locally advanced or metastatic SCAC.
Methods
Study design and participants
The CARACAS trial (NCT03944252) was a multicenter, open-label, ‘pick-the-winner’, randomized phase II trial promoted by the Gruppo Oncologico Nord Ovest Foundation (online supplemental file 2). The study involved 17 centers throughout Italy and was coordinated by Veneto Institute of Oncology IRCCS in Padua.
Supplemental material
Eligible patients aged at least 18 years were included; histologically proven diagnosis of SCAC was required, with an Eastern Cooperative Oncology Group (ECOG) performance score (PS) of 0–2 and a life expectancy of at least 12 weeks. Furthermore, patients were eligible if they had evaluable disease according to Response Evaluation Criteria In Solid Tumors (RECIST) criteria V.1.124 and if they had already received at least one previous line of treatment for metastatic disease; patients experiencing progression of disease within 6 months after the completion of chemoradiotherapy for non-metastatic disease were also eligible. Adequate bone marrow, renal, and hepatic function were required, with neutrophils ≥1.5×109 /L, platelets ≥100×109 /L, hemoglobin ≥9 g/dL, total bilirubin ≤1.5 × the upper-normal limits (UNL) of the normal values, creatinine clearance ≥30 mL/min according to the Cockcroft-Gault formula, or serum creatinine ≤1.5 × UNL. Importantly, HIV-positive patients were eligible if their CD4+cell count amounted to 300 cells per μL or more at the screening, if HIV viral load was undetectable, and if they were compliant with antiretroviral therapy. Patients with treated brain metastases were eligible if lesions were stable and asymptomatic for at least 3 months.
Patients with active autoimmune disease, or patients that required for any reason use of immunosuppressive treatment including chronic prolonged systemic corticosteroids (defined as corticosteroid use for ≥1 month) were excluded. Patients with active hepatitis B virus or hepatitis C virus infection at screening were also ineligible.
Patients with clinically significant cardiovascular disease, such as cerebral vascular accident/stroke, myocardial infarction, unstable angina, congestive heart failure (≥New York Heart Association Classification Class II), or serious cardiac arrhythmia requiring medication, were not allowed to participate.
Written informed consent to the study procedures and to molecular analyses was obtained from each patient before registration.
Randomization and masking
Patients considered eligible were randomized in a 1:1 ratio between two treatment arms: avelumab monotherapy (arm A) or cetuximab plus avelumab (arm B).
Registration and randomization procedures were centralized at Veneto Institute of Oncology and were performed by using an electronic web-based system. The randomization code consisted of a unique identification code and was used on all further documentation and correspondence, including electronic case record forms.
No stratification factors were adopted.
Procedures
In arm A, patients received avelumab 10 mg/kg intravenously on day 1 every 2 weeks; in arm B, patients received cetuximab 500 mg/m2 intravenously plus avelumab 10 mg/kg intravenously both on day 1 every 2 weeks. In both arms, treatment was administered until progression of disease, patient refusal or inacceptable toxicity. Treatment beyond progression was allowed only after discussion with the principal investigator of the trial.
The collection of tissue specimens was mandatory for study entry; a formalin-fixed paraffin embedded tumor block from primary tumor and/or metastasis was required during the screening phase. Histological diagnosis was centrally confirmed for all patients; HPV status was centrally assessed by means of HPV test performed with real time polymerase chain reaction in case of patients with missing information. Additional biopsy was recommended, but not mandatory, at the time of disease progression.
CT scan images were also collected for central review at the Veneto Institute of Oncology.
Outcomes
Outcomes were described separately in each arm since no formal comparison between the two study treatments was allowed. The primary endpoint of the study was overall response rate (ORR), defined as the percentage of patients achieving a complete (CR) or partial (PR) response, according to RECIST 1.1 criteria23 among the total of patients randomized in each arm.
Secondary endpoints were PFS, overall survival (OS), and safety profile description. Exploratory endpoints were translational analyses on biomarkers identified in blood, urine and tumor tissue samples.
