Article Text

Abstract
Background Bispecific T-cell engagers are an established therapeutic strategy for the treatment of hematologic malignancies but face several challenges when it comes to their application for the treatment of solid tumors, including on-target off-tumor adverse events. Employing an avidity-mediated specificity gain by introducing an additional binding moiety for the tumor-associated antigen can be achieved using formats with a 2+1 stoichiometry.
Methods Besides biochemical characterization and validation of target cell binding to cancer cells with different HER3 expression, we used in vitro co-culture assays with human peripheral blood mononuclear cells (PBMCs) and HER3-expressing target cells to determine T-cell activation, T-cell proliferation and PBMC-mediated cancer cell lysis of HER3-positive cell lines by the trivalent, bispecific antibodies.
Results In this study, we developed trivalent, bispecific antibodies comprising a silenced Fc region for T-cell retargeting to HER3-expressing tumor cells, combining a bivalent single-chain diabody (scDb) fused to a first heterodimerizing Fc chain with either an Fab or scFv fused to a second heterodimerizing Fc chain. All these HER3-targeting T-cell engagers comprising two binding sites for HER3 and one binding site for CD3 mediated target cell killing. However, format and orientation of binding sites influenced efficacy of target cell binding, target cell-dependent T-cell activation and T-cell-mediated target cell killing. Beneficial effects were seen when the CD3 binding site was located in the scDb moiety. These molecules showed efficient killing of medium HER3-expressing cancer cells with very low induction of cytokine release, while sparing target cells with low or undetectable HER3 expression.
Conclusion Our study demonstrates that these trivalent, bispecific antibodies represent formats with superior interdomain spacing resulting in efficient target cell killing and a potential advantageous safety profile due to very low cytokine release.
- immunotherapy
- immunotherapy
- active
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The ErbB family member HER3 has been reported to play an essential role in cancer progression, and elevated expression has been shown to correlate with worse overall survival.1 2 Furthermore, it has been demonstrated that upregulation of HER3 is an important resistance mechanism on epidermal growth factor receptor (EGFR) and HER2-targeted therapy.3–5 More than two dozen antibodies targeting HER3 are currently investigated in preclinical trials,6 7 mostly interfering with ligand binding and/or receptor dimerization.8 However, there is still no approved treatment targeting HER3. Since monoclonal antibodies9 10 as well as therapeutic approaches involving bispecific antibodies for dual targeting of HER3 and another member of the EGFR family11 have not shown improved therapeutic activity in clinical trials, therapeutic strategies such as HER3-directed antibody-drug conjugates12–14 have been developed, uncoupling the therapeutic activity from receptor signaling.
Major histocompatibility complex (MHC)-independent crosslinking of tumor cells and T-cells by bispecific antibodies represents a rapidly expanding treatment strategy in cancer therapies.15–17 Bispecific T-cell engagers are characterized by simultaneous binding of a tumor-associated antigen (TAA) and, in most cases, the CD3ε chain of the T-cell receptor (TCR)/CD3 complex, leading to the close apposition of target and effector cell resulting in activation of the T-cell. Secretion of cytokines and cytotoxic effector proteins by the T-cell eventually results in killing of the targeted tumor cell. Bispecific T-cell engagers are an established therapeutic strategy for the treatment of hematologic malignancies, for example, blinatumomab, a small bispecific T-cell engager (BiTE) targeting CD19 and CD3, approved for the treatment of acute lymphoblastic leukemia.18 However, bispecific T-cell engagers face several challenges when it comes to their application for the treatment of solid tumors, including the attack of non-tumor cells with low expression level of the TAA and/or systemic cytokine-associated adverse events.16 Recent studies have shown that an avidity-mediated specificity gain through bivalent binding to the TAA can be achieved using novel formats with a 2+1 stoichiometry.19–23 Additionally, formats in the 2+1 stoichiometry circumvent unspecific or non-targeted CD3-crosslinking and T-cell activation by monovalent binding to the trigger molecule CD3 on T-cells.17 24–26 We have recently demonstrated that a small trivalent, bispecific single-chain diabody (scDb)-scFv showed superior binding to target expressing tumor cells translating into more efficient target cell killing by peripheral blood mononuclear cells (PBMCs).27 Small bispecific formats such as BiTEs,28 dual-affinity re-targeting antibodies (DARTs)29 and scDb30 have been reported to mediate tight contacts between target cell and T-cells due to their small size and the short distance between the two binding sites, resulting in efficient T-cell activation. However, their pharmacokinetic properties are characterized by a very short serum half-life and continuous infusion is necessary.31 32
In the present study, trivalent, bispecific Fc-comprising anti-HER3×anti-CD3 antibodies were generated by combining an scDb molecule and an scFv or Fab fragment with a silenced heterodimerizing Fc part (scDb/scFv-Fc, scDb/Fab-Fc).33 Thus, the trivalent bispecific antibodies employ bivalent binding to the TAA HER3 and monovalent binding to CD3, combining the favorable properties of the scDb format with improved pharmacokinetic properties due to the introduced Fc part. We analyzed the effects of the different possible geometries on target cell binding, T-cell-mediated target cell killing and T-cell activation in vitro, demonstrating that these HER3-targeting T-cell engagers efficiently mediate target cell destruction independent of cytokine release, with a superior activity observed for the scDb/scFv-Fc format.
Materials and methods
Materials
Antibodies were purchased from BioLegend (PerCP/Cy5.5 anti-human CD3, 317336; PE anti-human CCR7, 353204; APC anti-human CD45RA; 304112), Miltenyi Biotec (anti-human CD4-VioBlue, 130-097-333; anti-human CD8-PE/Vio770, 130-096-556) or Dianova (goat IgG anti-human IgG (Fc)-RPE, 109-115-098). Human IFN-γ DuoSet ELISA kit (DY285) and Human IL-2 DuoSet ELISA kit (DY202) were obtained from R&D Systems. CellTrace CFSE Cell Proliferation Kit (C34554) was purchased from Thermo Fisher Scientific. FaDu, SKBR-3 and MCF-7 cells were obtained from different sources and cultured as described previously.34 LIM1215 were obtained from Merck KGaA (10092301-1VL) and cultured in RPMI-1640 (Thermo Fisher Scientific, 11875), 10% fetal bovine serum (FBS) (Pan Biotech, P30-3309). Density gradient centrifugation (Lymphocyte Separation Medium 1077, PromoCell, C-44010) was used to isolate human PBMCs from buffy coats of healthy donors (Blood Bank, Klinikum Stuttgart). PBMCs were cultivated in RPMI-1640, 10% FBS.
Antibody production and purification
Human anti-HER3 antibody 3–4334 and a humanized version of anti-CD3 UCHT127 were used to generate the different trivalent, bispecific antibodies. Genes encoding the different polypeptide chains (see figure 1) were cloned into the pSecTagAL1 vector (a modified version of pSecTagA (Invitrogen, Thermo Fisher Scientific, V90020)) and were produced in transiently transfected HEK293-6E cells (NRC Biotechnology Research Institute, Canada) using polyethylenimine (linear, 25 kDa, Sigma-Aldrich, 764604) as described previously.27 After 96 hours of incubation at 37°C and 5% CO2 shaking, supernatants were harvested and proteins were purified by Protein A affinity chromatography (Protein A Sepharose 4 Fast Flow, Pharmacia Biotech, Sweden, 17-0974-03) and subsequent size-exclusion fast protein liquid chromatography (FPLC) on a Superdex 200 10/300 GL column (phosphate-buffered saline (PBS) as mobile phase, 0.5 mL/min flow rate).
