Article Text

Abstract
Toll-like receptors (TLRs) are evolutionarily conserved molecules that specifically recognize common microbial patterns, and have a critical role in innate and adaptive immunity. Although TLRs are highly expressed by innate immune cells, particularly antigen-presenting cells, the very first report of a human TLR also described its expression and function within T-cells. Gene knock-out models and adoptive cell transfer studies have since confirmed that TLRs function as important costimulatory and regulatory molecules within T-cells themselves. By acting directly on T-cells, TLR agonists can enhance cytokine production by activated T-cells, increase T-cell sensitivity to T-cell receptor stimulation, promote long-lived T-cell memory, and reduce the suppressive activity of regulatory T-cells. Direct stimulation of T-cell intrinsic TLRs may be a relevant mechanism of action of TLR ligands currently under clinical investigation as cancer immunotherapies. Finally, chimeric antigen receptor (CAR) T-cells afford a new opportunity to specifically exploit T-cell intrinsic TLR function. This can be achieved by expressing TLR signaling domains, or domains from their signaling partner myeloid differentiation primary response 88 (MyD88), within or alongside the CAR. This review summarizes the expression and function of TLRs within T-cells, and explores the relevance of T-cell intrinsic TLR expression to the benefits and risks of TLR-stimulating cancer immunotherapies, including CAR T-cells.
- T-lymphocytes
- receptors
- immunologic
- immunotherapy
- receptors
- chimeric antigen
- review
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
A link between microbial toxins and regression of cancers has long been described. In the late 19th century, Coley observed that repeated local administration of bacterial toxins led to regression of cancers in some patients.1 Several decades later, BCG, an attenuated Mycobacterium bovis strain used as a tuberculosis vaccine, was found to elicit an antitumor effect,2 and remains in routine clinical use as an immunotherapy for bladder cancer.3 On rare occasions, leukemias spontaneously enter remission following bacterial or fungal infection, possibly related to an exuberant innate immune response elicited by sepsis.4
Janeway proposed that evolutionarily conserved ‘pattern recognition receptors’ (PRRs) recognizing common microbial characteristics are critical to immunological discrimination between ‘self’ and ‘non-self’. Importantly, he hypothesized that these PRRs are expressed on T-cells themselves, and that both PRR and T-cell receptor (TCR) stimulation may be important for full protective immunity.5 Medzhitov et al later detected mRNA encoding the gene we now refer to as Toll-like receptor 4 (TLR4), the archetypal PRR, within human T-cells, and found that a chimeric TLR4 construct resulted in activation of nuclear factor kappa B (NF-kB) within a human T-cell line.6 Ten human TLRs are now recognized, and their expression across a broad range of tissues and cell types described.7
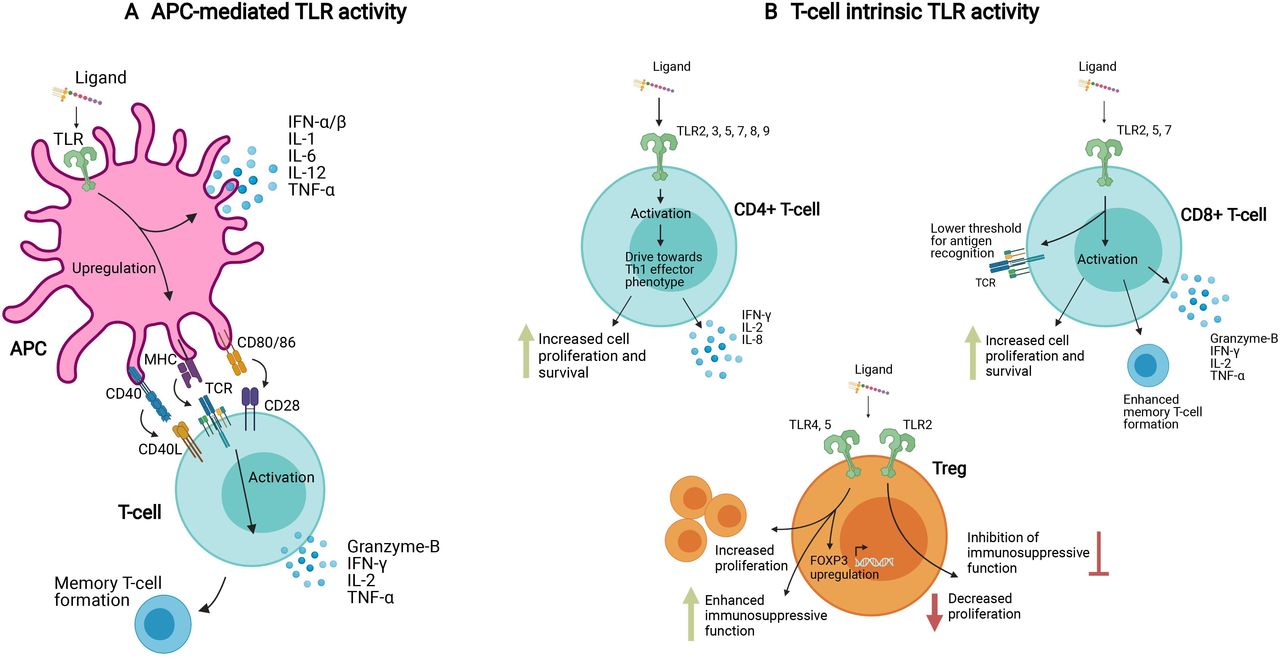
TLR ligands are important vaccine adjuvants, acting by binding to TLRs expressed on antigen-presenting cells (APCs), and resulting in APC activation, enhanced antigen presentation to T-cells and B-cells, and provision of costimulatory signals8 (figure 1A). Although recognized in the earliest report of human TLR expression, the role of T-cell-intrinsic TLRs is often overlooked.9 However, when the role of TLR expression on APCs versus T-cells is disentangled using knockout mouse models and T-cell transfer, it becomes evident that T-cell intrinsic expression of at least some TLRs is essential for full protective immunity, as Janeway had predicted (figure 1B).5 The distinction between APC-mediated TLR activity and T-cell intrinsic TLR activity in different T cell subsets is depicted in figure 1. It should be noted that in an immune response to infection, both mechanisms are likely to be active.