PFS was defined as the time from randomization to the first documentation of objective disease progression or death due to any cause, whichever occurred first. For alive patients free from progression at data cut-off, PFS was censored at the time of the last evaluable tumor assessment documenting absence of progressive disease.
OS was defined as the time from study enrolment to the date of death due to any cause. Patients still alive at the time of analysis were censored for OS on the last date they were known to be alive.
Duration of disease control was analyzed for all patients who had reached PR or stable disease (SD) and was defined as the time from the first response or stabilization of disease, whichever came first, to the first documentation of objective disease progression, or to the last evaluable tumor assessment.
Toxicities were reported according to the Common Terminology Criteria for Adverse Events V.4.03.
Statistical analysis
A Simon’s two-stage Mini-Max design25 was used for both arms in parallel.
The null hypothesis of a response rate of 5% (p0=0.05) was tested against alternative of a response rate of 20% (p1=0.20). Type I error rate was set at 0.05 (one sided) and power at 80%.
In the first stage, 13 patients had to be accrued in each arm. According to the two-stage mini-max design, in case of no responses in these 13 patients, the study would have been stopped. Otherwise, 14 additional patients would have been accrued for a total of 27 in each arm. The null hypothesis could have been rejected in case of at least 4 out of 27 patients in each arm had a tumor response according to RECIST V.1.1.
In order to assess the safety profile of the combination therapy, a safety run-in phase was also preplanned: the enrolment of new patients had to be temporary interrupted after the first six patients were randomly assigned to arm B. After six patients enrolled in arm B received two cycles of study treatment, a Safety Monitoring Committee completed a safety evaluation; as the study treatment combination was judged feasible and no major safety concerns arose, the enrolment was resumed.
The intention to treat (ITT) population included all randomized patients and was used to evaluate the endpoints related to activity and efficacy of the study treatment. The safety population included all patients who received at least one dose of the study medication designated according to the randomization arm.
The primary analysis of ORR was performed in the ITT population. For each arm, the proportion of responding patients was estimated and a one-sided 95% CI and a two-sided 95% CI were calculated using the Clopper and Pearson method based on the binomial distribution.
The median follow-up was calculated using the reverse Kaplan-Meier method. Survival endpoints (PFS, OS) were described using the Kaplan-Meier method, and Kalbfleisch and Prentice formula was applied to calculate the two-sided 95% CI. SAS software (V.9.4) and R software (V.4.0.3) were used for all statistical analyses.
Results
From September 18, 2018 to July 2, 2019, 62 patients were screened (figure 1). Screening failure was declared for two of them due to clinical conditions worsening: therefore, 60 eligible patients in total were randomized, 30 in each arm, 6 more than initially planned in order to counterbalance possible dropouts. Of note, target accrual was completed 7 months before the planned date of February 28, 2020.

Trial profile.
Baseline characteristics of patients are illustrated in table 1 and were well balanced between the two arms, with the only exception of female patients: 24 (80%) in arm A and 17 (57%) in arm B. Median age was 63 years (range 35–85 years). Twelve (40%) out of 30 patients in each arm had distant metastases; 7 (23%) in arm A and 10 (33%) in arm B had received two or more previous lines of treatment. Three (10%) patients in arm A and one (3%) patient in arm B had HIV infection.
Patients and disease baseline characteristics according to the randomly allocated treatment arm
As shown in online supplemental table S1, HPV status was centrally assessed for 56 (93%) patients, 28 in each arm. HPV-positive status was found in 25 (89%) patients in arm A and 26 (93%) in arm B. Out of 51 HPV-positive patients, HPV type was evaluable for 36 patients, 34 of them being HPV-16 positive (online supplemental table S1).