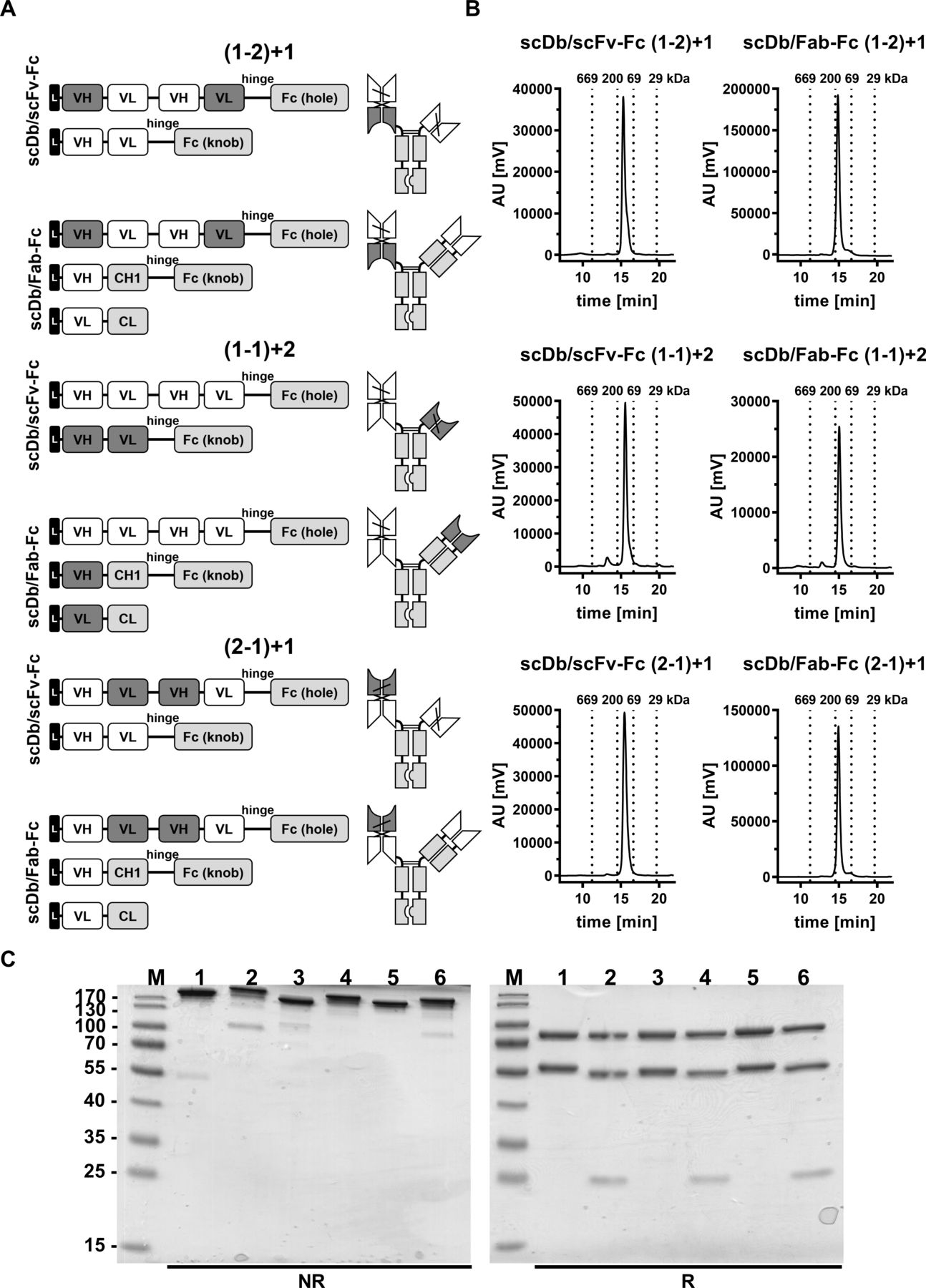
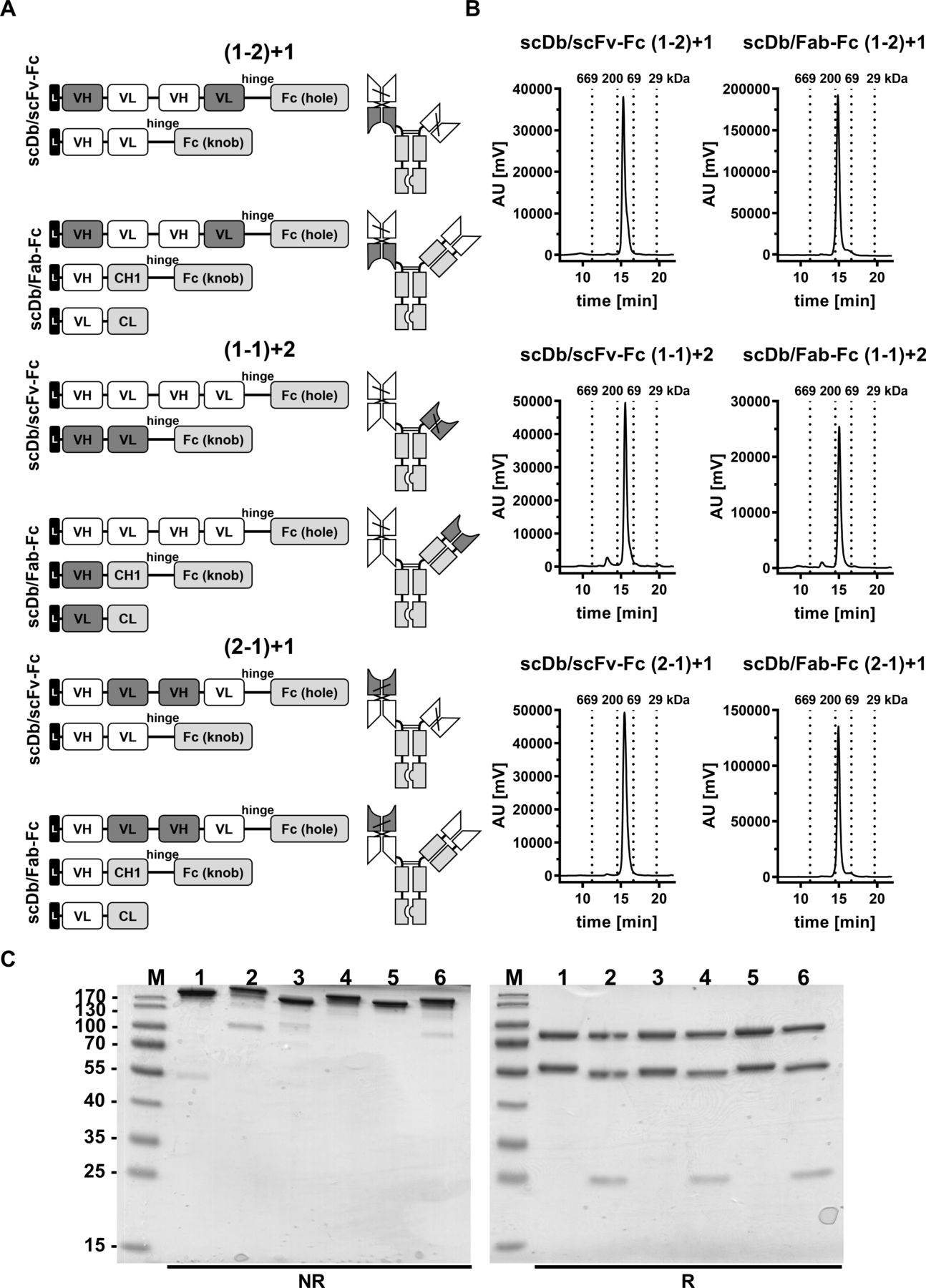
Biochemical characterization of scDb/scFv-Fc and scDb/Fab-Fc variants. (A) Composition and schematic illustration of trivalent, bispecific antibodies. Nomenclature: 1 refers to the HER3 binding site, 2 refers to the CD3 binding site. Numbers in brackets refer to the binding sites of the scDb moiety. Variable domains of HER3 and CD3 are shown in white and dark gray, respectively. Constant domains are shown in light gray. (B) Size-exclusion chromatography by high performance liquid chromatography using a Tosoh TSKgel SuperSW mAb HR column. (C) Sodium dodecylsulfate polyacrylamide gel electrophoreses analysis (12% PAA, 2 µg/lane, Coomassie blue staining) of (1) scDb/scFv-Fc (1-2)+1, (2) scDb/Fab-Fc (1-2)+1, (3) scDb/scFv-Fc (1-1)+2, (4) scDb/Fab-Fc (1-1)+2, (5) scDb/scFv-Fc (2-1)+1 and (6) scDb/Fab-Fc (2-1)+1 under reducing (R) and non-reducing (NR) condition. M, protein marker; AU, arbitrary units; CH1, constant heavy chain domain 1; CL, constant light chain domain; Fc, fragment crystallizable; scDb, single-chain diabody; VH, variable heavy chain domain; VL, variable light chain domain,
Biochemical characterization
SDS-PAGE analysis under reducing and non-reducing conditions stained with Coomassie Brilliant Blue G-250 was used to evaluate purified proteins. Oligomerization state of the proteins was determined using Waters 2695 HPLC and a TSKgel SuperSW mAb HR column (Tosoh Bioscience) at a flow rate of 0.5 mL/min with 0.1 M Na2HPO4/NaH2PO4, 0.1 M Na2SO4, pH 6.7 as mobile phase.27 Thyroglobulin (669 kDa, Sr 8.5 nm), β-amylase (200 kDa, Sr 5.4 nm), bovine serum albumin (67 kDa, Sr 3.55 nm) and carbonic anhydrase (29 kDa, Sr 2.35 nm) were used as reference proteins.
Cell binding
Target cells (1×105 cells/well) were incubated with a serial dilution of the trivalent, bispecific molecules in phosphate buffered saline-fetal bovine serum-sodium azide buffer (PBA; PBS, 2% (v/v) FBS, 0.02% (w/v) sodium azide) at 4°C for 1 hour. A PE-conjugated anti-human Fc antibody (Dianova) was used for detection of bound antibodies. Incubation and washing steps were performed in PBS, 2% FBS and 0.02% sodium azide. Fluorescence was determined using MACSQuant VYB or MACSQuant Analyzer 10 (Miltenyi Biotec) and data were analyzed using FlowJo (Tree Star). Relative median fluorescence intensities (rel. MFI) were calculated as followed: relative MFI = ((MFIsample−(MFIdetection−MFIcells))/MFIcells).
