Article Text

Abstract
Background Successful targeting of solid tumors such as breast cancer (BC) using chimeric antigen receptor (CAR) T cells has proven challenging, largely attributed to the immunosuppressive tumor microenvironment (TME). Myeloid-derived suppressor cells (MDSCs) inhibit CAR T cell function and persistence within the breast TME. To overcome this challenge, we have developed CAR T cells targeting tumor-associated mucin 1 (MUC1) with a novel chimeric costimulatory receptor that targets tumor necrosis factor–related apoptosis-inducing ligand receptor 2 (TR2) expressed on MDSCs.
Methods The function of the TR2.41BB costimulatory receptor was assessed by exposing non-transduced (NT) and TR2.41BB transduced T cells to recombinant TR2, after which nuclear translocation of NFκB was measured by ELISA and western blot. The cytolytic activity of CAR.MUC1/TR2.41BB T cells was measured in a 5-hour cytotoxicity assay using MUC1+ tumor cells as targets in the presence or absence of MDSCs. In vivo antitumor activity was assessed using MDSC-enriched tumor-bearing mice treated with CAR T cells with or without TR2.41BB.
Results Nuclear translocation of NFκB in response to recombinant TR2 was detected only in TR2.41BB T cells. The presence of MDSCs diminished the cytotoxic potential of CAR.MUC1 T cells against MUC1+ BC cell lines by 25%. However, TR2.41BB expression on CAR.MUC1 T cells induced MDSC apoptosis, thereby restoring the cytotoxic activity of CAR.MUC1 T cells against MUC1+ BC lines. The presence of MDSCs resulted in an approximately twofold increase in tumor growth due to enhanced angiogenesis and fibroblast accumulation compared with mice with tumor alone. Treatment of these MDSC-enriched tumors with CAR.MUC1.TR2.41BB T cells led to superior tumor cell killing and significant reduction in tumor growth (24.54±8.55 mm3) compared with CAR.MUC1 (469.79±81.46 mm3) or TR2.41BB (434.86±64.25 mm3) T cells alone. CAR.MUC1.TR2.41BB T cells also demonstrated improved T cell proliferation and persistence at the tumor site, thereby preventing metastases. We observed similar results using CAR.HER2.TR2.41BB T cells in a HER2+ BC model.
Conclusions Our findings demonstrate that CAR T cells that coexpress the TR2.4-1BB receptor exhibit superior antitumor potential against breast tumors containing immunosuppressive and tumor promoting MDSCs, resulting in TME remodeling and improved T cell proliferation at the tumor site.
- receptors
- chimeric antigen
- myeloid-derived suppressor cells
- tumor microenvironment
- immunotherapy
- cytotoxicity
- immunologic
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- receptors
- chimeric antigen
- myeloid-derived suppressor cells
- tumor microenvironment
- immunotherapy
- cytotoxicity
- immunologic
Introduction
Breast cancer (BC) is the most common malignancy in women worldwide and a leading cause of cancer-related deaths.1 Triple negative breast cancer (TNBC) is a heterogeneous group of tumors accounting for 15%–20% of all BC cases.2 3 However, patients with TNBC suffer the poorest outcomes with worst overall survival due to the lack of effective targeted therapies that are implemented for other BC subtypes.4 5 Thus, for TNBC, new targets and effective therapeutic strategies are urgently needed. Chimeric antigen receptor (CAR) T cell immunotherapy has demonstrated remarkable success in the treatment of hematological malignancies.6 7 However, CAR T cell treatment of solid tumors like BCs has proven more challenging, largely due to the hostile tumor microenvironment (TME) of solid tumors.8
The BC TME is characterized by a milieu of chemokines such as CXCL5, CCL2, and CCL5, which recruit bone marrow–derived myeloid cells that are subsequently polarized to myeloid-derived suppressor cells (MDSCs), due to the abundance of tumor suppressive cytokines (eg. granulocyte-colony stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and interleukin-6 (IL-6)).9–15 Indeed, in patients with BC, increased levels of both circulating and tumor-resident MDSCs have been correlated with advanced clinical stage, increased metastatic disease burden,16 17 and immune suppression. The presence of MDSCs has been associated with poor response to immunotherapies in patients with solid tumors.18 MDSCs inhibit effector T cell function through several mechanisms including (1) induction of T regulatory cells (Tregs), (2) production of reactive oxygen species, (3) secretion of anti-inflammatory cytokines (eg, IL-10 and TGFβ), (4) depletion of important amino acids (arginine and tryptophan) necessary for T cell proliferation through induction of arginase (Arg) and indoleamine 2,3-dioxygenase, and (5) expression of PD-L1.19 20 Additionally, MDSCs actively shape the TME through cross-talk with malignant BC cells and the surrounding stroma, leading to increased angiogenesis, tumor invasion, and metastasis by remodeling the extracellular matrix via production of matrix metallopeptidase-9 (MMP-9).21 22
Spurred by these facts, investigators have focused on discovering ways to eliminate MDSCs from the TME. Most strategies are non-specific and have significant side effects.23 24 However, a more targeted approach is the use of an agonist monoclonal antibody (mAb) against tumor necrosis factor (TNF)-related apoptosis induced ligand-receptor 2 (TR2), a receptor expressed on MDSCs that activates apoptosis on engagement with its soluble ligand, TRAIL.25 A clinical trial investigating a TR2 agonistic antibody (DS-8273a) demonstrated that treatment significantly reduced MDSC numbers in the peripheral blood and at the breast tumor site. This reduction in MDSCs was associated with longer progression-free survival.26
To target BC, our group has developed and validated CAR T cells directed toward a hypoglycosylated form of Mucin 1 (MUC1),27 which is selective to the tumor, thereby reducing issues related to on target off tumor toxicity.28–30
We sought to increase the potential for clinical benefit by enhancing the expansion and persistence of these MUC1 CAR T cells, while simultaneously remodeling the suppressive TME. To this end, we developed a novel chimeric costimulatory receptor termed TR2.41BB, which encodes the single chain variable fragment (scFv) of the TR2 agonist antibody DS-8273a followed by a 41BB endodomain.