Supplemental material
At cut-off date of September 21, 2020, at a median follow-up of 17.2 months (IQR: 15.7–18.2), median duration of therapy was 3.5 months in arm A and 4.6 months in arm B. Only two patients in each arm were still receiving treatment at the time of the data cut-off; the most common reason for treatment discontinuation was disease progression (87% in both arms).
Patients evaluated for RECIST 1.1 response were 57, since 3 patients (1 in arm A and 2 in arm B) died of disease before the first radiological re-evaluation.
After the first stage of enrolment, one patient in each arm achieved a PR. In the ITT population, of the 30 patients in each arm, 3 PRs (10%; one-sided 95% CI lower bound 2.8%; two-sided 95% CI 2.1 to 26.5) in arm A, and 5 PRs (16.7%; one-sided 95% CI lower bound 6.8%; two-sided 95% CI 5.6 to 34.7) in arm B were documented. None of the five PRs in arm B were observed in patients 28–30. All the PRs were confirmed by central review.
The blinded independent central review confirmed the Investigator-assessed results. Figure 2 depicts the radiological dynamics of tumor responses in each arm by means of waterfall plots (panels A-B), swimmer plots (panels C-D) and spider plots (panels E-F).
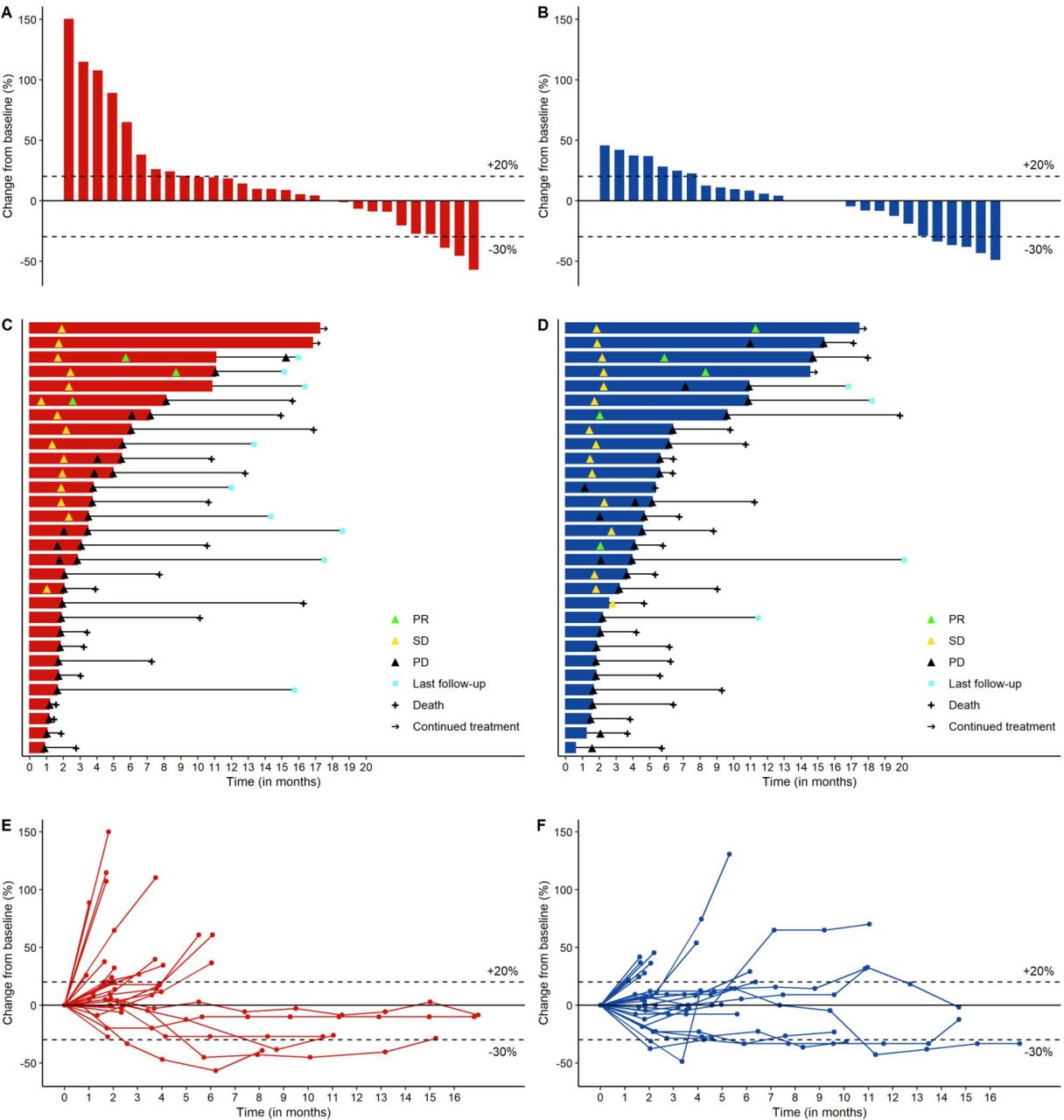
Plots describing response and duration of therapy in arm A (red color) and arm B (blue color). In sequence, waterfall plot (A,B) depicting RECIST V.1.1 responses and their depth in 57 out of 60 patients evaluated for response; swimmer plot (C,D) depicting time-on-treatment and tumor dynamics in 60 evaluable patients; spider plot (E,F) depicting the longitudinal assessment of RECIST V.1.1 response during treatment in 60 evaluable patients. PD, progressive disease; PR, partial response; SD, stable disease.