IL-2 / IFN-γ release assay
Previously seeded MCF-7 cells (2×104 cells/well) were incubated with a serial dilution of the trivalent, bispecific antibodies for 15 min at RT and 2×105 PBMCs/well (effector to target cell ratio: 10:1) were added subsequently. After 24 hours (interleukin (IL)-2) or 48 hours (interferon (IFN)-γ), IL-2/IFN-γ concentration in cell-free supernatants of the co-culture assay was determined using sandwich ELISA as described previously.27
T-cell proliferation
PBMCs were stained with carboxyfluorescein diacetate succinimidyl ester (CFSE) at 625 nM/1×106 cells/mL to analyze the proliferative effect on T-cells.27 MCF-7 cells (2×104 cells/well) were incubated with a serial dilution of the trivalent, bispecific antibodies for 15 min at RT and 2×105 CFSE-labeled PBMCs/well were added subsequently. Following an incubation for 6 days at 37°C and 5% CO2, fluorescence-conjugated antibodies directed against cell surface markers were used to label immune cells of interest and their proliferation was determined by multicolor flow cytometry analysis using MACSQuant Analyzer 10 (Miltenyi Biotec) as described previously.27
Cytotoxicity
Target cells (2×104 cells/well) were incubated with a serial dilution of the trivalent, bispecific antibodies for 15 min at RT before effector cells (PBMCs) were added in an effector to target cell ratio of 10:1.27 Supernatants were discarded after 3 days of incubation at 37°C and 5% CO2 and target cell viability was determined using crystal violet staining. Methanol (50 µL/well) was used to solve the staining and optical density measured at 550 nm using the Tecan spark (Tecan) as described previously.27
Statistics
All data are represented as mean±SD. Significances were calculated by GraphPad Prism V.7.0 and results were compared by t-test.
Results
Generation of bispecific antibodies
Trivalent, bispecific antibodies comprising a silenced Fc region and directed against HER3 and CD3 were generated by combining an scDb molecule fused to a first heterodimerizing Fc chain with either an Fab or scFv fragment fused to a second heterodimerizing Fc chain. All molecules were bivalent for HER3 (referred to as ‘1’ in the nomenclature) and monovalent for CD3 (referred to as ‘2’ in the nomenclature). Binding sites were arranged in all three possible configurations (figure 1A).