APC-mediated and T-cell intrinsic TLR activity. (A) TLR ligands can activate T-cells indirectly via APCs, where after ligand binding, TLR signaling via NF-kB induces an upregulation of MHC, costimulatory receptors, and proinflammatory cytokines. (B) TLR ligands can also induce T-cell activation directly via TLR molecules expressed on the T-cell surface. In CD4+ and CD8+ effector T-cells, this activation is characterized by proinflammatory cytokines, memory T-cell formation, enhanced sensitivity to antigens and suppression of regulatory T-cells. In regulatory T-cells, different TLR ligands can have contrasting effects, either enhancing or suppressing their function. These different mechanisms, APC-mediated and T-cell intrinsic, likely work in concert in response to systemic TLR ligand exposure. The T-cell intrinsic pathway can be specifically exploited in T-cell mediated treatments such as adoptive cell transfers. APCs, antigen-presenting cells; IL-1, interleukin 1; MHC, major histocompatibility complex; NF-kB, nuclear factor kappa B; TLR, Toll-like receptor; TNFα, tumor necrosis factor-α.
Cancer immunotherapies that exploit TLR signaling are in clinical development. Agonists for TLRs including TLR 2, 3, 4, 7, 8 and 9 have been heralded as promising immunotherapeutics for their capacity to stimulate T-cell immunity, both as monotherapies for cancer, and in conjunction with other treatment modalities, such as radiotherapy, chemotherapy or checkpoint inhibitors.10 More recently, TLR signaling domains and their downstream signaling adaptor myeloid differentiation primary response 88 (MyD88) have been incorporated into investigational chimeric antigen receptor (CAR) T-cells, providing the opportunity to specifically activate TLR signaling within tumor-recognizing T-cells.11–15
This review summaries the expression of TLR molecules by T-cell subsets, and explores their costimulatory, activating and regulatory functions. The relevance of this to cancer immunotherapies that exploit TLR signaling is discussed.
Introduction to the TLR family
The interleukin 1 receptor (IL-1R)/TLR superfamily is a group of cell surface receptors that play a crucial role in inflammation and disease.16 They are type I integral membrane receptors, located on the cell surface and within endosomes in both vertebrates and invertebrates.17 Originally discovered in Drosophila melanogaster as the transmembrane receptor Toll,18 a mammalian homologue was subsequently identified and defined as a TLR.6 Several more family members have since been discovered in mammals; to date 10 TLR genes have been identified in humans (TLR1–TLR10, figure 2) and thirteen in mice (TLR1–TLR13). Only twelve of the mouse TLRs are functionally expressed, as the murine TLR10 gene homolog has been disrupted by a retrovirus, resulting in an inactive pseudogene.19

Classical TLR signaling pathways in human innate immune cells. TLR molecules can signal through MyD88-dependent and independent pathways. In the MyD88-dependent pathway, activation of proinflammatory transcription factors AP1 and NF-kB is induced via IRAK1/4, TRAF6 and TAK1. In the MyD88-independent pathway, the adaptor molecule TRIF initiates signaling via TRAF3 and IRF, also resulting in the induction of proinflammatory gene transcription. AP1, activator protein 1; IRAK, IL-1R-associated kinase; IRF, interferon-regulatory factor; LPS, lipopolysaccharide; MyD88, myeloid Differentiation Primary Response 88; NF-kB, nuclear factor kappa B; TAK1, transforming growth factor-β-activated kinase-1; TIRAP, toll-IL 1 receptor domain containing adaptor protein; TLR, toll-like receptor; TRAF, tumor necrosis factor receptor-activated factor; TRAM, TRIF-related adaptor molecule; TRIF, toll/IL-1R domain-containing adaptor-inducing IFN-β.
TLR proteins are PRRs, which recognize pathogen associated molecular patterns (PAMPs) or endogenous damage associated molecular patterns and initiate immune responses. This results in the activation of downstream proinflammatory transcription factors such as NF-κB and interferon (IFN) regulatory factors20 (figure 2). All TLRs are composed of three parts: a leucine-rich N-terminal ligand binding domain, a single-pass transmembrane domain and a conserved C-terminal intracellular toll/IL-1R (TIR) signaling domain, which interacts with various adaptor proteins, primarily MyD88.8 TLRs are expressed at varying levels in many different cell types, both immune and non-immune, although most cells express only a subset of TLRs, and often at low levels. Hematopoietically derived sentinel cells, such as macrophages, neutrophils, and dendritic cells, express virtually all TLRs at relatively high levels, with some variation between cell subsets.21 For this reason, TLRs are traditionally associated with innate immune cells.