DCR in the ITT population was 50% (two-sided 95% CI 31.3 to 68.7) in arm A and 57% (two-sided 95% CI 37.4 to 74.5) in arm B. Durable responses and disease stabilizations were observed in both arms: median duration of disease control was 4.2 (two-sided 95% CI 1.8 to 8.6) in arm A and 4.6 (two-sided 95% CI 1.9 to 9.1) in arm B (table 2).
Response and disease control on the ITT population
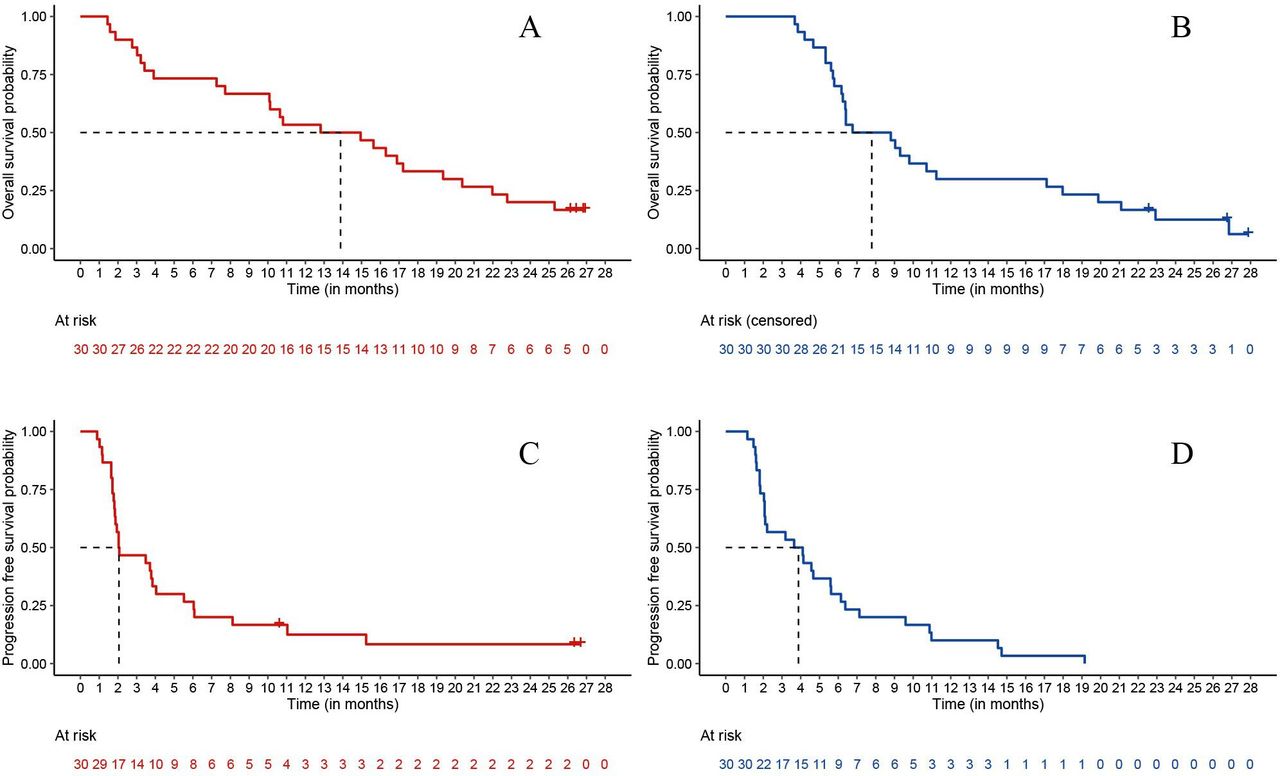
At the cut-off date of September 21, 2020, a total of 55 progression and 43 death events occurred. Median PFS (mPFS) was 2.0 months (two-sided 95% CI 1.8 to 4.0) in arm A and 3.9 months (two-sided 95% CI 2.1 to 5.6) in arm B; median OS (mOS) was 12.8 months (two-sided 95% CI 7.7 to 16.9) in arm A and 7.8 months (two-sided 95% CI 6.2 to 11.2) in arm B.
Updated OS and PFS were obtained at cut-off date July 15, 2021: after a median follow-up of 26.7 months (IQR 26.5–26.9), median PFS was 2.0 months (two-sided 95% CI 1.8 to 4.0) in arm A and 3.9 months (two-sided 95% CI 2.1 to 5.6) in arm B (figure 3A,B); median OS was 13.9 months (two-sided 95% CI 7.7 to 19.4) in arm A and 7.8 months (two-sided 95% CI 6.2 to 11.2) in arm B (figure 3C,D). At the cut-off date of July 15, 2021, a total of 56 progression and 52 death events occurred.

{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier curves depicting PFS (A,B) and OS (C,D) in arm A (red color) and arm B (blue color). Note: given the non-comparative nature of the trial, p value was not provided. Data updated at July 15, 2021.