The trivalent, bispecific antibodies were produced in transiently transfected HEK293-6E cells and purified by protein A affinity chromatography (online supplemental figure S1) followed by a preparative size-exclusion chromatography step (SEC) using FPLC to remove high-molecular and low-molecular weight species (figure 1B). Protein purity was confirmed by SDS-PAGE analysis under reducing and non-reducing conditions, revealing one band under non-reducing conditions showing correct assembly of the antibodies (figure 1C, left panel). SDS-PAGE under reducing conditions revealed two bands at ~80 kDa and ~56 kDa for all scDb/scFv-Fc molecules corresponding to the calculated molecular mass of 80 kDa for the scDb-Fc(hole) chain and 54 kDa for the scFv-Fc(knob) chain (figure 1C, right panel). For the scDb/Fab-Fc molecules, three bands at ~80 kDa, ~55 kDa and ~25 kDa were observed, corresponding to the scDb-Fc(hole) (80 kDa), the heavy chain (knob) (52 kDa) and the light chain (26 kDa) (figure 1C, left panel). In analytical SEC, all trivalent, bispecific antibodies eluted as one major peak confirming purity and integrity of the proteins. A molecular mass of ~134 kDa for the scDb/scFv-Fc molecules and ~157 kDa for the scDb/Fab-Fc molecules was determined in accordance with the calculated molecular mass of 129 kDa and 150 kDa, respectively (figure 1B). In summary, all six trivalent, bispecific antibody configurations could be produced in mammalian cells and assembled into intact proteins with yields between 1.2 and 16 mg/L (online supplemental table S1).
Supplemental material
Binding to HER3-expressing target cells
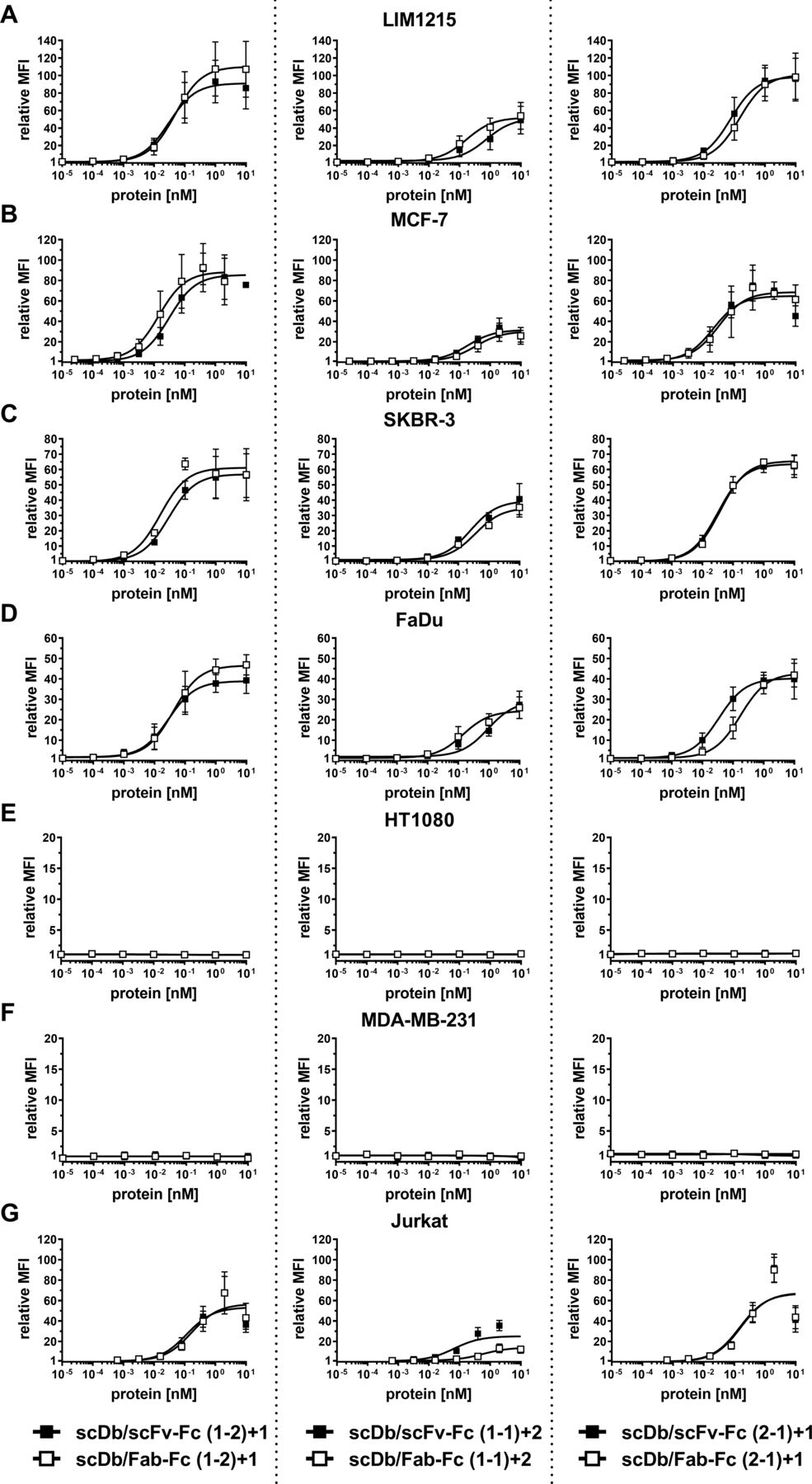
A panel of cell lines expressing varying HER3 receptor levels was used to determine tumor cell binding. On the tumor cell lines LIM1215 (~20,000 HER3/cell), MCF-7 (~18,000 HER3/cell), SKBR-3 (~14,000 HER3/cell) and FaDu (~3000 HER3/cell), the scDb/scFv-Fc molecules in the (1-2)+1 and the (2-1)+1 configuration (see figure 1) showed similar binding with EC50 values in the low picomolar range, while the scDb/scFv-Fc molecule in the (1-1)+2 configuration showed a 10-fold to 15-fold lower binding capacity (figure 2A–D, table 1). A similar trend was observed for the scDb/Fab-Fc molecules, with a twofold to fivefold reduced binding for the (1-1)+2 configuration. No binding for all trivalent, bispecific molecules was observed on the HER3-negative cell lines HT1080 and MDA-MB-231 (figure 2E,F). All trivalent, bispecific molecules showed binding to the CD3-expressing Jurkat cell line. Here, the scDb/Fab-Fc molecules in the (1-1)+2 configuration showed sixfold reduced binding compared with the scDb/scFv-Fc in the same configuration, while all other molecules showed similar binding in the low nanomolar range (figure 2G, table 1). In summary, scDb/Fab-Fc and scDb/scFv-Fc in the (1-2)+1 and (2-1)+1 configuration showed strong binding to HER3-expressing cell lines, while both formats in the (1-1)+2 configuration showed reduced binding.