Cell surface receptors
TLR1, 2, 4, 5 and 6, all located on the cell surface, specialize in the detection of ligands in the extracellular space, contributing to self/non-self-discrimination through recognition of microbially derived molecules. TLR1 forms a functional heterodimer with TLR2 to recognize triacylated lipopeptides from Gram-positive bacterial products, and TLR2 recognizes a wide range of structurally diverse PAMPs from bacteria, yeast, fungi, parasites and viruses.22 TLR2 ligands include lipopeptides from a variety of pathogens, and peptidoglycan and lipoteichoic acid from Gram-positive bacteria.8 Depending on the PAMP ligand, TLR2 can function as a homodimer or heterodimerize with TLR1, TLR6, or other non-TLR molecules.23 TLR4 was the first mammalian TLR identified.6 It recognizes lipopolysaccharide (LPS), a component of Gram-negative bacteria outer membranes.24 25 TLR5 responds to flagellin, which is produced by virtually all Gram-negative and Gram-positive bacteria.26 TLR6 forms a functional heterodimer with TLR2 to recognize diacylated lipopeptides from gram-positive bacteria, mycoplasma, fungi and some viruses.22 Until relatively recently, TLR10 was the only human TLR without a known ligand or biological function. It has since been discovered that TLR10 is a modulatory receptor with an inhibitory role, working through complex mechanisms that are not yet fully understood, but that include forming heterodimers with TLR2.27
Intracellular receptors
TLR3, 7, 8, and 9 are located intracellularly on the surface of endosomes and lysosomes, their ligand binding domains projected into the interior of the organelles. The ligands that they recognize, primarily viral nucleic acids, are not in all cases unique to microbes. As such, the self/non-self-discrimination for these TLRs is mediated by the location of their ligands rather than their molecular nature.28 Viral, but not host, nucleic acids are located in cell endosomes/lysosomes. These are accessible to intracellular TLRs through processes such as autophagy and intracellular compartment fusion.29 30TLR3 responds to double-stranded RNA,31 and TLR7 and TLR8 are both activated by single-stranded RNA from viruses.32 TLR9 recognizes unmethylated CpG motifs that are present in bacterial DNA.33 TLR11 and 12, two endosomal TLRs unique to murine species, have recently been shown to be activated by the actin-binding protein profilin, which is produced by the parasite Toxoplasma gondii.34 35 TLR11 is also capable of activation via flagellin produced by Salmonella and Escherichia coli.36 TLR13, the final murine TLR, is present on endosomal surfaces and is activated by an unmethylated motif in bacterial RNA.37
TLR signaling pathways
TLR signaling pathways in mammals are partly homologous to those of the IL-1R family, with both pathways interacting with the adaptor molecule MyD888 (figure 2). For signaling to occur, the TLR must either homodimerize or heterodimerize in order for the protein to undergo the conformational changes required for adaptor molecule binding.38 TLRs transmit a signal through their TIR, initiating a signaling cascade that is MyD88 dependent for all TLRs except TLR3. Some molecules, such as the adaptor Toll-IL-1R domain containing adaptor protein (TIRAP), are only involved in the signaling pathways of certain TLRs.39
After binding to the TLR TIR, MyD88 recruits the serine/threonine kinases IL-1R-associated kinases (IRAKs), primarily IRAK4 and IRAK1, which interact with tumor necrosis factor receptor-activated factor 6 (TRAF6) on phosphorylation.40 This interaction initiates distinct signaling pathways, resulting in the activation of proinflammatory transcription factors, NF-kB and/or activator protein 1 (AP1).20
TLR3, the only TLR to signal in a MyD88-independent manner, instead recruits the adaptor molecule Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF) to its TIR domain.41 TLR4 is able to signal through both MyD88- and TRIF-dependent pathways, the latter pathway involving the TRIF-related adaptor molecule (TRAM), which interacts with TRIF exclusively in the TLR4 pathway.42 It is likely that differences in the signaling molecules recruited contribute to the variations in functional immune responses induced by different TLR ligands, alongside other characteristics such as cell subset-specific TLR expression patterns and spatial localization of TLRs. For example, the ‘smooth’ and ‘rough’ variants of LPS can initiate different signaling cascades—either MyD88- or TRIF-dependent—allowing cells to respond differentially to bacteria containing different LPSs.43
Canonical TLR signaling pathways have principally been studied in innate cells due to their relatively high levels of TLR expression. There are fewer reports of the pathways involved in TLR signaling within T-cells, which express lower levels of certain TLRs, although limited information suggests that signaling pathways may differ. For example, it has been established that within APCs, signaling in response to the TLR2 ligand Pam3CSK4 requires the recruitment of MyD88, TIRAP and IRAK4. However, in T-cells from either MyD88- or IRAK4-deficient mice, TLR2 activation can elicit a partial immune response via c-Jun N-terminal kinase, p38 and an unknown adaptor molecule, suggesting an alternative TLR2 signaling pathway in T-cells.44 It has also been shown that in T-cells, the TLR2 signaling pathway converges with TCR signaling at the level of Erk1/Erk2 and Akt activation, to enhance TCR signal-dependent functions.45
TLR expression within T-cells
Although expressed particularly highly by APCs, TLRs are also expressed by a wide range of immune and non-immune cells, including B-cells, T-cells, NK cells, epithelial cells and fibroblasts.7 46 T-cell intrinsic TLR expression has been well reported, but studies vary widely in terms of species and mouse strain, analysis of CD4+ versus CD8+ T-cell subsets, T-cell activation status, memory vs naïve phenotype, and mRNA vs protein analysis, etc. Table 1 summarizes the T-cell intrinsic TLR expression data from literature cited in this review. A filled symbol for mRNA, protein, or functional data indicates that evidence of these forms of TLR expression has been found in the T-cell subsets examined. An empty symbol indicates that no TLR expression was detected in the various studies cited here, although further research may update this.