In total, 11 patients received therapy beyond progression, 6 in arm A and 5 in arm B (figure 2C,D). The decision to continue therapy beyond first progression was justified by the possibility of pseudoprogressive disease and was always discussed with the patient and with the principal investigator, carefully evaluating the tolerability showed during the treatment.
Online supplemental table S2 shows the data on further treatments received after experimental therapy are reported: 39% of patients in each arm received at least one further treatment, in most cases represented by chemotherapy.
The most common adverse event (AE) was fatigue in arm A (17% of patients) and skin and subcutaneous disorders in arm B (87% of patients). No grade 3–4 treatment-related AEs were observed in arm A; the most common grade 3–4 treatment-related AEs in arm B were skin and subcutaneous disorders, hypomagnesemia and aspartate aminotransferase/alanine aminotransferase increase, each of them being observed in 6% of patients (online supplemental table S3). Only 2% and 5% of avelumab administrations in arms A and B, respectively, and 5% of cetuximab administrations were delayed due to AE. Dose modifications due to AE did not occur in arm A; in arm B, avelumab and cetuximab doses were modified due to AE in 2% and 5% of the total administrations, respectively; two patients (7%) in arm B permanently interrupted the treatment due to treatment-related AE; no permanent interruptions due to treatment related AE were observed in arm A (online supplemental table S4). No toxic deaths were reported.