We hypothesized that engagement of this TR2.41BB receptor with TR2 expressed on TME-resident MDSCs would lead to both MDSC apoptosis and CAR T cell costimulation, promoting superior antitumor effects.
Materials and methods
Donor and cell lines
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers at Gulf Coast Regional Blood Center. The 293T and human BC cell lines (MDA-MB-231, MDA-MB-453, SUM-159, and BT-20) were obtained from the American Type Culture Collection (ATCC) and were grown in Dulbecco’s Modified Eagle Medium (GE Healthcare Life Sciences) supplemented with 10% FBS (HyClone, Waltham, Massachusetts, USA) and 2 mM L-Glutamax (Gibco BRL Life Technologies, Gaithersburg, Maryland, USA). All cell lines were maintained in a humidified atmosphere containing 5% carbon dioxide (CO2) at 37°C.
Generation of retroviral constructs and retroviral supernatant
We used a codon-optimized second-generation CAR with specificity against tumor-associated MUC1 comprised of an HMFG2 scFv, linked to a IgG2-CH3 spacer domain followed by a CD28 transmembrane and costimulatory endodomain and CD3 ζ chain in a SFG retroviral backbone.27 To generate the costimulatory TR2.41BB receptor, an SFG-based retroviral vector was engineered to encode an scFv derived from the TR2 (DS-8273a) mAb26 fused to an IgG2-CH3 spacer domain, followed by a 41BB costimulatory endodomain. The SFG retroviral vector encoding the HER2-specific CAR was previously described, consisting of the HER2-specific scFv FRP5, a M13 hinge, a CD28 transmembrane, and a CD28.ζ signaling domain.31 Retroviral supernatant encoding either CAR or the TR2.41BB receptor was produced using 293T cells as previously described.32 Briefly, cells were cotransfected using GeneJuice transfection reagent (Novagen) with 3.75 µg of CAR.MUC1, TR2.41BB, or CAR.HER2 retroviral vectors, 3.75 µg of Peg-Pam-e plasmid containing the sequence for the MoMLV gag-pol, and 2.5 µg of the RDF plasmid containing the sequence for the RD114 envelope. Retroviral supernatant was collected at 48 and 72 hours post-transfection, filtered using a 0.45 mm filter, and stored at −80°C.
Generation of MDSCs
CD14+ cells within PBMCs were isolated using anti-CD14 magnetic microbeads and LS column separation (Miltenyi Biotech) per the manufacturer’s instructions. These cells were then cultured in complete RPMI media (10% FBS and 2 mM L-Glutamax) for 7 days in the presence of GM-CSF (20 ng/mL) and IL-6 (20 ng/mL). GM-CSF was added on days 1, 3, and 5, whereas IL-6 was added only on day 533 34 (online supplemental figure 1). The cells were then harvested on day 7, and flow cytometry was performed to assess the phenotype (CD33+CD11b+HLA-DRlowCD14+CD15-).
Supplemental material
Generation of CAR T cells
The CD14 negative PBMC fraction was used to generate CAR T cells using a previously described protocol.27 To generate CAR.MUC1 and TR2.41BB coexpressing cells, activated T cells were sequentially transduced with CAR.MUC1 and TR2.41BB on days 3 and 4, respectively. Transduction efficiency was measured 3 days post transduction via flow cytometry. CAR.HER2 and TR2.41BB coexpressing cells were generated in the same manner.
Flow cytometry
Cells were stained with ~2 µL of antibodies for 20 min at 4°C, washed (PBS, Sigma-Aldrich) and acquired on the Gallios flow cytometer (Beckman Coulter). Analysis was performed using Kaluza flow analysis software (Beckman Coulter). Details for antibodies used are in table 1.
Antibodies used for flow cytometry
Chromium-release assay
The cytotoxic potential of effector T cells was measured in a standard 4-hour or 6-hour chromium-51 (51Cr)-release assay using effector:target (E:T) ratios of 40:1, 20:1, 10:1, and 5:1. Effector T cells (Non-transduced (NT), CAR.MUC1, TR2.41BB, CAR.HER2, CAR.MUC1.TR2.41BB, and CAR.HER2.TR2.41BB) were coincubated with 51Cr-labeled target cells in triplicate wells of a V-bottomed 96-well plate. Targets included BT-20, MDA-MB-231, MDA-MB-453, SUM-159, and MDSCs (5–6 hour assay). At the end of their respective incubation periods at 37°C and 5% CO2, supernatants were harvested, and radioactivity was counted in a gamma counter. Percentage of specific lysis was calculated as follows: % specific cytotoxicity=[experimental release (cpm)–spontaneous release (cpm)]/[maximum release (cpm)–spontaneous release (cpm)]×100.
Suppression assay
The suppressive function of MDSCs was measured by their ability to inhibit the proliferation of autologous T cells. Fresh T cells were isolated from PBMCs of autologous donors using anti-CD3 microbeads and magnetic LS column separation (Miltenyi Biotech). T cells were then labeled with 1 µM CFSE (5,6-carboxyfluorescein diacetate succinimidyl ester) and stimulated with anti-CD3/CD28 mAbs (Invitrogen) and IL-2 (200 U/mL, R&D Systems). Stimulated T cells were seeded at a concentration of 0.5×106 cells/well alone and with autologous MDSCs at 1:1 and 1:2 ratios in a 24-well plate. After 3 days of culture, cells were harvested and stained with CD4-PacBlue (clone 13B8.2) and CD8-APC (clone B9.11) antibodies obtained from BD Biosciences. T cell proliferation was assessed as CFSE dilution determined by flow cytometry.
Immunohistochemistry (IHC)
Paraffin-embedded tumor tissues from tumor only and tumor +MDSC-treated mice were sectioned. Sections (5 µm) were deparaffinized in xylene, dehydrated and rehydrated in ethanol, and subjected to steamer antigen retrieval. The sections were then processed with the following antibodies: mouse anti-CD31 (R&D Systems) and human anti-αSMA (Leica). A total of 10–14 fields of view were analyzed for each group.