Expression and function of TLR molecules in murine and human T lymphocytes
In humans, TLR expression has been identified in T-cells in several research studies, although there is variation between reported findings. This may relate in part to between-donor variation, and in part due to technical differences between studies. Several studies have found that human CD4+ T-cells express mRNA for almost all TLRs at varying levels47–49 while activated CD8+ T-cells express higher levels of TLR2, 3 and 5,48 50–53 and in a rheumatoid arthritis setting, TLR4.54
The expression of TLR mRNA and proteins by murine CD8+ T-cells has been explored in some detail; mRNA encoding TLRs can be found at varying levels both naïve and activated CD8+ T-cells.55 TLR surface expression and function, however, appears to be restricted to TLR2 and its heterodimer partners, TLR1 and TLR6. Activation of CD8+ T-cells is associated with significant enhancement of surface TLR2 expression and function.56 Within murine CD4+ T-cells, mRNA of virtually all TLRs is expressed, however the difference in expression between cell subsets is significant.57 For example, it has been shown that antigen experienced Tregs express mRNA for TLR1, 2, 4, 5, 6, 7, and 8 at varying but detectable levels, whereas naïve Tregs express significantly lower levels of TLR4, 5, 7, and 8, but maintain high levels of TLR1, 2, and 6.58 Protein expression is relatively broad,59 although as for CD8+ T-cells, TLR2 surface expression is significantly upregulated on CD4+ T-cell activation.60
The differences in surface expression of TLR molecules within T-cells is not the only determining factor of TLR function within that cell subtype; the expression of downstream molecules in the TLR pathways is equally important in determining function. For example, it has recently been shown that differences in the expression of TIRAP, which is induced by TCR or IL-2R activation and regulated by mammalian target of rapamycin complex 1 (mTOR), may underpin the variation in responses to TLR2 ligands between naïve and antigen-experienced T-cells.61
Together, these reports indicate broad expression of TLR mRNA in T-cells of both mice and humans, with protein expression and function restricted to a narrower range of predominantly cell surface-expressed TLRs, particularly in CD8+ T-cells. It is notable that the majority of studies report differences between mRNA and protein expression, emphasizing the importance of protein-level and functional analyses alongside mRNA quantitation when exploring the role of TLRs in T-cell subsets.
Functions of T-cell intrinsic TLRs
Several functional roles for T-cell-intrinsic TLRs have been established: enhancement of T-cell effector function; enhancement of T-cell proliferation and survival; and modification of regulatory function.
TLRs as costimulatory molecules
Cosignaling molecules, both stimulatory and inhibitory, have a crucial role in the regulation of T-cell activation, differentiation, cytotoxic function and memory formation. Following peptide/Major Histocompatibility Complex (MHC) binding, a balance between costimulatory signals received via CD28 and ICOS and inhibitory signals from PD-1 and CTLA4 will determine the magnitude and nature of the T-cell response.62 In addition to the conventional cosignaling molecules, a number of alternative pathways have been identified. One of these non-classical signaling routes is through TLR pathways—the engagement of which has been repeatedly shown to enhance the activation, effector function and memory formation of T-cells.
TLR costimulation of CD4+ T-cells
Many studies have reported TLR costimulatory activity in CD4+ T cells, predominantly focusing on the effects of TLR2. This is in large part due to the robust evidence that surface expression of TLR2 in human T-cells is enhanced by TCR activation.51 The majority of studies report that TLR2 costimulation directs T-cells toward a Th1 effector phenotype, although there are several reports in the literature of TLR2 ligands driving human and mouse T-cells toward a Th963 or Th1764 65 phenotype in some contexts.
In naïve human CD4+ T-cells, both adult and neonatal, TLR2 engagement has been shown to enhance the production of IL-2 and key Th1 cytokines, while simultaneously reducing the production of suppressive IL-10.66 This effect was more pronounced in neonatal cells, enabling them to produce levels of IFN-γ and IL-2 equivalent to adult cells. This points to the potential of TLR2 ligands as neonatal vaccine adjuvants.66 In one study of activated human CD4+ T-cells, elevated levels of Th1 cytokines were produced in response to the TLR2 ligand Pam3CSK4.51 Furthermore, it was shown that CD4+ memory (CD45RO+) T-cells produced higher levels of cytokines in response to TLR2 ligands than did CD4+ naïve (CD45RA+) T-cells. Interestingly, it was also shown that even without TCR engagement, TLR2 activation along with IL-2 or IL-15 stimulation increased proliferation and IFN-γ production in memory T-cells in vitro. This suggests a TCR-independent role for TLR2 in T-cell memory formation or maintenance. In the absence of APCs, the TLR2 ligand Pam3CSK4, the TLR5 ligand flagellin, and the TLR7/8 ligand R-848, can all enhance proliferation and the production of IFN-γ, IL-8, and IL-10 in human CD4+ T-cells. This can be both TCR-dependent stimulation (anti-CD3 mAb) and TCR-independent (anti-CD2 mAb or IL-2).49 This effect is enhanced in memory T-cells compared with naïve T-cells, and was not seen with ligands for TLR3 (poly I:C) or TLR4 (LPS).
In mice, there is also abundant evidence of TLR activity in CD4+ T cells. In one report, TCR-independent TLR2 engagement on mouse Th1 effector cells resulted in IFN-y production, cell proliferation and survival even in the absence of TCR stimulation. This suggests that TLR2 engagement on cells has the potential to directly trigger effector function.44 TLR2 engagement on chronically activated mouse Th1 cells has also been shown to play a role in the reinvigoration of exhausted T-cells during chronic infection.67 On engagement of TLR2 on chronically activated Th1 cells, CD4+ T-cells significantly alter expression of T-bet, IFN-γ, IL-2 and the antiapoptotic molecule Bcl-2, reduce expression of PD-1 and Lag-3, and exhibit an enhanced ability to activate B-cells. Ultimately, these observations resulted in reduced lung pathology and improved disease control in models of chronic tuberculosis infection. Interestingly, in one paper looking at activated CD4+ murine T-cells, it was reported that treatment with the TLR3 ligand poly I:C and the TLR9 ligand CpG directly enhanced T-cell survival, an effect which was not seen with ligands for TLR4, or with an alternative TLR2 ligand peptidoglycan,57 rather than the more conventional Pam3CSK4. TLR2 expression was not observed in this study, in contrast with several other papers reporting increased TLR2 in T cells following activation.