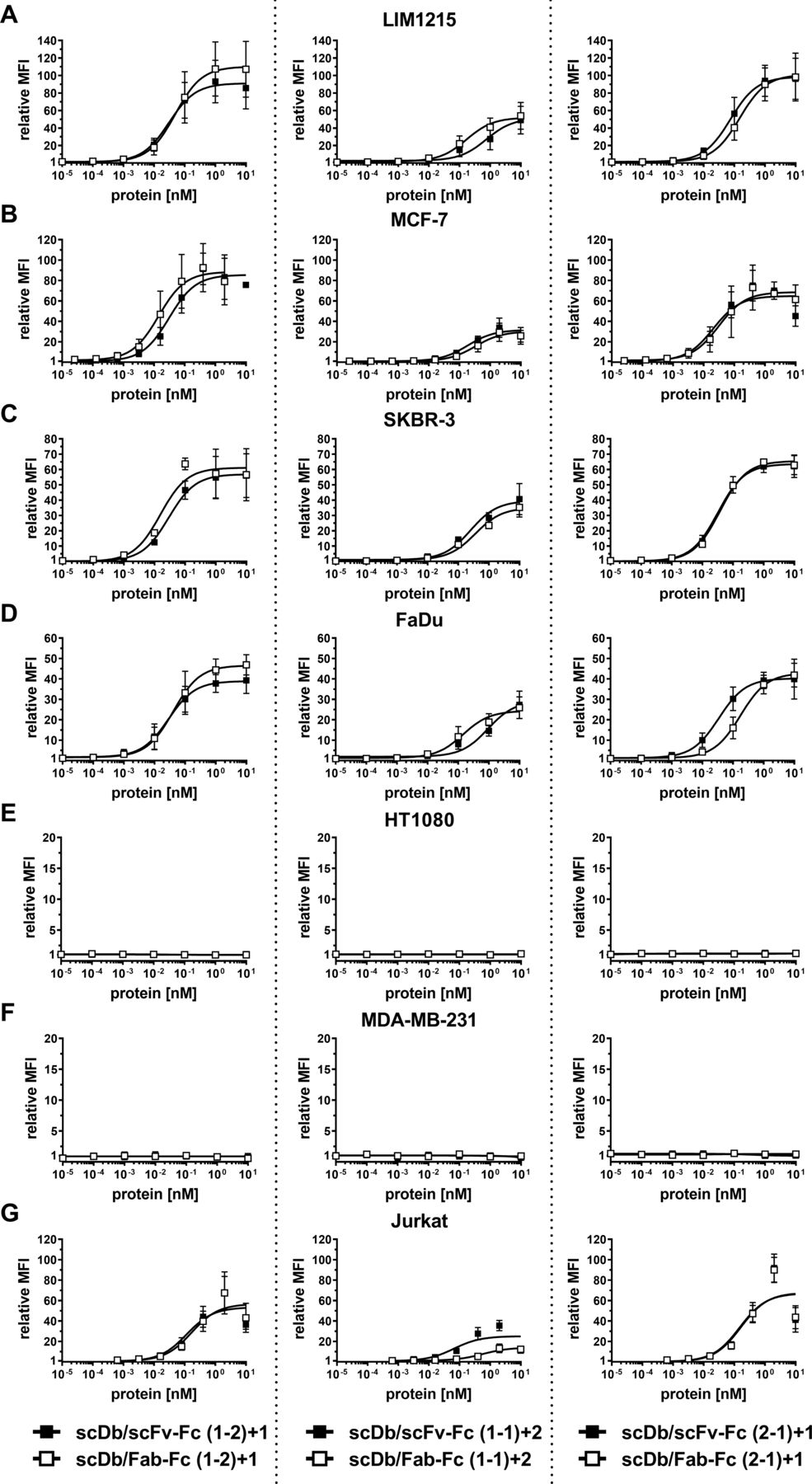
Binding properties of scDb/scFv-Fc and scDb/Fab-Fc variants. Binding to (A) LIM1215, (B) MCF-7, (C) SKBR-3, (D) FaDu, (E) HT1080, (F) MDA-MB-231 and (G) CD3-expressing Jurkat cells was analyzed in flow cytometry. A PE-labeled anti-huFc mAb was used to detect bound protein. Mean±SD, n=3. MFI, median fluorescence intensities; scDb, single-chain diabody.
Binding to HER3-expressing tumor cells of scDb/scFv-Fc and scDb/Fab-Fc variants
Activation of effector T-cells by trivalent, bispecific molecules
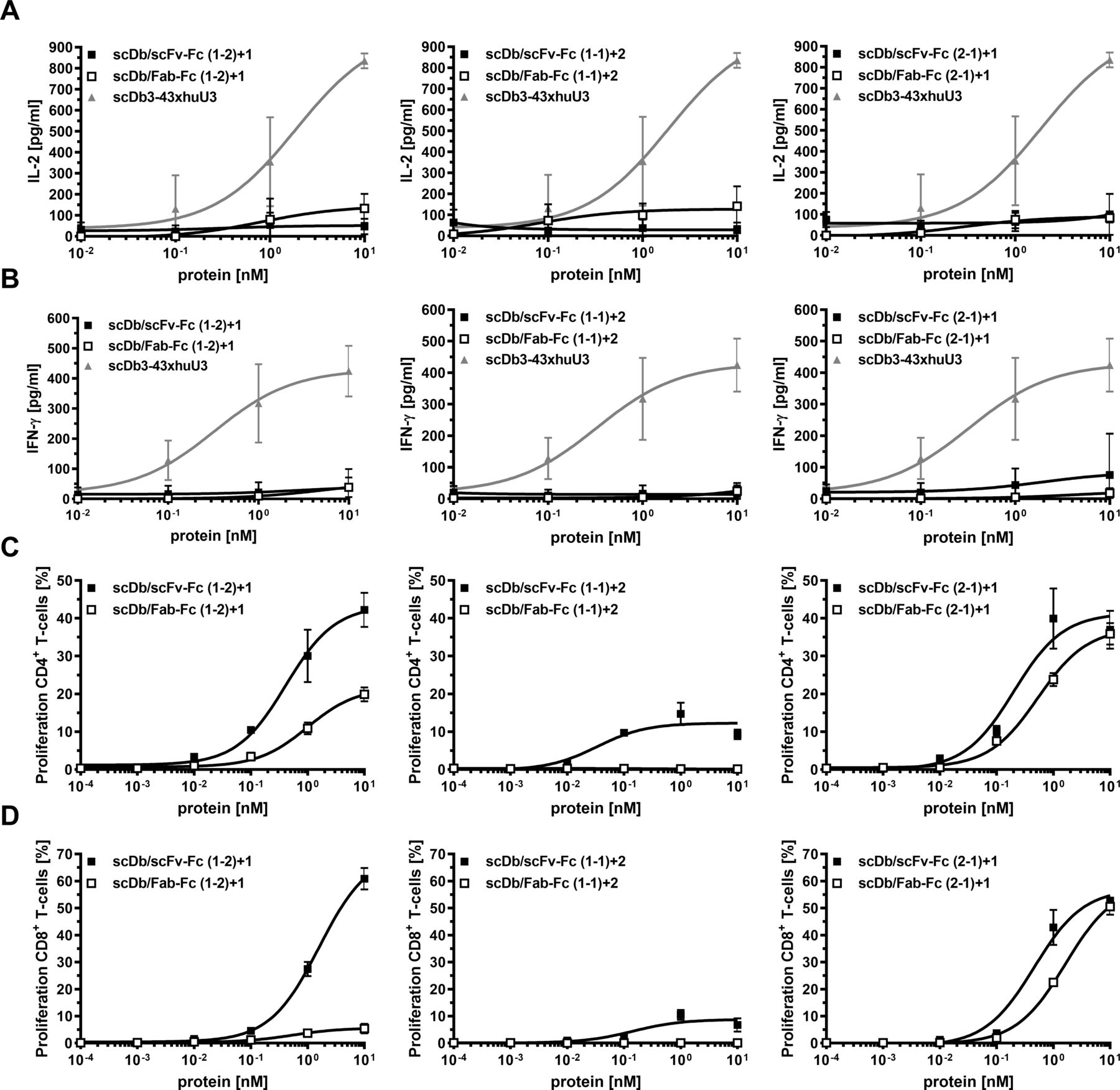
Co-culture assays were used to demonstrate simultaneous binding of the trivalent, bispecific molecules to T-cells and target cells resulting in T-cell activation. First, cytokine release (IL-2 and IFN-γ) was determined. No or only a marginal induction of IL-2 release was observed for all molecules (figure 3A). Similar results were obtained for the IFN-γ release (figure 3B). An scDb directed against HER3 and CD327 was included in these experiments, showing a strong cytokine release in both assays. Next, we investigated proliferation of CD4+ and CD8+ T-cells. All molecules in the scDb/scFv-Fc format mediated proliferation of CD4+ and CD8+ T-cells. Regarding proliferation of CD4+ T-cells, superior activity was observed for the scDb/scFv-Fc format compared with the scDb/Fab-Fc format for all configurations (figure 3C, table 2). In line with this, the scDb/scFv-Fc formats also showed higher proliferative capacity on CD8+ T-cells (figure 3D, table 2). However, the difference between the scDb/scFv-Fc and the scDb/Fab-Fc format was more pronounced for the (1-2)+1 configuration for CD8+ T-cell proliferation, as the scDb/Fab-Fc (1-2)+1 only showed a low induction of proliferation. Additionally, the effect of the trivalent, bispecific molecules on the proliferation and the frequency of T-cell subpopulations was investigated. Treatment with the scDb/scFv-Fc (2-1)+1 and scDb/Fab-Fc (2-1)+1 mainly led to proliferation of central memory (TCM) and effector memory (TEM) CD4+ T-cells (figure 4A, left panel). Similar results were obtained for the scDb/scFv-Fc (1-2)+1. However, the scDb/Fab-Fc (1-2)+1 mainly induced proliferation of naive (TN) CD4+ T-cells. Low to no proliferation was observed for the two molecules in the (1-1)+2 configuration. Similarly, treatment with the scDb/scFv-Fc (1-2)+1, scDb/scFv-Fc (2-1)+1 and scDb/Fab-Fc (2-1)+1 mainly led to the proliferation of TCM and TEM CD8+ T-cells. Regarding the frequency of T-cell subtypes, no differences for the CD4+ T-cells was observed. However, scDb/Fab-Fc (1-2)+1 and scDb/scFv-Fc (1-1)+2 mediated an increased frequency of CD8+ effector T-cells, while all other molecules shifted the population toward CD8+ effector memory T-cells (figure 4B). In summary, the scDb/scFv-Fc molecules were more potent in mediating proliferation of T-cells compared with the scDb/Fab-Fc format with a low or neglectable induction of cytokine release for all trivalent, bispecific molecules.

Activity of scDb/scFv-Fc or scDb/Fab-Fc variants on cytokine release, T-cell activation and proliferation. (A) IL-2 and (B) IFN-γ release mediated by scDb/scFv-Fc or scDb/Fab-Fc variants. Peripheral blood mononuclear cells were co-cultured with MCF-7 cells in the presence of a serial dilution of the scDb/scFv-Fc or scDb/Fab-Fc variants. Cytokine release was determined after 24 hours (IL-2) or 48 hours (IFN-γ) using sandwich ELISA. Proliferation of (C) CD4+ and (D) CD8+ T-cells was measured by carboxyfluorescein diacetate succinimidyl ester dilution in flow cytometry. Mean±SD, n=3. IFN-γ, interferon-γ; IL-2, interleukin 2; scDb, single-chain diabody.