Determination of apoptosis
PBMCs (resting or activated) were labeled with CFSE (Biolegend) and cocultured in the presence of NT T cells or T cells transduced with CAR.MUC1, TR2.41BB, or CAR.MUC1.TR2.41BB. Cells were harvested at 6 and 24 hours, and apoptosis was evaluated using an Annexin-V-PE apoptosis detection kit with 7-AAD (Biolegend) by flow cytometry. Cells were washed twice with cold PBS and resuspended in Annexin V binding buffer. They were then stained with 5 µL of Annexin-V-PE and 5 µL of 7-AAD viability staining solution for 15 min at room temperature in the dark prior to flow cytometric analysis.
ELISA
NT or TR2.4-1BB transduced T cells were cultured alone (negative control) or in the presence of recombinant TR2 protein or anti-CD3 and anti-CD28 (positive control). The cells were then harvested at 120 min and the nuclear fraction was extracted using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit from Thermo Fisher Scientific.
An ELISA for human NFκB was performed using the NFκB p65 Transcription Factor Assay Kit (ab133112, Abcam) following manufacturer’s instructions.
Supernatants from the suppression assay wells were harvested on day 3, and ELISA for human IFNγ was performed using the IFNγ ELISA Kit (EHIFNG; Thermo Fisher Scientific) following manufacturer’s instructions. Absorbance for the above ELISAs was measured at 450 nm with a TECAN Infinite M200 Pro plate reader.
Western blot assay
Harvested cells (resting and activated T cells and MDSCs) were washed three times with ice-cold PBS and protein lysate was extracted by RIPA lysis buffer containing protease inhibitor cocktail for 20 min on ice, followed by centrifugation at 12,500×g for 20 min at 4°C. The protein concentration of this lysate and the above nuclear extract was determined using the BCA protein assay kit (Thermo Fisher Scientific). Equivalent amounts of protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, and then transferred to a polyvinylidene difluoride membrane (Bio-Rad). After blocking with 5% (w/v) milk overnight at 4°C, the blots were incubated with primary antibodies (anti-cFLIP) (clone 7F10, Enzo Life Sciences), anti-p65 (clone F6, Santa Cruz Biotechnology), anti-PCNA (clone PC10, Santa Cruz Biotechnology), and anti-GAPDH (clone 6C5, Sigma Aldrich) overnight at 4°C. The membranes were washed three times and further incubated with a corresponding secondary antibody (IRDYE 680RD goat anti-mouse, Licor) for 2 hours at room temperature. The membrane was imaged on Licor, and the cFLIP expression was normalized to GAPDH while the p65 expression was normalized to PCNA.
In vivo studies
Animal experiments followed a protocol approved by Baylor College of Medicine Institutional Animal Care and Use Committee. For the in vivo cell-derived xenograft (CDX) mouse model, 6-week to 8-week-old female NSG (NOD.CgPrkdcscid Il2rgtm1Wjl/SzJ, Jackson Laboratories, stock #005557) mice were injected with 5×106 SUM-159 cells, genetically modified with a retroviral vector encoding a green fluorescent protein firefly luciferase (GFP.ffLuc), fusion gene (SUM-159.GFP.ffLuc), with/without 5×106 MDSCs suspended in 50% DPBS/50% matrigel subcutaneously into the left fourth mammary fat pad. Tumor burden was monitored using the IVIS Lumina In Vivo Imaging System (Caliper Life Sciences, Hopkinton, Massachusetts, USA) 15 min after intraperitoneal injection with 100 µL of D-luciferin (15 mg/mL). Tumor size was measured biweekly via caliper and tumor volume (mm3) was calculated as follows: length×width×width/2. For the in vivo CAR T cell therapy study, the same xenograft mouse model was used. Once the tumors reached ~100 mm3 (~2 weeks), animals were injected intravenously on day 14 with 1.5×106 CAR.MUC1, TR2.41BB, or CAR.MUC1.TR2.41BB T cells, and tumor volume was measured. For the MDA-MB-231 CDX mouse model, NSG mice were implanted with 5×106 MDA-MB-231 cells with/without 5×106 MDSCs. Once tumors reached ~100 mm3 (~2 weeks), animals were injected intravenously on day 14 with 1.5×106 GFP.ffLuc+ CAR.MUC1, TR2.41BB, or CAR.MUC1.TR2.41BB T cells. T cell expansion and persistence was monitored using the IVIS Lumina In Vivo Imaging System. All in vivo analysis was performed using Living Image software (Caliper Life Sciences, Hopkinton, Massachusetts, USA). For the MDA-MB-453 HER2+ CDX mouse model, 5×106 MDA-MB-453 cells were subcutaneously injected into the left fourth mammary fat pad of NSG mice. On day 14, 5×106 MDSCs were intratumorally administered to a subset of these mice. On day 21, once tumors reached ~100 mm3 (~3 weeks), mice were injected intravenously with 1.5×106 GFP.ffLuc+ CAR.HER2, TR2.41BB, or CAR.HER2.TR2.41BB T cells. All parameters were measured as described above.
Data sources
Bulk transcriptomic data were analyzed from two independent publicly available human BC datasets; METABRIC (microarray), PMID: 22 522 925 and CPTAC-BRCA (RNA-seq), PMID: 33 212 010. The mRNA quantification for the TR2 gene was obtained.
Statistical analysis
All statistical analysis was performed using the GraphPad Prism V.8.01 statistical software. The results were expressed as the mean of arbitrary values±SEM. Statistical significance between groups was assessed by two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons, or unpaired two-tailed t-test where a p value less than 0.05 denoted statistical significance.