TLR costimulation of CD8+ T-cells
As well as providing CD4+ T-cell costimulation, TLR engagement, especially TLR2, can enhance the cytotoxic activity of CD8+ T-cells. In cord blood-derived human CD8+ T-cells, the TLR2 ligand Pam3CSK4, as well as the TLR5 ligand flagellin, significantly enhanced proliferation, memory formation and cytokine production (IL-2, IFN-γ, and TNF-α) in the presence of TCR stimulation.68 This effect was enhanced when both ligands were used in combination. Additionally, TLR3 on the surface of human CD8+ effector T-cells can act as a functional costimulatory molecule by increasing production of IFN-γ.53
There are also several reports of TLR costimulation of murine CD8+ T cells. For example, in CD8+ T-cells from TCR-transgenic mice, TLR2 engagement via Pam3CSK4 resulted in increased cell proliferation and survival, increased IFN-γ production and granzyme-B secretion.55 In addition to this, TLR2 engagement can enhance effector functions of the CD8+ T-cells by lowering the threshold of activation for additional costimulatory signals from APCs. Moreover, TLR2 activation on CD8+ T-cells, lowers the antigen density required for optimal activation.69 This resulted in the proliferation of effector cells even in environments of low antigen density, ultimately leading to the generation of functional memory T-cells in response to a suboptimal TCR signal and only partial activation. The mechanism behind the lowering of antigen threshold needed for activation has been in part attributed to TLR2 signaling synergising with the TCR to prolong the t1/2 of IFN-y mRNA.70 These results have been reaffirmed with the use of TLR2-/- mice, which have a decreased frequency of CD8+ memory T-cells in comparison to wildtype (WT) mice, further suggesting that TLR2 has a role in the maintenance and formation of memory T-cells.69 When further probed, this was shown to work through a TLR2-dependent mechanism, which increases the IL-7 induced proliferation of memory CD8+ T-cells. In the context of viral infection, TLR2 signaling was shown to promote the survival of activated mouse CD8+ T-cells.71 Furthermore, cells lacking TLR2-MyD88 signaling exhibited a drastically reduced ability to differentiate into long-lived memory T-cells. A more recent study investigating the contribution of TLR7 stimulation to murine CD8+ T-cell functions found that the TLR7 ligand R-848 (Resiquimod) could enhance T-cell effector function in vitro, when in combination with CD3 activation.72 This occurs through activation of the MyD88/Akt-mTOR signaling pathway.
TLRs as modulators of regulatory T-cell function
Several independent studies have reported that stimulation of TLR2 in T-regulatory cells can dampen their immunosuppressive capabilities. In humans, both TLR2 ligand Pam3CSK4 and a TLR2/6 heterodimer ligand FSL-1 reduced the suppressive function of naïve (CD4+CD25high FOXP3lowCD45RA+) and memory/effector (CD4+CD25high FOXP3highCD45RA-) Tregs, by skewing them toward a Th17 effector phenotype.64 Another study found that engagement of TLR2 on human CD4+CD25+ Tregs caused a reduction in Treg-mediated suppression of responder T-cells.73 Conversely, a TLR5 ligand delivered with anti-CD3 activation enhanced suppressive activity and expression of the Treg-associated transcription factor FOXP3 in human CD4+CD25+ T regulatory cells.50
A similar variability in TLR-mediated effects on Treg suppressive function has been observed in mice. The discovery that there are significantly fewer CD4+CD25+ Treg cells in TLR2-/- mice compared with WT control mice was one of the first indications of a link between Tregs and TLR2.74 The TLR1/2 ligand Pam3CSK4, when combined with TCR stimulation, results in a temporary loss of the suppressive function of CD4+CD25+ Tregs in vitro.75 The same effect was not seen with TLR4 or TLR9 ligands. Similar results were seen in another study of murine CD4+CD25+ Tregs and CD4+CD25- effector T-cells, where Pam3CSK4 administration alongside CD3 activation resulted in a transient loss of suppressive activity by Tregs as well as effector T-cell resistance to the suppressive effect of Tregs.60 In contrast to the above studies, in C57BL/6 mice, a subset of regulatory CD45RBlowCD25+CD4+ T-cells exhibited enhanced suppressive abilities and proliferation after administration of the TLR4 ligand LPS.58
Together, these results provide evidence for a significant and sometimes crucial role for TLR signaling within T-cells. Direct activation of TLR molecules on T-cells can enhance their proliferation and effector function. The capacity of TLRs to enhance function of T-cells that typically exhibit suboptimal responses will be of interest in specific settings, such as the vaccination of neonates.66 76 Moreover, the capacity of TLR2 stimulation to lower the TCR signal threshold for optimal T-cell activation may be exploited in settings of low antigen density, such as within some tumors.69 The variability in the response of T-regulatory cell subsets to different TLR ligands, observed in both humans and mice, is an important consideration in the development of therapeutics. Overall, the specific impact of TLR stimulation on T-cell subsets can be expected to vary by T-cell subset, by TLR agonist, and in varying immune contexts.