Discussion
In patients with SCAC, no standard treatments are currently available after failure of standard first-line chemotherapy. In particular, no randomized trials in second line have been published up today.
Unfortunately, the access to clinical trials in this setting is indeed limited by the rarity of the disease, the poor life expectancy and the clinical conditions often compromised by the aggressive behavior of this malignancy, its complications and associated comorbidities such as HIV infection. Therefore, investigations on new treatment options in this setting clearly address a major unmet clinical need. Coupling the above reported consideration with a robust biological rationale, we conceived the present study to explore the activity of the anti-PD-(L)1 monoclonal antibody (MoAb) avelumab alone or in combination with the anti-EGFR MoAb cetuximab.
CARACAS was designed as a non-comparative, randomized phase II trial with a ‘pick-the-winner’ strategy based on ORR. The primary endpoint was met in the combo arm, where ORR 17% was observed (the prespecified threshold for further investigation), but not in the avelumab alone arm; disease control duration was 4–5 months. The activity and favorable safety profile observed in avelumab monotherapy arm is in line with the outcomes previously reported for anti-PD-1 agents.10 11
Tumor shrinkage as well as durable disease stabilizations in the combination arm led the basis for encouraging results in terms of PFS. It could be estimated from the Kaplan-Meier model that about 30% of patients remained free from progression for at least 6 months, with a 10% not progressing at 1 year, corroborating the finding of potential meaningful impact of this therapeutic strategy.
We would be cautious in the interpretation of OS results due to the small sample size and potential confounding effect of minor unbalances such as a higher percentage of women in arm A, which is a well-known positive prognosticator in SCAC.26 Other factors that might have influenced the OS are the slightly unbalanced burden of disease at the time of enrolment or number of previous lines and treatments between the two arms (table 1). In addition, looking at the shape of the OS curves the median value of OS is the widest point of distance, but globally the OS outcomes are similar as the curves cross at 6 months and proceed very close after 18 months. No major differences in poststudy treatments were observed, as 11 patients received further treatments in both arms.
Worth noting, one patient underwent poststudy radical surgery after a prolonged disease stabilization with avelumab monotherapy and pathologic CR on primary tumor was documented at the histological examination, with minimal residual nodal localizations (data not shown). This unique case does not affect a formal read of results: in our study, anti-PD-L1 monotherapy did not reach the preplanned threshold for ORR.
Our results confirm the already documented limited activity of anti-PD-L1 monotherapy in SCAC: however, in other squamous neoplasms, more pronounced benefit of anti-PD-L1 alone were observed, especially when compared with monochemotherapy. In non-small cell lung cancer with squamous histology (SqNSCLC), the study CheckMate 017 tested monochemotherapy versus nivolumab alone in patients with disease progressed after first line, significantly improving PFS, OS and ORR.27 As well, in head and neck squamous cell carcinomas (HNSCC), both nivolumab28 and pembrolizumab29 conferred better OS compared with monochemotherapy in platinum-resistant disease, irrespectively of PD-L1 expression. Importantly, in addition to squamous histology, HNSCC shares with SCAC the correlation with oncogenic viruses. In SCAC, HPV represents a fundamental driver and one of the most important prognosticators; its predictive role in immune checkpoint blockade therapy, however, has not been fully explored. In HNSCC, the phase II HAWK study demonstrated notable activity of durvalumab in platinum resistant disease, being response rate and survival numerically higher in HPV +tumors compared with HPV ones;30 the biologic rationale of a higher activity of anti-PD-L1 in HPV+malignancies might be related to the observed upregulation of PD-L1 and PD-L2 on fibroblasts in these malignancies.31 To what extent we could translate these observations concerning HNSCC on SCAC is still debated. In SCAC, HPV+combined with evidence of high TILs in tumor microenvironment showed better relapse-free survival (RFS) after chemoradiotherapy; linear correlation between HPV infection and TILs infiltration was also described.32 This observation throws light on new settings in which testing PD-L1 blockade in SCAC, like the association with standard chemoradiotherapy; furthermore, it gives an intriguing prompt toward patients’ selection for ICB, another gray area that might account for disappointing results of immunotherapy in SCAC.