Activity of scDb/scFv-Fc or scDb/Fab-Fc variants on proliferation and composition of T-cell subpopulations. (A) Proliferation of naive (TN, CD45RA+, CCR7+), central memory (TCM, CD45RA−, CCR7+), effector (TE, CD45RA+, CCR7−) and effector memory (TEM, CD45RA−, CCR7−) subpopulations of CD4+ T-cells and CD8+ T-cells was determined by carboxyfluorescein diacetate succinimidyl ester dilution in flow cytometry. (B) Composition of CD4+ and CD8+ T-cell subpopulation was measured in flow cytometry. Mean±SD, n=3. scDb, single-chain diabody.
T-cell proliferation mediated by scDb/scFv-Fc and scDb/Fab-Fc variants
Cancer cell lysis of HER3-positive cell lines by trivalent, bispecific antibodies
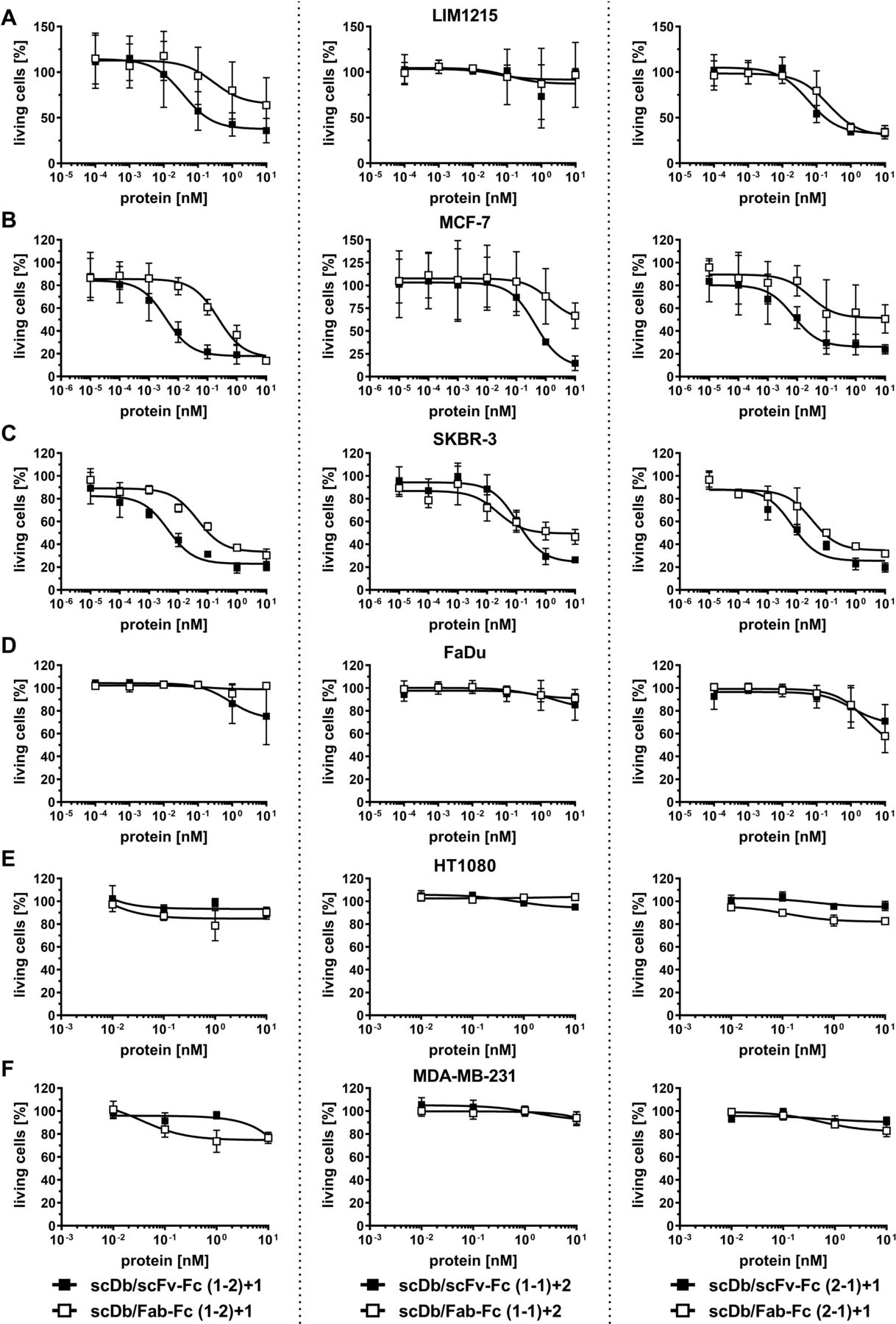
Cell lines with medium (LIM1215, MCF-7, SKBR-3), low (FaDu) and undetectable (HT1080, MDA-MB-231) HER3 expression were used to determine the cytotoxic effects of PBMCs on target cells mediated by the trivalent, bispecific antibodies. All trivalent, bispecific antibodies in the (1-2)+1 and (2-1)+1 configuration were able to redirect unstimulated PBMCs to lyse target cells with medium HER3 expression in a concentration-dependent manner. Regarding potency (EC50 value in cell killing), scDb/scFv-Fc (1-2)+1 and scDb/scFv-Fc (2-1)+1 mediated superior target cell killing compared with the scDb/Fab-Fc in the same configuration on the medium HER3-expressing cell lines (figure 5A–C). In general, the differences between the scDb/scFv-Fc and the scDb/Fab-Fc format were more pronounced in the (1-2)+1 configuration (13-fold to 88-fold) than in the (2-1)+1 configuration (4-fold to 7-fold) (figure 5A–C, table 3). For the trivalent, bispecific molecules in the (1-1)+2 configuration, a 10-fold to 40-fold lower cytotoxic capacity compared with the molecules in the (1-2)+1 and (2-1)+1 configuration was observed. Additionally, predominant target cell killing for the scDb/scFv-Fc (1-1)+2 compared with the respective scDb/Fab-Fc format was only observed on the MCF-7 cell line (fourfold) while no target cell lysis was observed on the LIM1215 cells with the scDb/scFc-Fc (1-1)+1 and scDb/Fab-Fc (1-1)+2. Importantly, only a slight cytotoxic effect was observed on the low expression FaDu cell line for all trivalent, bispecific molecules at the highest concentration (figure 5D) while cell viability of the HER3-negative HT1080 and MDA-MB-231 cells remained unaffected (figure 5E,F). Furthermore, analyzing efficacy (maximum target cell lysis at 10 nM trivalent, bispecific antibody) revealed 50%–80% killing of LIM1215, MCF-7 and SKBR-3 cells for the molecules in the (1-2)+1 configuration. Treatment with the scDb/scFv-Fc in the (1-1)+2 and (2-1)+1 configuration led to lysis of 70%–85% of MCF-7 and SKBR-3 cells while the respective scDb/Fab-Fc molecules only showed 30%–70% killing. On the LIM1215 cell line, the molecules in the (2-1)+1 configuration showed an efficacy of 70% while no killing was observed for the (1-1)+2 antibodies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cytotoxic potential of peripheral blood mononuclear cells (PBMCs) stimulated with scDb/scFv-Fc or scDb/Fab-Fc variants. (A) LIM1215, (B) MCF-7, (C) SKBR-3, (D) FaDu, (E) HT1080 and (F) MDA-MB-231 cells were incubated with a serial dilution of scDb/scFv-Fc or scDb/Fab-Fc variants in the presence of PBMCs in an effector:target cell ratio (E:T) of 10:1. Cell viability was determined using crystal violet staining after 3 days. Mean±SD, n=3. scDb, single-chain diabody.
T-cell mediated killing of tumor cells by scDb/scFv-Fc and scDb/Fab-Fc variants
Discussion
In this study, we report the generation of novel Fc-comprising trivalent, bispecific antibody molecules capable of redirecting T-cells to HER3-expressing cancer cells. Deploying avidity effects by utilizing a 2+1 stoichiometry resulted in potent T-cell-engaging trivalent, bispecific antibodies, allowing to differentiate between cells of medium and low HER3 expression.