TLR agonists as cancer immunotherapies
In the late 19th century, Coley observed that repeated administration of certain bacterial toxins produced a moderate antitumor effect in some patients.1 A closer look into Coley’s toxins found that it was the polysaccharide fraction from the Serratia marcescens bacteria inducing tumor necrosis.77 We now define this component of the outer membrane of Gram-negative bacteria as LPS, a TLR4 agonist. Unfortunately, due to the toxicity of LPS, the tolerable dose is typically too low to induce a robust antitumor effect in patients with cancer, and therefore, is not a viable treatment, although it suggests there is potential for TLR activation in anticancer therapy.78 Much work has been done since to provide further insight into the anti-tumor effects of TLR ligands.78–81
BCG, an attenuated Mycobacterium bovis strain, was developed as a tuberculosis vaccine, however it was proposed relatively early on to have anticancer properties after an observation that administering it into tumors had a moderate anticancer effect.1 Almost a century on, this was confirmed in successful trials using BCG as a treatment for bladder cancer,82 83 and it is still used to this day. Peptidoglycan, an important component of the Mycobacterium cell wall and a TLR2/4 agonist, is thought to drive BCG’s local anti-tumor effect through the induction of a substantial inflammatory response.78 Peptidoglycan potentiates the maturation of dendritic cells, leading to increased proinflammatory cytokine secretion and immune cell infiltration.84
The most well defined antitumor effect of TLRs is their ability to stimulate the adaptive immune system, either directly or via APCs, and enhance its ability to act against aberrantly expressed antigens present in cancers. So far, several TLR agonists, most notably those activating TLR2, 3, 4, 7, 8 and 9, have been heralded as promising novel immunotherapeutic agents due to their ability to initiate T-cell immunity, both as monotherapies and in conjunction with other treatments.10 85 Agonists can also be delivered alongside antigen as vaccine adjuvants, for example by conjugating TLR ligands to antigenic peptides.86 87
In humans, both the TLR1/2 ligand Pam3CSK4 and the TLR7 ligand gardiquimod have been used as antitumor vaccine adjuvants to boost T-cell responses. These combinations have been shown to induce tumor-specific human CD8 +T cells with reduced PD-1 expression and improved anti-tumor activity in human xenograft mouse models.88 TLR agonists, including imiquimod (TLR7), CpG 7909 (TLR9) and Poly I:C (TLR3) have shown therapeutic potential in various cancers when administered locally as monotherapies, however, their use as systemic therapies are limited by toxicity.10
In mice, stimulation of TLR2 and/or TLR1/2 heterodimer in T-cells has shown recent promise as an anticancer therapy. In mouse models of lung carcinoma, leukemia and melanoma, systemic treatment with the TLR1/2 agonist bacterial lipoprotein (BLP) leads to dose-dependent tumor regression and long-lasting protection against tumor re-challenge.89 Moreover, it was shown through the use of Severe Combined Immunodeficient (SCID) mice lacking T-cells or B-cells that this effect was at least partially mediated by T-cells themselves, as well as through TLR-expressing APCs. BLP reduced the suppressive function of FOXP3+ T-regulatory cells and enhanced the effector function of tumor specific T-cells. Systemic injection of BLP has also been shown to enhance the antitumor effects of adoptive T-cell therapy in a study of glioma-bearing mice.90 Results showed enhanced long-term survival and immune protection in comparison to mice receiving adoptive T-cell transfer without BLP. These effects were not seen when either TLR2-/- mice or TLR2-/- transferred T-cells were used. This suggests that the full antitumor effect of BLP in this study requires TLR2 expression by both the T-cells themselves and by other immune cells in the tumor microenvironment.
In a different study, Pam3CSK4 stimulation of mouse cytotoxic T lymphocytes enhanced antitumor activity in a an ovalbumin-expressing mouse model of melanoma.91 Adoptive transfer of OT-1 (ovalbumin-specific CD8+) T-cells followed by Pam3CSK4 injection resulted in an anti-tumor effect, whereas adoptive transfer of TLR2-/- OT-1 T-cells plus Pam3CSK4 had a minimal anti-tumor effect. Another study reported that in TCR transgenic ‘pmel’ mice, TLR2-stimulated, melanoma gp100 antigen-targeting CD8 T-cells responded to significantly lower tumor antigen levels and were more cytotoxic than TLR2-/- or MyD88-/- pmel T-cells.92 This enhanced antitumor activity was attributed to both increased effector function and increased survival of the T-cells.
Beyond the T-cell intrinsic effects of TLR signaling illustrated here, a recent review has described a broad pattern of TLR expression in human and mouse NK cells. TLR2, 3, 4, 5, 7 and 9 agonists can stimulate NK cell cytotoxicity and IFN-γ production resulting in antitumor activity.93 This combined body of evidence implicates NK cells, along with T-cells and APCs, in the response to TLR-mediated cancer immunotherapies.
Clinical trials
As of August 2021, there were over 200 currently active or completed cancer-related clinical trials using TLR ligands, inhibitors or TLR signaling domains listed in the National Institutes of Health’s National Clinical Trials (NCT) database. The vast majority of these trials target the TLR pathway in combination with other treatments such as radiotherapy and chemotherapy (NCT01421017), immune checkpoint inhibition (NCT02643303) and dendritic cell vaccines (NCT01204684).94–98 Listed in table 2 are late-stage clinical trials for various TLR ligands (completed phase 2 and above). While this list illustrates the vast number of existing clinical trials for TLR ligands as cancer therapies, it is by no means exhaustive. There are many phase 1 and early-stage phase 2 trials listed in clinical trial registries that are currently active across all TLRs, for agonists and inhibitors listed below as well as other novel ligands.
Phase 3 and completed phase 2 clinical trials targeting the TLR pathway for treatment of cancer
The number of later-phase trials listed in table 2 highlights the keen interest in TLR ligands as cancer therapies. A major caution for the use of systemic TLR agonists is that although these compounds can result in direct and indirect T-cell activation, in certain circumstances, TLR activation may also promote tumor growth.99 Several studies have shown that not only is the expression of certain TLRs elevated in both human and mouse tumors, but that the activation of these TLRs can enhance tumor progression and worsen disease prognosis.100 101 TLR2 activation in particular has been shown to have a direct tumor-stimulating effect in certain malignancies, promoting tumor cell survival, proliferation and metastatic capabilities. It can also enhance resistance to chemotherapeutic agents through its activation of NF-κB.101–104 Similar results have been seen with TLR4 activation, where stimulation with LPS resulted in increased tumor cell survival, proliferation and metastatic potential.105 106 This effect was also observed with TLR5 and TLR9 agonists.107–109
Overall, there is strong evidence that administration of TLR agonists can have either a protumor or antitumor effect, depending on the ligand and the tumor type. Evidently, the tumor-stimulating effect is not driven by TLRs expressed on the T-cells, but rather by the TLRs expressed on the cancer cells themselves. As such, there is a substantial advantage to being able to exclusively harness TLR action on T-cells, without stimulating TLRs on the tumor cells. This holds particular potential with TLR2, which has immense T-cell activation potential, but can induce a protumor effect in various cancers.