Experimenting new strategies is of crucial importance to improve efficacy of PD-L1 blockade in SCAC. In other squamous malignancies, anti-PD-L1 agents have been successfully tested in association with chemotherapy. In SqNSCLC, in the KEYNOTE-407 trial, adding the anti-PD-1 pembrolizumab to standard chemotherapy doublets significantly improved PFS and OS, primary endpoints of the study, irrespectively of PD-L1 expression;33 as well, the anti-PD-L1 atezolizumab showed benefit in PFS when in association with chemotherapy in the IMpower 131 trial.34 Both studies were conducted in chemotherapy-naïve patients.
In the first-line setting in SCAC, modified DCF chemotherapy with or without atezolizumab is under investigation by a randomized, non-comparative phase II trial.35 Also, carboplatin-paclitaxel with the anti-PD-1 MoAb retifanlimab or placebo is tested by the phase III, placebo-controlled, double-blind InterAACT2 trial (NCT04472429). Regarding the combination with biological agents, a recent study did not show signals of synergistic activity when adding bevacizumab to atezolizumab in patients with previously treated SCAC.36
To our knowledge, the CARACAS randomized study is the first to explore the value of combining an anti-EGFR agent and immunotherapy in patients with advanced SCAC. Thanks to the non-overlapping AEs of immune checkpoint inhibitors and anti-EGFR agents (with the only exception of potential infusion-related reactions), it was possible to include patients with ECOG PS2, heavily pretreated disease and HIV infection. Despite that, the safety profile of the combination was manageable.
Based on our results, avelumab-cetuximab therapy is worthy of further investigation by future phase III trials. In our opinion, there are the major challenges to be faced in order to better focus future efforts. First is patients’ selection and molecular complexity. An important issue in SCAC is the urgent need of translational research on biomarkers that may identify patients who can benefit from PD-L1 blockade. Exploratory translational analyses are currently ongoing on blood, urine and tissue samples collected at the baseline and after PD from patients enrolled in our trial: by means of next generation sequencing, several biomarkers will be tested in order to isolate predictors of response to the treatments delivered. In detail, we will focus on TMB and on markers of immune escape, such as PD-L1 expression; we will also test microsatellite status, in order to obtain data on the frequency of microsatellite instability in advanced squamous cell anal carcinoma (aSCAC) and to eventually investigate on the role of microsatellite instability as predictor of response to immunotherapy in this malignancy.
Another challenge in SCAC is to find driver mutations by investigating on molecular characterization. In aSCAC, low TMB has been reported, in line with other HPV-related squamous tumors: this observation suggests that other factors could underlie the immunogenicity of these tumors.37 Furthermore, in HNSCC, significant association was described between objective response to immunotherapy and high TMB in HPV and Epstein-Barr virus (EBV)-negative tumors; on the contrary, this association was not confirmed in HPV+ or EBV+cases0.38 This observation supports the theory that in virus-related squamous neoplasms, viral neoepitopes more than somatic antigens might play a decisive role in the process of immune-escape.
A second crucial challenge is to provide the oncologic community with robust evidence regarding the potential impact of these innovative treatments on quality of life. The present study did not assess quality of life nor patient reported outcomes. However, it would be important to have this matter extensively addressed in further developments for measuring how new treatments translate into specific symptoms control and clinical benefit of patients with advanced SCAC.
The third point raised by our results is if immune checkpoint inhibition should be better developed in earlier lines of treatment as a combination with recommended chemotherapy regimens like cisplatin-fluoropyrimidines or carboplatin-paclitaxel, or as a chemotherapy-free option in patients considered chemotherapy-ineligible or chemorefractory.