Results
TR2.41BB costimulatory receptor specifically targets TR2 and induces 41BB signaling
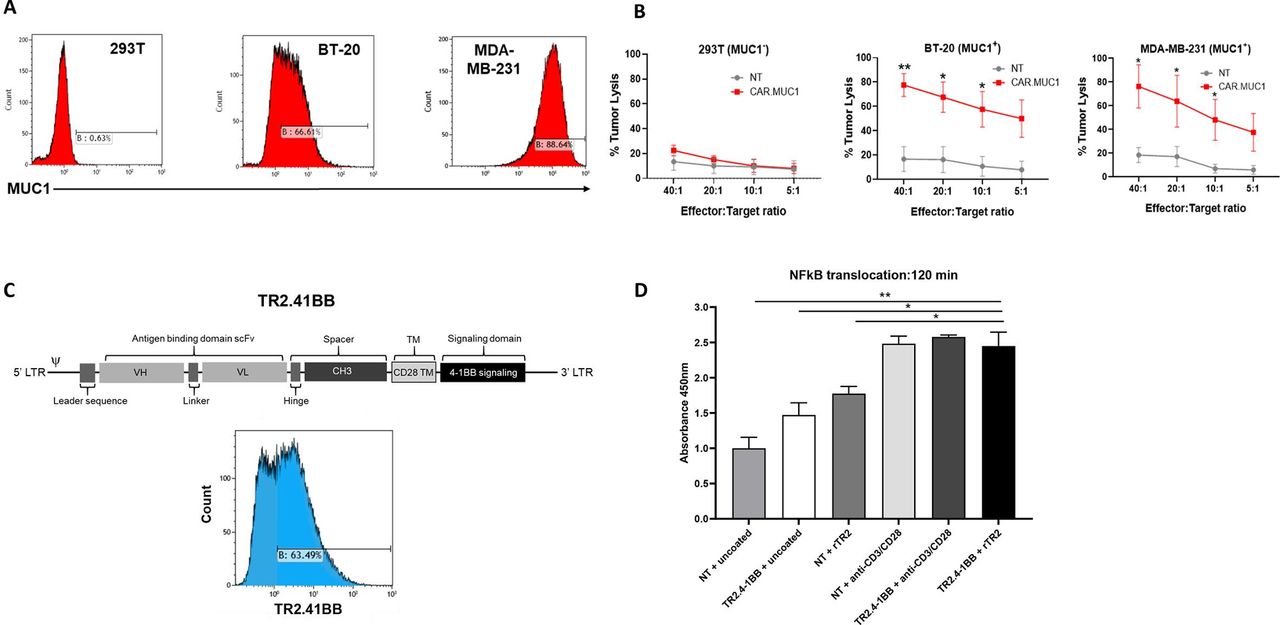
We used a retroviral vector previously generated and validated by our group encoding a second generation human, codon optimized CAR directed toward MUC1 to target TNBC.27 We confirmed that CAR.MUC1 transduced T cells specifically eliminate MUC1 expressing BC cell lines (BT-20 and MDA-MB-231), without killing the MUC1 negative cell line 293T (figure 1A,B).

The novel TR2.41BB costimulatory receptor induces 41BB signaling on TR2 engagement. (A) Mucin 1 (MUC1) expression on triple negative breast cancer (TNBC) cell lines BT-20, MDA-MB-231, and the MUC1- cell line 293T. (B) In vitro cytolytic function of control (non-transduced (NT)) and CAR.MUC1 T cells assessed in a 5-hour 51Cr-release assay at effector:targets of 5:1 to 40:1 using MUC1 + targets (BT-20, MDAMB-231) and MUC1- target (293T). Data represent mean±SEM (n=5). (C) Schematic representation of the TR2.41BB construct and transgenic expression of TR2.41BB T cells detected using biotinylated TR2. (D) NT and TR2.41BB transduced T cells were cultured alone or in the presence of rTR2 or anti-CD3 and anti-CD28. Cells were harvested at 120 min, the nuclear fraction was harvested, and an ELISA was performed to measure the translocation of NFκB into the nucleus. Data represent mean±SEM (n=3). Statistics: unpaired two-tailed t-test (B), two-way analysis of variance followed by Tukey’s multiple comparisons (D); *p<0.05; **p<0.01.
To overcome the suppressive TME, we set out to generate a chimeric receptor based on the TR2-specific agonist antibody DS-8273a26 designed to induce MDSC apoptosis while simultaneously providing T cells with 41BB costimulation (TR2.41BB) (figure 1C). To establish that a DS-8273a scFv-based chimeric receptor specifically recognizes TR2, we first designed a receptor that in addition to the 41BB costimulatory domain also included a T-cell activating, CD3ζ domain (TR2.41BB.CD3ζ). TR2.41BB.CD3ζ T cells specifically killed K562 target cells that had TR2 knocked-out and transduced with only the ectodomain of TR2, demonstrating TR2 specificity of the DS-8273a scFv in the context of our receptor design (online supplemental figure 2a,b).
We next sought to determine the functionality of our TR2.41BB costimulatory receptor. One readout of 41BB signaling is nuclear translocation of NFκB. Using an ELISA assay, we observed that ligation of TR2.41BB receptor with recombinant TR2 resulted in translocation of NFκB into the T cell nucleus at levels similar to polyclonal stimulation (absorbance: 2.48±0.10 and 2.44±0.19, NT and TR2.41BB, respectively, mean±SEM, n=3) (figure 1D) similar results were obtained on performing a western blot (online supplemental figure 3a,b). Collectively, these results demonstrate that the TR2.41BB construct binds TR2 and induces downstream T cell costimulatory signaling through the 41BB domain.
MDSCs promote tumor burden, vascularization, and stroma formation in TNBC cell line-derived xenograft (CDX) mouse models
MDSCs were generated from PBMCs of healthy donors by differentiating CD14 + monocytes in vitro33 34 (online supplemental figure 1). Differentiated MDSCs expressed defined monocytic MDSC phenotypic markers: CD33+, CD11b+, HLA-DRlow, and CD14+,35 36 and high surface levels of our TR2 target (online supplemental figure 4a). We confirmed that these MDSCs suppressed the proliferation and IFNγ production of autologous activated T cells (online supplemental figure 4b–d).