Costimulation of gene-modified T-cells via TLR signaling
Gene-modified T-cells, expressing a specific TCR or CAR directed against a specific antigen, afford a unique opportunity to both redirect and to modify the function of T-cells. Tumor-specific CARs typically incorporate one or more costimulatory domains in the intracellular region of the construct, alongside the TCR signaling domain, CD3ζ. Costimulatory domains must be carefully considered, as different signaling proteins can change crucial functional aspects of the CAR T-cells with the same antigen-recognition domain, including their kinetics, cytotoxicity and safety profile.110 Although CAR costimulatory molecules have traditionally been confined to the immunoglobulin (Ig) superfamily members CD28 or ICOS, or the tumor necrosis factor receptor (TNFR) superfamily members, 4-1BB and OX40, TLR signaling domains may also represent useful costimulatory partners for CD3ζ.11–13
In one study, the TLR adaptor molecule MyD88 was employed alongside CD40 in an inducible costimulatory complex consisting of a chemical inducer of dimerization binding domain, and coexpressed with a first-generation HER2-targeting CAR construct in T-cells.14 These inducible MyD88/CD40 CAR T-cells exhibited superior T-cell proliferation, cytokine production and tumor killing ability in comparison to second-generation CAR T-cells that did not contain the inducible MyD88/CD40 molecule both in vitro and in xenograft models. This same inducible system, adapted to target PSCA, is currently being used in a phase 1/2 clinical trial (NCT02744287). Recently, work has been published on CD19- and CD123-targeting MyD88/CD40 CAR T-cells, this time with the MyD88 and CD40 domains tethered to the CAR molecule15 (figure 3B). These signaling domains have been shown to successfully enhance CAR T-cell proliferation in vivo.
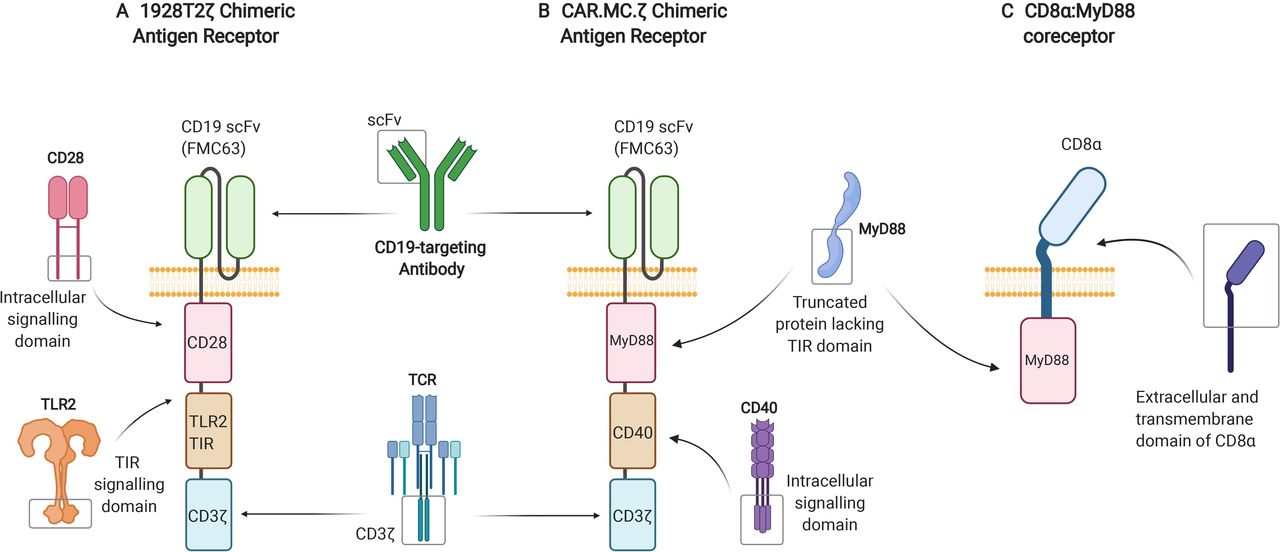
{kind=link}
{kind=link}
{kind=link}
Depiction of CAR and coreceptor constructs utilizing the TLR signaling pathway. (A) Third-generation, CD19-targeting, 1928T2z CAR construct incorporating the intracellular TIR signaling domain derived from TLR2, alongside the intracellular signaling domain of CD28 and CD3ζ. (B) Third-generation, CD19-targeting, CAR.MC.ζ construct incorporating a truncated version of MyD88 lacking its TIR domain and the intracellular signaling portion of CD40 alongside CD3ζ. (C) CD8α:MyD88, a synthetic coreceptor that fuses together the extracellular and transmembrane domains of CD8α and the intermediate and death domains of MyD88. CAR, chimeric antigen receptor; MyD88, myeloid differentiation primary response 88; TIR, toll/interleukin-1 receptor; TLR, Toll-like receptor.