In conclusion, the CARACAS trial met its primary endpoint in the cetuximab-avelumab combination arm, thus supporting the biological rationale of dual EGFR and PD-L1 blockade in patients with advanced SCAC; avelumab showed a manageable safety profile both as single-agent and as combination with cetuximab in this patients’ population. Given these encouraging results and the absence of standard treatments for this disease in lines after the first, randomized clinical trials are necessary to clearly establish the efficacy of avelumab-based therapy. The CARACAS experience, on top of the positive results, is a good example of feasibility of academic research in a challenging setting for the conduction of innovative clinical studies. The enrolment in our trial was completed several months before the expected and this is expression of the great unmet need represented by the scarcity of therapeutic options for advanced SCAC.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Approval for the protocol was granted through the institutional Ethic Committee from each participating center. The trial was conducted according to the Declaration of Helsinki.
Acknowledgments
We wish to thank Filippo Venturini, Federica Alberghini and Julien Bonnefoux, all of Merck Serono S.p.A., Rome, Italy, an affiliate of Merck KGaA, Darmstadt, Germany, for their unevaluable support to the project. We also thank Giovanna Magni for her contribution to data analysis. A special thanks goes to the GONO Foundation team, in particular Laura Delli Ponti, and to the Independent Safety Data Monitoring Committee, composed by Giuseppe Aprile, Chiara Cremolini, and Daniele Santini.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SL conceived the study and was involved in patients’ treatment, data interpretation, supervision on the study, writing, editing and reviewing the manuscript; she was also responsible for the overall content as the guarantor. AAP was involved in patients’ treatment, data collection and curation, data interpretation, supervision and writing. AS-B was involved in investigation and contributed to edit and review the manuscript. VV was involved in data curation and project administration. FL was involved in data interpretation and editing and reviewing of the manuscript. FP contributed in conceiving the study and was involved in patients’ treatment, data interpretation, writing, editing and reviewing the manuscript. All other authors were involved in investigation; each author read and approved the final work. SL, AAP and FP had access and verified the raw data.
Funding This work was partially supported by Merck Serono S.p.A., Rome, Italy, an affiliate of Merck KGaA, Darmstadt, Germany, as part of an alliance between Merck KGaA and Pfizer. Merck KGaA, Darmstadt, Germany and Pfizer reviewed the manuscript for medical accuracy only before journal submission. This work was also supported by Gruppo Oncologico Nord Ovest (GONO) foundation.
Disclaimer The authors are fully responsible for the content of this manuscript, and the views and opinions described in the publication reflect solely those of the authors.
Competing interests SL was advisor and part of speakers’ bureau of Amgen, Merck Serono, Lilly, Bristol-Myers Squibb, Servier, Roche, Pierre-Fabre, GSK; had advisory or consulting role for AstraZeneca, Incyte, Daiichi-Sankyo; received research grant by Bayer. AAP reported grants from Bayer. FM reported honoraria from Servier. VF reported personal fees from Merck KGaA, Amgen, Servier, Sanofi. MS reported personal fees from MSD, Merck, Eisai, Sanofi, Bayer, Servier. SM reported personal fees from Merck Serono. AS-B reported personal fees from Amgen, Bayer, MSD, Sanofi, Servier. MF reported grants from QED Therapeutics, grants and personal fees from Astellas Pharma, personal fees from Tesaro-GSK, Diaceutics. VZ had consulting role for Bristol-Myers Squibb, MSD, Italfarmaco, Eisai; was part of speakers’ bureau of Roche, Astellas Pharma, Bristol-Myers Squibb, Servier, AstraZeneca, MSD, Janssen, IPSEN. Received research funding from Bayer, Roche, Lilly, AstraZeneca, BMS, IPSEN, Astellas Pharma; received travels accommodation from Bayer, Roche and Servier. All of the above-mentioned financial relationships were outside this work. PDB reported personal fees from GONO Foundation during the conduct of the present study. All other authors had no conflict of interests to disclose.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.