We established a CDX model in vivo using the SUM-159 TNBC cell line, details are provided in the Materials and methods section (figure 2A). Compared with mice that received tumor cells alone, mice that received MDSCs had significantly higher tumor volume (from 311.32±54.75 mm3 to 769.95±98.03 mm3, p=0.01) and weight (from 1.06±0.20 g to 1.56±0.15 g, p=0.03) (figure 2B,C). This difference was not as significant when we measured bioluminescence of the tumor cells (4.35×1010±1.54×1010 p/s vs 7.08×1010±4.90×109 p/s for SUM-159 and SUM-159 +MDSCs, respectively) (figure 2D,E).

Introduction of myeloid-derived suppressor cells (MDSCs) into a triple negative breast cancer (TNBC) cell-derived xenograft (CDX) mouse model leads to a higher tumor burden and a marked increase in tumor growth by promoting vascularization and accumulation of fibroblasts. (A) Schematic of in vivo experiment in which NSG (NOD.CgPrkdcscid Il2rgtm1Wjl/SzJ) mice were transplanted with GFP.ffLuc-labeled SUM-159 cells with or without MDSCs. (B) Bioluminescence imaging and (C) quantification of mice assessed weekly to monitor tumor burden and (D) tumor volume measured by calipers and (E) tumor weight quantification (mean±SEM, n=5/group). (F) NSG mice coinjected with SUM-159 cells and MDSCs were sacrificed on 7, 14, and 21 days, flow cytometry was performed to assess the presence of MDSCs at tumor site, (G) quantification of MDSCs within primary tumor. (H) Representative images of tumor sections from mice transplanted with SUM-159 cells in the presence or absence of MDSCs stained to detect vascularization (CD31 Ab). Total of 10 fields of view were analyzed for each group using ImageJ. (I) Quantification of CD31 + staining. (J) Representative images of tumor sections from mice transplanted with SUM-159 cells in the presence or absence of MDSCs stained to detect fibroblast accumulation (αSMA Ab). Total of 14 fields of view per mouse were analyzed for each group using ImageJ. (K) Quantification of alpha-smooth muscle actin (α-SMA)+ staining. Data represent mean±SEM (n=7). Statistics: unpaired two-tailed t-test (B, C, E, I, and K); *p<0.05; **p<0.01.
MDSC persistence at the tumor site was determined by coinjecting NSG mice with SUM-159 cells and MDSCs at a 1:1 ratio. We then sacrificed these mice on days 7, 14 and 21 post injection and performed flow cytometry to track MDSC (CD11b+CD33+) presence at the tumor site. MDSCs were detected at day 7 post engraftment with a sevenfold decrease in numbers on day 14 and complete loss of detection on day 21 (figure 2F,G).
To further investigate the marked increase in tumor volume observed in the presence of MDSCs, we performed IHC staining of the tumors to quantify vascularization and fibroblast accumulation through expression of CD31 and alpha-smooth muscle actin (α-SMA), respectively. Tumors coinjected with MDSCs exhibited more angiogenesis and accumulation of fibroblasts/stroma (figure 2H–K). These observations recapitulate known mechanisms of MDSCs,37 38 and likely account for the greater increase in tumor volume without equivalent increase in tumor cell bioluminescence in mice that received MDSCs.
Together, our results demonstrated that in vitro generated MDSCs closely resemble MDSCs found in patients with BC, allowing us to use these MDSCs in further functional studies of our TR2.41BB receptor.
TR2.41BB rescues CAR.MUC1 cytotoxic activity in the presence of MDSCs
We achieved high rates of transgene expression in activated T cells transduced or cotransduced with retroviral constructs encoding CAR.MUC1 and TR2.41BB, as determined by flow cytometry (87.3%±4%, 80.8%±2.7%, and 74%±5.5% for CAR.MUC1, TR2.41BB, and CAR.MUC1.TR2.41BB transduced T cells (figure 3A), respectively).

The TR2.41BB construct is able to rescue the cytotoxic activity of CAR.MUC1 in the presence of myeloid-derived suppressor cells (MDSCs). (A) Transgenic expression of CAR.MUC1.TR2.41BB T cells detected using anti-IgG2-CH3 spacer and biotinylated TR2, respectively, and quantitation of CAR.MUC1.TR2.41BB on T cells. Data represent mean±SEM (n=3). (B) CAR.MUC1 T cells were cultured with BT-20 (MUC1+) cells in the presence or absence of MDSCs and cytotoxic function was assessed in a 5-hour 51Cr-release assay. Data represent mean±SEM (n=7). (C) CAR.MUC1, TR2.41BB, and CAR.MUC1.TR2.41BB T cells were cultured with BT-20 and 51Cr-labeled MDSCs and the percentage of MDSC lysis was assessed in a 5-hour 51Cr-release assay. Data represent mean±SEM (n=5). (D) CAR.MUC1, TR2.41BB, and CAR.MUC1.TR2.41BB T cells were cultured with 51Cr-labeled BT-20 targets in the presence of MDSCs and cytotoxic function was assessed in a 5-hour 51Cr-release assay. Data represent mean±SEM (n=5). Statistics: two-way analysis of variance followed by Tukey’s multiple comparisons (B, C, and D); *p<0.05; **p<0.01.
T cell differentiation, activation, and exhaustion were assessed by flow cytometry 14 days post transduction. There were no significant differences in the memory T cell phenotype between the different constructs and NT cells with similar percentages of naïve and central memory subsets and a skewed ratio toward T cell effector memory (online supplemental figure 5a). No significant differences were noted in activation and exhaustion marker expression, increased levels of TIM3 and CD25 were observed across all T cell products (online supplemental figure 5b,c). Surface expression of transgenes was also similar across each condition (online supplemental figure 5d).