Another study generated third generation CARs containing the TIR domain of TLR2 alongside CD28 and CD3ζ: one targeting CD19 (1928zT2) and another targeting mesothelin (m28zT2).12 The CD19-targeting and mesothelin-targeting CARs harboring the TLR2 TIR domain displayed enhanced expansion, persistence and effector function both in vitro and in vivo compared with second generation CD28 signaling CARs with no TLR2 domain. In a phase I clinical trial (NCT02822326), one patient with relapsed B-cell ALL receiving a single dose of 5×104 cells/kg of 1928T2z CAR T-cells experienced complete eradication of the leukemia.12 Subsequently, a further three patients diagnosed with relapsed or refractory ALL were treated with between 5×104 and 1×106 1928T2 z CAR T cells/kg and achieved complete remission without serious adverse events.11 Building on these results, a phase I clinical trial using a 1928T2z CAR (figure 3A) to treat relapsed or refractory non-Hodgkin’s B-cell lymphoma is currently being undertaken at our own center (‘ENABLE’, NCT04049513).13Further research has recently been conducted with the aim of making these CAR T-cells more effective against solid malignancies. When engineered to secrete human IL-7 and CCL19, glypican-3-specific (g28T2z) and mesothelin-specific (m28T2z) CAR T-cells displayed enhanced tumor clearance in both xenograft models and a phase I clinical trial (NCT03198546).111
In addition to CARs, TLR pathway signaling domains are showing success in other synthetic T-cell stimulatory molecules. CD8α:MyD88, a synthetic coreceptor that fuses together the extracellular and transmembrane domains of CD8α and the intermediate and death domains of MyD88 (figure 3C), is being used as another way of activating the TLR signaling pathway in T-cells.112 On antigen binding, the CD8α portion of the coreceptor interacts with the TCR and initiates TLR pathway activation through the fused MyD88 intracellular domain. This results in increased effector function and decreased exhaustion of the T-cells, which has been shown to provide an advantage against weak tumor antigenicity and suppressive tumor microenvironments.
The field of CAR-NK cells is growing rapidly, having shown early promise in research and clinical settings.113 Noting the impact of TLR agonists on NK cell function, CAR constructs containing TLR pathway signaling domains could also provide a functional advantage to CAR NK-cells, although this area of research is yet to be explored.
Benefits of T-cell and NK-cell intrinsic TLR signaling include enhanced proliferation and antitumor activity in response to lower levels of antigen. TLR signaling in CAR T-cells leads to qualitative improvement in phenotype and function, which may enable the use of lower doses of gene-modified T-cells for adoptive cell therapy. Restricting TLR signaling to the T-cells themselves avoids any potential growth-promoting impacts of TLR agonists on tumor and microenvironment. It should be noted that this approach is not without risk. Enhancement of T-cell cytokine production by TLR signals could plausibly present an increased risk of cytokine release syndrome, a key CAR T-cell toxicity. Furthermore, an activating point mutation of MYD88 has been implicated as a driver mutation in some B-cell lymphomas,114 115 and a theoretical concern is that constitutive TLR signaling might carry a risk of malignant transformation of the gene-modified T-cells. This highlights the need for careful monitoring of phase I safety trials for these new therapies. The use of inducible costimulatory domains, or incorporation of TLR TIRs within the CARs so TIR dimerization is driven by antigen-engagement, could mitigate against constitutive MyD88 activity. Safety measures of broad applicability to gene-modified cellular therapies include use of late-generation lentiviral vectors with an established safety record, incorporation of ‘safety switches’ that can deplete gene-modified cells, and recipient enrolment to cellular therapy registries that capture rare or long-latency toxicities.116 Finally, the relative merits of employing MyD88 and TLR TIR domains within gene-modified T-cells, compared with costimulatory domains derived from Ig or TNFR superfamily members, are yet to be fully determined, and benefits of specific costimulatory domains may prove to be specific to the CAR T-cell target or to the malignancy being treated.110
Concluding remarks
Since Janeway first demonstrated T-cell intrinsic expression of TLR4 in 1989, understanding of the critical role of TLRs within T-cells has expanded substantially. It is now clear that many TLRs can be expressed by, and are functional within, both human and murine T-cells. TLR agonists can lower the threshold for TCR activation, enhance T-cell proliferation and cytokine production, promote T-cell memory formation, and modify the suppressive functions of Tregs.
T-cell intrinsic TLR expression is essential for the full effects of some systemically-administered TLR ligands, including classes of agent that are in clinical trials as cancer immunotherapies. The observation that TLR stimulation can promote tumor growth in some circumstances emphasizes the need for caution when administering TLR agonists systemically. Careful selection of agonist and route of administration, informed by TLR expression in the tumor and its microenvironment, may be crucial. Furthermore, the effect of combining TLR ligands with existing T-cell-based cancer immunotherapies, particularly checkpoint blockade, needs to be clarified.
T-cell-intrinsic TLR signaling clearly plays a key role in the quality and longevity of adaptive immune responses. In previous studies, this fact has often been overlooked in favor of focusing on the functions of TLR signaling in innate immune cells, which typically express much higher levels of TLRs. The burgeoning field of gene-modified adoptive cell therapies, including CAR T-cells, affords a new opportunity to specifically trigger TLR signaling within T-cells. Further research will determine the safety and efficacy of TLR signaling domains in CAR T-cells, compared with other costimulatory domains. Based on an in-depth review of the literature along with our own experience, we conclude that the appreciation of both T-cell intrinsic and extrinsic effects of TLR signaling is essential for those planning, and interpreting the results of, clinical trials involving TLR-activating cancer immunotherapies.
Ethics statements
Patient consent for publication
Acknowledgments
We wish to thank the Thompson Family Foundation and the Grady Grant for their generous sponsorship of the Malagahan Institute’s CAR T-cell research programme, as well as the John and Margaret Hunn Education Trust for their support of Yasmin Nouri. Figures were created using BioRender.com.
References
Footnotes
Contributors RP and RW conceived the manuscript; YN, RP and RW wrote the manuscript; all authors approved the final manuscript.
Funding The authors received funding from the Health Research Council of New Zealand (grants 19-816 to RW and RP, and 19-139 to RW), Freemasons New Zealand, a research fellowship from the Keith & Faith Taylor Charitable Trust (RP) and a PhD scholarship from Leukemia & Blood Cancer New Zealand (YN).
Competing interests RP and RW are employees of the Malaghan Institute of Medical Research, a registered charity and sponsor of a trial of chimeric antigen receptor T-cells incorporating a TLR2-derived domain; none of the authors have a proprietary or intellectual property interest in the product; no other competing interests to declare.
Provenance and peer review Not commissioned; externally peer reviewed.