A critical step of T-cell-mediated eradication of tumor cells is the formation of a tight immune synapse between tumor cell and T-cell.35 Here, the close proximity of target and effector cell can be facilitated by the small, compact and rigid scDb format enforcing the formation of a cytolytic synapse similar to bispecific T-cell-engaging tandem scFv molecules (BiTE format).36–38 Moreover, our study showed that the molecular geometry and the arrangement of the different binding moieties impact the potency of bispecific antibodies.
For a FynomAb, comparing different geometries by fusing the TAA binders either N-terminally to an anti-CD3 mAb or C-terminally to the Fc resulted in a more than 20-fold higher potency for the N-terminal fusion attributed to the favorable closer proximity between TAA and TCR binding.39 Similarly, others used a monovalent tumor-targeting IgG scaffold and fused an anti-CD3 scFv to the C-terminus of either the light chain of the TAA arm or the non-targeting binder.40 They showed that the fusion to the TAA arm benefits from smaller interdomain spacing resulting in higher in vitro and in vivo potency. In line with this, our data show that positioning the TAA binding sites and the CD3 moiety on opposing arms of the molecule in the (1-1)+2 configuration resulted in more than 30-fold lower potency in target cell killing.
Additionally, superior target cell killing by the scDb/scFv-Fc format might also be attributed to the closer proximity of target cell and T-cell due to the smaller scFv compared with the Fab. Furthermore, prior studies have demonstrated that bispecific antibodies targeting epitopes proximal to the membrane elicited more potent T-cell killing compared with those binding distal epitopes.38 41–43 Targeting membrane-proximal epitopes augments the formation of cytolytic synapses by promoting target clustering and exclusion of the negative regulatory protein CD45 from the immune synapse,42 resulting in increased T-cell activation and higher potency in target cell lysis.38 41 42 44 45 Accordingly, the potency of the trivalent, bispecific antibodies might not only be attributed to the format, configuration and interdomain spacing but also to the anti-HER3 antibody targeting a membrane-proximal epitope.34
While the trivalent, bispecific antibodies investigated in this study showed potent killing of HER3-expressing tumor cells, only very low levels of released cytokines were observed. Growing evidence suggests that cytotoxic activity is uncoupled from released cytokines, supported by the definition of two activation thresholds and the expendability of cytokine release for target cell lysis.46 While a low number of TCR:peptide-MHC complexes is sufficient to trigger T-cell-mediated target cell killing, a high number of complexes is necessary for cytokine secretion. One explanation could be the separated but intertwined signaling pathways of the TCR differentially regulating release of cytokines and lytic effector molecules.47–49 Thus, it has been shown that T-cells maintain their cytolytic activity despite a lack of cytokine release in response to treatment with bispecific T-cell engagers.50
Moreover, anti-CD3 antibodies have been identified, which, when deployed in a bispecific T-cell-engaging molecule, efficiently kill tumor cells accompanied by minimal cytokine release.47 High apparent affinity toward the TAA and a low apparent affinity toward CD3 are not only beneficial for reducing cytokine release but are also important in driving antibody distribution toward the tumor and not to the T-cell compartments, such as bone marrow and lymph nodes.51 Avidity-mediated specificity gain through bivalent binding to HER3 combined with monovalent CD3-binding and improved pharmacokinetic properties due to the introduced Fc part presumably should thus result in beneficial safety properties of HER3-targeting T-cell engagers.