To determine if the cytotoxic activity of CAR.MUC1 T cells was attenuated in the presence of MDSCs, we measured their cytolytic function in a 5-hour 51Cr-release assay toward MUC1+ BT-20 cell line cocultured with or without MDSCs. Indeed, we observed that presence of MDSCs resulted in a nearly 25% reduction in the cytotoxic potential of the CAR.MUC1 T cells (from 67.99±3.55% to 43.90±4.90%, p=0.003 for 40:1 E:T ratio) (figure 3B). We next sought to determine whether the TR2.41BB construct induced apoptosis of MDSCs. Using MDSCs as targets, we observed that the TR2.41BB transduced T cells induced almost 40% apoptosis of MDSCs at a 40:1 E:T ratio, which was not observed in the conditions with NT or CAR.MUC1 T cells. Interestingly, CAR.MUC1.TR2.41BB cotransduced T cells induced almost 60% MDSC lysis at the same ratio (figure 3C). We assessed if coexpression of TR2.41BB construct with CAR.MUC1 on T cells could rescue their ability to kill tumor cells in the presence of MDSCs. Results showed that the addition of TR2.41BB increased the cytolytic function of CAR.MUC1 T cells from 43.90±4.90% to 77.64±11.76%, p=0.014 at a 40:1 E:T ratio, with this nearly 30% increase in cytotoxic potential observed across each of the E:T ratios (figure 3D). This demonstrates the efficacy of our dual targeting approach in vitro.
Due to the proximity of tumor and MDSCs in these co-culture experiments, we did observe some bystander killing of the MDSCs by CAR-MUC1 T cells and of the tumor cells by TR2.41BB T cells. This was expected and could be attributed to the known bystander effects of IFNγ/TNFα and/or FAS/FASL.39–41
To determine the safety of TR2.41BB receptor, we assessed the surface expression of TR2 on resting and activated PBMCs at 24, 48, and 72 hours by flow cytometry. Both resting and activated T cells showed high TR2 surface expression (online supplemental figure 6a,b). Next, percentage of apoptotic resting and activated T cells after co-culture with transduced T cells was assessed as described in the Materials and methods section. We did not observe any significant increase in the percentage of apoptotic T cells cultured in the presence of the TR2.41BB T cells compared with the NT or CAR.MUC1 groups (online supplemental figure 6a-c). Using western blot, we show that both resting and activated CD4 + and CD8+ T cells express high levels of cFLIP compared with MDSCs (online supplemental figure 6d,e). Thereby confirming results from earlier reports that resistance to TR2-mediated apoptosis was due to increased expression of cellular FLICE-like inhibitory protein (cFLIP), a negative regulator of apoptosis.42 43 These results demonstrate that TR2.41BB does not induce toxicity in resting or activated T cells.
Expression of TR2.41BB enhances the in vivo expansion and persistence of CAR.MUC1 T cells and augments their antitumor activity despite the presence of MDSCs
To determine the efficacy of our CAR T cell therapy in vivo, we used the MDA-MB-231 CDX model, details are provided in the Materials and methods section (figure 4A). T cells were detected at the primary tumor site in all conditions, with CAR.MUC1.TR2.41BB T cells demonstrating improved T cell proliferation and persistence in the presence of MDSCs compared with CAR.MUC1 or TR2.4-1BB T cells alone (2.32×108±1.07×108p/s vs 3.06×107±2.39×107p/s, p=0.02 and 1.01×108±7.02×107p/s, p=0.05 for CAR.MUC1 and TR2.41BB, respectively, on day 28 after T cell injection) (figure 4B,C). The inhibitory function of MDSCs in vivo was evident in this model, as mice receiving CAR.MUC1 T cells in the absence of MDSCs demonstrated better tumor control than mice that received tumors and MDSCs. This effect was rescued by introduction of the TR2.41BB receptor, whereby in the presence of MDSCs, only CAR.MUC1.TR2.41BB T cells significantly decreased the rate of tumor growth (181.71±22.98 mm3) compared with either CAR.MUC1 or TR2.41BB alone (410.01±29.04 mm3, p=0.001 and 363.77±42.17 mm3, p=0.01, respectively, on day 28 after T cell injection) (figure 4D).

Expression of TR2.41BB enhances CAR.MUC1 T cell expansion, persistence, and antitumor activity leading to decreased metastasis in vivo in the presence of myeloid-derived suppressor cells (MDSCs). (A) Schematic of in vivo experiment in which NSG (NOD.CgPrkdcscid Il2rgtm1Wjl/SzJ) mice were transplanted with MDA-MB-231 cells with or without MDSCs and then treated with GFP.ffLuc-labeled NT, CAR.MUC1, TR2.41BB, or CAR.MUC1.TR2.41BB T cells. (B) Bioluminescence imaging indicating expansion and persistence of T cells and quantification of (C) bioluminescence at the tumor site and (D) tumor volume measured by calipers. Mean±SEM, n=4/group. (E) Schematic of in vivo experiment in which NSG mice were transplanted with GFP.ffLuc-labeled SUM-159 cells with or without MDSCs and treated with NT, CAR.MUC1, TR2.41BB, or CAR.MUC1.TR2.41BB T cells. (F) Tumor volume measured by calipers. Mean±SEM, n=5/group. Statistics: two-way analysis of variance followed by Tukey’s multiple comparisons (C, D, and F); *p<0.05; **p<0.01; ***p<0.001. CAR, chimeric antigen receptor; MUC1, Mucin 1; NT, non-transduced.
We tested our CAR T cell therapy in a second BC orthotopic xenograft model, using SUM-159.GFP.ffLuc tumor cells (figure 4E). Similar to the MDA-MB-231 model, the CAR.MUC1.TR2.41BB T cells exhibited superior tumor cell killing and resulted in a significant reduction in tumor growth (24.54±8.55 mm3) compared with either CAR.MUC1 (469.79.9±81.46 mm3, p=0.003) or TR2.41BB (434.86±64.25 mm3, p=0.0004) T cells, on day 28 after T cell injection (figure 4F).