Non-clinical safety and tolerability of CD3 bispecific antibodies can be evaluated using studies in pharmacological relevant species, that is, cynomolgus monkey, which might require to substitute the CD3 binding site with that from a cynomolgous CD3-reactive antibody. Possible side effects include cytokine release syndrome (CRS) and neurotoxicity even at low doses.52 Therefore, safety and tolerability are rather determined based on the anticipated biological effect not adverse effects.53–55
Our results are in accordance with recent findings for an Fc-less trivalent, bispecific scDb-scFv fusion protein directed against HER3 and CD3 that, compared with a bivalent bispecific scDb, mediated a strongly increased cytotoxicity toward medium HER3-expressing target cells, which correlated with increased target cell binding, while cytokine release was unaffected.27 Thus, cytotoxicity mediated by the best (1-2)+1 scDb/scFv-Fc molecule was only reduced 3-fold to 13-fold to that of the scDb-scFv fusion protein described in the previous study, for example, with an EC50 value of 3 nM for the (1-2)+1 scDb/scFv-Fc and 1 nM for scDb-scFv for MCF-7 and 3 nM vs 41 nM for LIM1215, respectively.27 Of note, only medium HER3-expressing cells were lysed by the (1-2)+1 scDb/scFv-Fc molecule, while low HER3-expressing cells remained basically unaffected. This reduction in T-cell-mediated killing by the scDb/scFv-Fc and scDb/Fab-Fc molecules might be caused by the presence of the Fc-region affecting formation of a tight immunological synapse. In another study, the 2+1 stoichiometry in a T-cell-dependent bispecific antibody targeting HER2 and CD3 has been shown to increase tumor selectivity by efficiently targeting HER2-positive target cells, while sparing low HER2-expressing cells.22 Accordingly, the bivalent binding mode of a carcinoembryonic antigen (CEA)-specific T-cell-engaging bispecific antibody (TCB) translated into selective killing of high CEA-expressing cancer cells while sparing normal epithelial cells resulting in a wide safety window.20 Moreover, a correlation between valency and retention time at the tumor was described for a (scFv’)2 further showing that valency directly affects tumor localization.56 Hence, increased avidity can result in increased selectivity, thus allowing to discriminate between target cells expressing low and medium or high target antigens and consequently may lower the risk of on-target off-tumor adverse events.19 43
Although the used HER3 antibody (3–43) is cross-reactive for murine HER3, the CD3 antibody moiety in the trivalent bispecific molecules derived from UCHT1 does not bind to the murine CD3. Therefore, different strategies could be pursued to evaluate in vivo efficacy of bispecific T-cell-engaging antibodies. Immunodeficient mice engrafted with human tumors57 and human immune cells58 are a widely used model to study efficacy of T-cell-retargeting antibodies.20 59–61 In addition to human tumor cell lines, fresh biopsy tissue can be engrafted into immunodeficient mice.62 Inoculating human PBMCs into a immunodeficient mouse leads to vigorous response of the human B-cells and T-cells to murine tissue antigen, causing potentially fatal graft versus host disease (GVHD) and severely limits the ability of the human immune cells in their response to exogenous antigen.63 64 To eliminate the risk of GVHD and to better recapitulate human disease, xenograft models reconstituted with a human hematopoietic system through the engraftment of human cord blood CD34+ hematopoietic stem cells have been developed.65 However, this mouse model is hampered by the maturation of human T-cells in the murine thymus with murine MHC and the lack of human costimulatory molecules.66 67 Compared with cross-species xenograft models, syngeneic mouse models can offer a more natural tumor environment since mice possess an intact immune system. Additionally, syngeneic models enable the possibility of linger and repeated dosing regimens. However, the use of syngeneic mouse models requires the development of surrogate molecules accurately representing their human analog.68 Additionally, murine tumor cells have to be manipulated to express the human antigen bearing the risk of poor engraftment efficacy.
Redirecting T-cells to tumor cells has been reported to be associated with a number of challenges probably limiting or reducing the target cell killing potency, especially in the context of solid tumors. Particularly, disadvantages are associated with the recruitment of naive, exhausted or regulatory T-cells,69 70 while the tissue-resident memory T-cell phenotype (CD8+CD69+CD103+) has been shown to express high levels of cytotoxic molecules and is associated with a good clinical outcome in cancer.71 Central memory T-cells have been shown to mostly traffic to secondary lymphoid tissue and exhibit a high proliferative capacity,72 whereas T-cells of the effector memory subtype preferentially localize in non-lymphoid tissue and demonstrated a rapid development of effector functions.73 Xu et al74 demonstrated that the frequency of a T-memory stem cell phenotype in the bulk population of CAR-T-cells positively correlates with their subsequent in vivo expansion. In line with this, treatment with the trivalent, bispecific antibodies used in this study mainly led to the proliferation of central memory (CD45RA−CCR7+) and effector memory (CD45RA−CCR7−) CD8+ T-cells. Additionally, the CD8+ T-cell population shifted toward effector or effector memory T-cells, whereas only low proliferation of naive CD8+ T-cells was observed.
The current study demonstrates that the format, geometry and orientation of binding sites influence the potency of bispecific antibodies for T-cell retargeting. For the effective formation of immune synapses resulting in T-cell-mediated tumor cell lysis, the scDb/scFv-Fc format with small interdomain spacing in the (1-2)+1 and (2-1)+1 configuration showed efficient killing of medium HER3-expressing cancer cells while sparing target cells with low HER3 expression with an additional preferable safety profile due to low cytokine secretion.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Acknowledgments
We would like to thank Nadine Heidel and Sabine Münkel for excellent technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors NA, LK and OS performed cloning, protein expression and purification, biochemical analysis, binding studies and bioactivity assays. NA, OS and RK analyzed and interpreted the data. NA, OS and RK were responsible for experimental design and supervised the work. NA, OS and RK wrote the manuscript. All authors read and approved the final manuscript. RK acts as guarantor.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests NA, OS and RK are named inventors on patent applications covering the HER3 antibody and scDb-based T-cell engagers.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.