We observed greater tumor clearance in mice receiving CAR.MUC1.TR2.41BB T cells compared with the other groups as evidenced in both the primary tumor and the failure to develop metastases (online supplemental figure 7).
Combined expression of TR2.41BB and CAR.HER2 on T cells enhances antitumor potential in a HER2+ BC model
To test the benefit of the TR2.41BB expression in CAR T cells targeting another BC antigen, we took advantage of our previously generated HER2-CAR.31 Similar to CAR.MUC1 T cells, the in vitro cytotoxic potential of CAR.HER2 T cells against HER2 +MDA MB-453 cells was attenuated in the presence of MDSCs (63.27%±6.23% vs 84.87±2.68% for 40:1 E:T ratio, p=0.05) (figure 5A). Consistently, expression of the TR2.41BB construct restored the cytotoxic activity of CAR.HER2 T cells in the presence of MDSCs (from 63.40±6.13% to 90.40±4.97% for 40:1 E:T ratio, p=0.041) (figure 5B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Combined expression of TR2.41BB and CAR.HER2 on T cells enhances antitumor potential in a HER2 +BC model. (A) CAR.HER2 T cells were cultured with MDA-MB-453 (HER2+) cells in the presence or absence of myeloid-derived suppressor cells (MDSCs) and cytotoxic function was assessed in a 5-hour 51Cr-release assay. Data represent mean±SEM (n=3). (B) CAR.HER2, TR2.41BB, and CAR.HER2. TR2.41BB T cells were cultured with 51Cr-labeled MDA-MB-453 in the presence of MDSCs and cytotoxic function was assessed in a 5-hour 51Cr-release assay. Data represent mean±SEM (n=3). (C) NSG (NOD.CgPrkdcscid Il2rgtm1Wjl/SzJ) mice were transplanted with GFP.ffLuc-labeled MDA MB-453 cells with or without MDSCs and tumor volume was measured by calipers (mean±SEM, n=3–5/group). (D) NSG mice were transplanted with MDA-MB-453 with or without MDSCs and treated with GFP.ffLuc-labeled NT, CAR.HER2, TR2.4-1BB, or CAR.HER2.TR2.41BB T cells and tumor volume was measured by calipers (mean±SEM, n=5/group). Statistics: two-way analysis of variance followed by Tukey’s multiple comparisons (A, B, and D); unpaired two-tailed t-test (C); *p<0.05. BC, breast cancer; CAR, chimeric antigen receptor; NT, non-transduced.
We next assessed the efficacy of the CAR T cells in a HER2+CDX model. As in the TNBC CDX model, the inclusion of MDSCs mediated increased tumor growth (figure 5C). For the CAR T cell study, since the MDA-MB-453 cell line is slow growing and MDSCs are undetectable by day 21, we engrafted 5×106 MDA-MB-453 cells and then administered 5×106 MDSCs intratumorally 14 days later, details are provided in the Materials and methods section. In the presence of MDSCs, CAR.HER2.TR2.41BB T cells led to reduced tumor volume (134.27±27.6 mm3) compared with CAR.HER2 (253.82±71.05 mm3) or TR2.41BB (432.24±29.71 mm3, p=0.01 on day 14 after T cell injection) T cells (figure 5D). Thus, we demonstrate in both TNBC and HER2+ BC models that CAR T cells coexpressing our TR2.41BB receptor exhibit strong antitumor effects and modulate the TME, leading to sustained T cell persistence at the tumor site.
Discussion
We have demonstrated that coexpressing TR2.41BB receptor on CAR T cells augments CAR-T cell responses targeting either MUC1 or HER2 against orthotopic tumors in three distinct BC models. The TR2.41BB receptor eliminates MDSCs, thereby modulating the TME while simultaneously delivering a second costimulatory signal, leading to improved T cell survival, proliferation, and persistence at the tumor site.
We have previously validated a second-generation CAR which targets the highly tumor specific form of MUC1, expressed on greater than 80%–90% of BCs.44 In this body of work, we sought to simultaneously target MDSCs, thereby modifying the hostile TME and skewing it toward conditions that support and nurture CAR T cell effector function. Our results confirm that MDSCs hinder CAR T cell killing of tumor cell targets. An in vitro short-term cytotoxicity assay demonstrated markedly reduced killing of MUC1+ BT-20 cells by CAR.MUC1 T cells in the presence of MDSCs. In BC xenograft mouse models, tumor-bearing mice coinjected with MDSCs developed larger tumors, despite no detection of MDSCs by day 21 at the tumor site. These tumors were highly vascularized and enriched in stromal formation, with a propensity toward metastases. These results align with the role MDSCs play in enhancing neo-angiogenesis,45–48 recruiting stromal cells49 and inducing metastases via basal membrane remodeling.50–52 Also, the presence of MDSCs at the tumor was associated with failure of CAR T cells to control tumor growth. These findings further highlight the need to eliminate these cells in order to improve CAR T cell therapies for BC.
Because MDSCs highly resemble normal myeloid cells, specifically targeting and eliminating them from the TME without toxicity is extremely challenging. Flow cytometry analysis of MDSCs in this study confirm high expression of TR2 on their surface, supporting the idea of engaging this receptor to induce MDSC apoptosis. Condamine et al reported that targeting TR2 significantly reduced MDSC viability and improved immune responses in tumor-bearing mice.25 The clinical trial conducted by Dominguez et al reported that DS-8273a, a mAb against TR2, was able to selectively eliminate MDSCs in patients with solid tumors, and that this compound had no safety concerns.26 However, this approach had limited clinical efficacy on its own. This is consistent with results from other trials targeting TRAIL receptors for the treatment of cancer by using recombinant TRAIL or anti-TRAIL-R antibodies.53–66 The failure may be attributed to reduced potential to induce clustering of TRAIL-Rs and stabilization of higher-order DISCs (death-inducing signaling complex).67 Also, engagement with decoy, non-apoptosis inducing TRAIL-Rs (TRAIL-R3, TRAIL-R4, and OPG) may lessen the apoptosis-inducing capacity of any recombinant form of TRAIL. To overcome these obstacles, we sought to deliver a molecule with high TR2 specificity on the surface of the T cells, thereby facilitating formation of TR2 clusters through cell–cell interactions.
We have therefore developed a novel immunotherapeutic strategy to target both BC cells and MDSCs by genetically modifying CAR T cells to coexpress a costimulatory receptor (TR2.41BB) that enhances their effector functions and modifies the TME. This receptor not only induces apoptosis of MDSCs at the tumor site through activation of TR2 but also delivers a second costimulatory signal to the CAR T cells through a 41BB endodomain. As a result, T cells exhibit improved persistence and proliferation at the tumor site. We chose to include two costimulatory domains, CD28 for the CAR construct and 41BB for the costimulatory receptor, based on previous results demonstrating a synergy between CD28 and 41BB costimulation.68 69 Using our strategy, the CAR provides signals 1 and 2 on encountering a TAA (tumor-associated antigen) and the engagement of our costimulatory construct with TR2 on MDSCs provides a second costimulatory signal, thereby optimally activating the T cells.
As TR2 is also expressed on normal resting and activated T cells,70 we wanted to address whether there were concerns associated with the safety of using this construct. Despite having increased TR2 expression, neither resting nor activated T cells underwent increased apoptosis when they were cultured in the presence of transgenic T cells expressing TR2.41BB. In line with the earlier reports, these cells were protected due to upregulated levels of the protein cFLIP, a regulator protein that resembles caspase-8 but lacks the protease activity necessary for apoptosis. It is known that competitive binding of cFLIP to Fas-associated death domain can inhibit caspase-8 activation in a dominant negative manner thereby preventing apoptosis.42 43 Hence, TR2.41BB is unlikely to be toxic to normal resting or activated cells. However, certain types of BCs are reported to be sensitive to TR2-mediated apoptosis, and this sensitivity was correlated with reduced levels of cFLIP.71 RNA expression data of TR2 by PAM50 subtype from two publicly available human BC datasets yielded TR2 expression across all BC subtypes with expression being higher in basal versus luminal B subtype among both datasets (online supplemental figure 8). Additionally, it is known that suppressive tumor-associated macrophages (TAMs) in mammary carcinoma express TR2.72 Therefore, TR2.41BB transgenic T cells could have beneficial effects by inducing apoptosis of not only MDSCs but TAMs and BC cells sensitive to TR2-mediated apoptosis.
We observed that transgenic expression of TR2.41BB on T cells induced apoptosis of MDSCs on engagement with TR2 via the extrinsic death receptor pathway.25 Introduction of TR2.41BB on CAR.MUC1 T cells further enhanced MDSC apoptosis and restored the ability of CAR T cells to kill BC cells in vitro and control tumor growth in vivo. We also found that incorporation of the TR2.41BB receptor on CAR.MUC1 T cells was associated with increased T cell expansion and persistence at the tumor site compared with mice receiving MUC1 CAR T cells alone in the presence or absence of MDSCs. Improved persistence of TR2.41BB CAR T cells is likely due to the costimulatory signaling via the 41BB endodomain. Furthermore, the group that received CAR.MUC1.TR2.41BB T cells demonstrated control of primary tumor growth and absence of metastatic potential. To further demonstrate the potential of using this receptor to enhance other CAR T cell approaches, we confirmed that addition of TR2.41BB to CAR.HER2 T cells yielded similar beneficial results in our in vitro and in vivo studies against a HER2+ BC cell line.
Conclusion
In this study, we demonstrate the feasibility of a novel transgenic TR2.41BB receptor to improve CAR T cell therapy against BC. Our data highlight that MDSCs in humans are known to be suppressive and their loss may add further benefit even beyond the dual stimulation achieved in our mouse models by colligation of MUC1 and TR2-specific CARs. The enhanced intratumoral proliferation of CAR T cells in our in vivo models underpin the potential of clinically translating this strategy to treat patients with BC.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
Patient consent for publication not required. Mice were housed and treated in accordance with Baylor College of Medicine Animal Husbandry and Institutional Animal Care and Use Committee guidelines (AN-5639).
Acknowledgments
Our sincere gratitude to Dr Jonathan Lei Thomas for his contribution and help with the RNA transcriptomics data. We would like to thank the Baylor College of medicine histology and pathology core. We are also grateful to Texas Children’s Hospital Small Animal Imaging Facility, Texas Children’s Hospital Flow Cytometry Core Laboratory. We would sincerely like to thank Cancer Prevention and Research Institute of Texas (CPRIT) (Grant ID: RR170024) and the CPRIT Scholar in Cancer Research.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors The study was conceived by VH. VH and SAN designed the experiments. SAN, JJ, and AA performed experiments and SAN analyzed data. VH, AL, JV, SG, and NW characterized and optimized retroviral constructs. VH, RP, and SN optimized in vitro MDSC generation. SN and JJ performed in vivo experiments, and SAN analyzed data. PB provided material and technical help. MK provided technical help and expertise for in vitro and in vivo studies. SN and VH wrote the manuscript. VH, PS, LK, AL, PB, NW, MK, SG, RP and AA critically reviewed the manuscript. All authors read and approved the final manuscript. VH is the guarantor for the above study.
Funding This work was supported by the Cancer Prevention and Research Institute of Texas (CPRIT) (Grant ID: RR170024).
Competing interests VH, SAN, AL, and JV have a pending patent application for work in this manuscript: BLG 20-052. VH has another pending patent: BLG 11-013. AL and JV are co-founders and equity holders of Allovir and Marker Therapeutics. SG has patent applications in the fields of T-cell and/or gene therapy for cancer. He has a research collaboration with TESSA Therapeutics, is a DSMB member of Immatics, and on the scientific advisory board of Tidal. VH and PS have received the Susan Komen Foundation grant. VH has stocks in Allovir and Marker Therapeutics. SG has grants or contracts from the following: NIH, NCI, CIRM, ACGT, ALSF, CKC, Assisi Foundation, ALSAC.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.